
Abstract
Key message
We characterized the Ammopiptanthus nanus transcriptome, identified a large number of orthologs and predicted the divergence time between A. nanus and A. mongolicus, the only two species in genus Ammopiptanthus.
Abstract
Plants in the genus Ammopiptanthus are relic trees of the Tertiary period that have survived dramatic climate change in the Central Asia dryland. Currently, the only two remaining Ammopiptanthus species, A. nanus and A. mongolicus, are distributed in distinct arid regions. Comparative analysis between the two species will facilitate our understanding of speciation and their different adaptation mechanisms. However, lack of nucleotide sequence data hindered the functional genomics research on A. nanus and comparative transcriptome analysis between the two Ammopiptanthus species. Here, we utilized RNA-Seq to characterize the A. nanus transcriptome and then performed a comparative transcriptome analysis between the two species. A total of 69,990 transcripts were assembled from 93 million clean reads of A. nanus, and more than 78% of these transcripts were annotated. A batch of miRNA precursors was predicted and thousands of potential simple sequence repeats were identified. A total of 6606 pairs of putative orthologs with full-length coding sequences were obtained using reciprocal blast. Based on the rates of nonsynonymous (Ka) to synonymous (Ks) substitutions, the divergence time between A. nanus and A. mongolicus was calculated to be approximately 0.70 ± 0.62 Mya. We identified 15 rapidly evolved genes that likely experienced positive selection; these genes are mainly involved in plant defense, epigenetic regulation, and plant development. Our results provide a comprehensive transcriptomic dataset for A. nanus and advance understanding on the evolutionary adaptation of the two species to their individual arid habitats.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Approximately one-third of the Earth’s land surface is desert. In China, the desert regions occupy 22% of total land area (Ge et al. 2005). With the ongoing global warming, desertification has been—and will continue to be—a global issue that threatens human lives and activities worldwide (Wang and Liu 2013). Conservation of the genetic resources of endemic desert plants is an essential part of global efforts to curb desertification. Ammopiptanthus, the only genus with an evergreen broad-leaf habit in the desert and arid regions of Central Asia, plays an important role in maintaining desert ecosystems. Understanding the molecular mechanism of adaptation to desert environments in plants in the genus Ammopiptanthus would be helpful in protecting the environment of the desert regions.
The genus Ammopiptanthus (Leguminosae) comprises two species with high levels of morphological similarity: A. mongolicus (Maxim. ex Kom.) Cheng f. and A. nanus (M. Pop.) Cheng f. Both A. nanus and A. mongolicus grow in dry desert areas and are narrowly distributed species. The habitats of these two Ammopiptanthus plants are located in regions with the same latitude (39–40′), but approximately 2500 km apart. Both habitats are stony and/or sandy deserts where the annual precipitation ranges from 100 to 160 mm, and have similar annual average temperatures (7–8 °C). However, their habitats have different annual average sunshine (A. mongolicus: 3300–3800 h; A. nanus: 2400–2600 h) and different altitude levels (A. mongolicus: 1000–1200 m; A. nanus: 2100–2700 m). The two Ammopiptanthus plants are relic trees of the Tertiary period. According to fossil evidence, Central Asia, the habitat of the ancestral species of genus Ammopiptanthus, has undergone dramatic climate change over time. As the weather became drier and colder, the overwhelming majority of broad-leaf trees became extinct, and the distribution range of the genus Ammopiptanthus became smaller, probably resulting in habitat fragmentation that may have promoted the speciation of the two Ammopiptanthus species. Thus, comparative analysis between the two Ammopiptanthus species will facilitate our understanding of differential habitat adaptation and speciation.
Comparison of the transcriptomes of closely related species promotes the understanding of how genomic variation translates into morphological variation and helps to identify the associated selective pressures. The advent of RNA-Seq (whole-transcriptome shotgun sequencing using next generation sequencing technology) has opened the door to unprecedented large-scale and cross-species comparative transcriptome analyses (Wang et al. 2009; Necsulea and Kaessmann 2014). RNA-Seq allows for the comparison of the variation in the coding sequences (CDS) of ortholog pairs expressed in different species, subsequently leading to the identification of the rapidly evolved genes and providing clues to explain the evolution of divergent phenotypes. Such a strategy has been successfully adopted in the comparative analysis of frogs (Yang et al. 2012), lizards (Yang et al. 2014, 2015a), primroses (Zhang et al. 2013a), holly mangrove (Yang et al. 2015b), poplars (Zhang et al. 2013b), and ramies (Cheng et al. 2015).
Owing to the high academic value and ecological importance of the Ammopiptanthus plants, many studies have been conducted on their physiological and anatomical traits (Liu and Qiu 1982), drought resistance mechanisms (Wang et al. 2007; Xu et al. 2002), characterization of the putative stress tolerance-related genes (Chen et al. 2011; Wei et al. 2011, 2012a, b), and analysis of gene and miRNA profiling under drought and cold stress conditions using next generation sequencing (Zhou et al. 2012; Gao et al. 2015, 2016; Wu et al. 2014; Pang et al. 2013). However, almost all of these studies were carried out in A. mongolicus. Few studies were performed in A. nanus, despite the ecological significance of A. nanus in Central Asia. In addition, although the genetic diversity and geographic differentiation between the two Ammopiptanthus species and within each species were investigated in a previous study (Ge et al. 2005), a comparative study at the transcriptome level has not yet been reported, possibly due to the lack of A. nanus nucleotide data available in the public databases.
In the present study, we established a high-quality transcriptome dataset for A. nanus using the Illumina sequencing platform, and a large number of protein-coding genes and miRNA precursors were annotated from the assembled transcripts. A batch of orthologous genes that might be under positive selection were identified by comparing their transcriptomes. These results provide a comprehensive genomic resource for functional genomics research on A. nanus in the future, and enrich the current knowledge about the origin of the two Ammopiptanthus species and their adaptive evolution to their habitats in Central Asia.
Materials and methods
Ethics statement
The A. nanus seed collection and research activities were scientifically conducted under permits issued by Wuqia Forestry Bureau. The experimental procedures were approved by the Ethics Committee for Plant Experiments of Minzu University of China and the State Forestry Administration, China.
Plant materials
Ammopiptanthus nanus seeds, collected from the desert region in Wuqia County, Xijiang Autonomous Region, China, were surface-sterilized with ethanol and soaked in water for 48 h at 25 °C. The surface-sterilized seeds were sown in commercial pots (9-cm diameter) containing vermiculite and perlite (1:1, w/w), in a greenhouse at approximately 25 °C and 35% relative humidity under a photosynthetic photon flux density of 120 μmol m− 2 s− 1 with a photoperiod of 16 h light and 8 h dark. The seedlings were watered at 3-day intervals with half-strength Hoagland’s solution. 2–3 week after germination, roots and leaves of the seedlings were harvested and used for Illumina transcriptome sequencing.
Transcriptome sequencing
Total RNA was extracted using TRIzol reagent (Invitrogen, Burlington, Canada) according to the manufacturer’s protocol. The quality and purity of RNA samples were assessed using the RNA 6000 Nano LabChip kit and a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, USA) with the RNA integrity number (RIN) > 7.0. Magnetic beads with Oligo (dT) (Invitrogen, Burlington, Canada) were used to isolate mRNA from 10 µg total RNA. Following purification, the mRNA was cleaved into short fragments with divalent cations under elevated temperature. Then, the cleaved RNA fragments were reverse transcribed to create the final cDNA library in accordance with the protocol for the RNA-Seq sample preparation kit (Illumina, San Diego, USA). The average insert size for the paired-end libraries was 300 bp (± 50 bp). We performed the paired-end sequencing on an Illumina HiSeq 2000 (LC Sciences, USA) following the vendor’s protocol.
De novo transcriptome assembly and annotation
The sequenced raw reads were subjected to a quality check using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and then cleaned by removing reads with adaptor sequences, reads with unknown nucleotides larger than 5%, and low quality reads (reads with more than 15% of bases with Phred quality score of ≤ 20). Sequencing reads were de novo assembled using Trinity software (Grabherr et al. 2011) under default parameters and with a k-mer size of 25. Assembly quality was assessed by length distribution analysis with custom Perl scripts. N50 number, average length, max length, and contig number during different length intervals were all calculated. Contigs shorter than 200 bp were discarded from all assemblies. The assembled transcriptome sequences were named assembled transcripts.
All assembled transcripts were compared with the NCBI Nr, Swiss-Prot, Pfam, KEGG, COG, and GO databases (Ashburner et al. 2000) using BLASTX with a typical cut-off e-value of 1e − 5 to search for homologs. The putative CDS for each transcript were predicted using GENSCAN software (Burge and Karlin 1997). The putative transcription factor genes were identified by aligning the assembled transcripts to the peptide sequences of Glycine max TFs in PlantTFDB (http://planttfdb.cbi.pku.edu.cn/). A BLASTP search was performed, and an e-value of 1e − 5 was used as the threshold. For miRNA precursor prediction, the assembled transcripts were aligned to the stem-loop precursors of miRNAs in A. mongolicus and G. max with a cut-off e-value of 1e − 10, qualified secondary structures of miRNA precursors were determined according to the criteria defined in miRBase (Kozomara and Griffiths-Jones 2011).
Assessment of transcriptome assembly
Three assessment tools were used to estimate the quality and completeness of our transcriptome assembly. First, the Benchmarking Universal Single Copy Orthologs (BUSCO) v2 (Simão et al. 2015) were used to identify universal single copy orthologs (USCOs) in our transcriptome assembly, as a measure of the completeness. BUSCO analysis was performed using the plant dataset (embryophyta_odb9, creation date: 2016-11-01). Second, we used TBLASTX to query the list of 357 eukaryotic UCO protein sequences from Arabidopsis (http://compgenomics.ucdavis.edu/compositae_reference.php) (Kozik et al. 2008) with an e-value threshold of 1e − 10. BLAST results were parsed to determine the number of A. nanus transcripts that showed a positive hit to the UCO sequences with amino acid alignments of at least 30 residues. Third, the transcriptome coverage was assessed by comparing the A. nanus transcripts with the PlantTribes database (Wall et al. 2008). In this analysis, 959 shared single copy tribes from Arabidopsis thaliana, Populus trichocarpa, Vitis vinifera, and Oryza sativa (APVO) (Duarte et al. 2010) were compared with the A. nanus transcripts using TBLASTX and an e-value cut-off of 1e − 6.
EST-SSR marker identification
MISA (http://pgrc.ipk-gatersleben.de/misa/) was used to identify the potent simple sequence repeats from expressed sequence tag (EST-SSR) markers in all unique sequences. Dinucleotide repeats of more than six times, and trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeats of more than five times were considered the search criteria for simple sequence repeats (SSRs) in the MISA script.
Identification of orthologous contigs and estimation of substitution rates
We employed the reciprocal best-hit method in BLASTN to identify potentially orthologous sequences between A. nanus and A. mongolicus (Altenhoff and Dessimoz 2009; Zhang et al. 2013a). Pairs of transcripts that were each other’s best hit (e-value < 10− 10) and not less than 200 bp were retained. To obtain pairs of orthologous genes with higher confidence, the putative pairs of orthologous unigenes were further aligned against the G. max protein dataset. If the two transcripts in a putative ortholog pair mapped to different proteins via BLASTX, they were removed. The transcriptome sequences of A. mongolicus used for ortholog pair screening were assembled in a previous study (Gao et al. 2016).
To identify genes undergoing selection, we estimated the rates of nonsynonymous and synonymous substitution between A. nanus and A. mongolicus. The KaKs_Calculator was employed to estimate Ka, Ks, and Ka/Ks using the YN method (Zhang et al. 2006). A Ka/Ks ratio of 0.5 was used as the cut-off to identify genes under positive selection and ortholog pairs with Ks > 0.1 were excluded to avoid potential paralogs (Elmer et al. 2010). When genes with Ka/Ks > 0.5 were found, their sequences were checked manually to guarantee their accuracy. For validation, five positively selected genes (PSGs) across two species were randomly selected and their CDS were sequenced to confirm the corresponding mutation sites. Divergence in the CDS sites between A. nanus and A. mongolicus was calculated using the K2P model (Graur and Li 2000; Zhang et al. 2013a).
Results
Sequencing and assembly of the A. nanus transcriptome
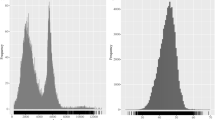
To obtain a high-coverage de novo transcriptome assembly of A. nanus, we constructed two cDNA libraries from the leaves and roots of A. nanus seedlings and performed transcriptome sequencing using the Illumina HiSeq2000 platform. In total, approximately 41 and 53 million paired-end 125 bp reads were generated from leaves and roots, respectively (Table 1). All read data were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the Short Read Archive (SRA) under the accession number SRR2886792. After trimming adapters and filtering out low quality reads, more than 93 million reads were obtained in total. These reads were assembled, using Trinity software, into 69,990 transcripts with a mean size of 1353 bp (Table 2). The size distribution of the assembled transcripts is shown in Fig. 1.

Length distribution of the assembled transcripts
Functional annotation of the A. nanus transcriptome sequences
For functional annotation, the assembled transcripts were aligned against the Nr (non-redundant protein sequences in NCBI), Swiss-Prot, Pfam, Kyoto encyclopedia of genes and genomes database (KEGG), clusters of orthologous groups of proteins (COG), and gene ontology (GO) databases using the BLASTX algorithm. A typical cut-off value of e < 10− 5 was used. As a result, 54,916 assembled transcript sequences (78.46%) were successfully annotated by at least one database (Table 3). The mapping rates of the assembled transcripts against the Swiss-Prot and Nr protein databases were 50.38 and 78.07%, respectively. The results indicated that the assembly represented a substantial portion of the entire A. nanus transcriptome, and the majority of the assembled transcripts were protein-coding genes.
The annotation rate of the assembled transcripts (the number of annotated transcripts vs. the total number of assembled transcripts) was positively correlated with the length of the assembled transcripts. More than 92% of the assembled transcripts over 700 bp in length had homology matches in the Nr database, whereas less than 50% of the assembled transcripts shorter than 400 bp had significant matches (Fig. 2). The low percentage of BLAST hits for the short assembled transcripts may be partially due to the lack of a known conserved functional domain. Alternatively, these transcripts might represent non-coding RNAs.

The annotation rates of the assembled transcripts in different length distribution ranges
GO assignments were used to classify the functions of the A. nanus transcripts. Based on sequence homology, the 69,990 assembled transcripts were assigned GO terms. The annotated GO terms were classified into 50 functional groups that were distributed under the three main categories, including biological process, cellular component, and molecular function (Fig. 3). In the biological process category, “regulation of transcription”, “DNA-dependent”, and “proteolysis” were the major GO terms. In the cellular component category, “integral to membrane”, “nucleus”, and “plasma membrane” were the top three well-represented GO terms. Within the molecular function category, “ATP binding” “protein serine/threonine phosphatase complex”, and “protein binding” were the top three GO terms.

GO classification of the assembled transcripts
The assembled transcripts were also aligned to the COG database to predict and classify their possible functions. A total of 46,837 out of 69,990 sequences had COG functional classifications, which were grouped into 25 functional categories (Fig. 4). The top three categories were “General function prediction only” (6198 transcripts, group R), “Signal transduction mechanisms” (3956 transcripts, group T), and “Posttranslational modification, protein turnover, and chaperones” (3026 transcripts, group O). “Cell motility” (9 transcripts, group N), “Extracellular structures” (75 transcripts, group W) and “Nuclear structure” (129 transcripts, group Y) were the smallest COG categories.

COG function classification of the assembled transcripts
We further performed a systematic analysis of gene function by assigning the assembled transcripts to biochemical pathways in the KEGG database. A total of 23,653 transcripts were annotated in the KEGG database (Supplementary Data 1), which were associated with 2818 KO entries, 36 KEGG level-two pathways, and 269 KEGG pathway terms (Fig. 5). Among these pathways, “Starch and sucrose metabolism” was the largest group of transcripts (647 transcripts, ko01100), followed by “Ribosome” (644 transcripts, ko03010), “Purine metabolism” (634 transcripts, ko00230), “Endocytosis” (506 transcripts, ko04144), “Glycolysis/Gluconeogenesis” (452 transcripts, ko00010), “MAPK signaling pathway” (451 transcripts, ko04010), “Ubiquitin mediated proteolysis” (432 transcripts, ko04120), and “Phenylpropanoid biosynthesis” (391 transcripts, ko0090). The top six level-two pathways were “Carbohydrate Metabolism”, “Amino Acid Metabolism”, “Signal Transduction”, “Transport and Catabolism”, “Cell Growth and Death”, and “Lipid Metabolism”.

KEGG function classification of the assembled transcripts
Assembly quality assessment and sequence conservation of the A. nanus transcripts
We first assessed the quality and completeness of our assembled transcripts by comparing their sequences to a core set of plant genes using BUSCO. The result showed that, of 1440 BUSCO groups searched, 83.47% were “complete”, 5.21% were “fragmented”, and the remaining 11.32% were “missing”. Ultraconserved orthologs (UCOs, available at http://compgenomics.ucdavis.edu/compositae_reference.php) and APVO sequences represent a highly conserved set of genes and have been widely used as important indicators for gene detection and sampling breadth. The two different assessment tools were used to estimate the transcriptome coverage of our assembly using the TBLASTX algorithm. We identified all 357 (100%) UCOs from the assembled transcripts. We detected 931 (97.1%) of the 959 shared single copy tribes represented in the PlantTribes database.
To evaluate the amino acid sequence conservation of the A. nanus transcripts, we analyzed the species distribution of the assembled transcripts by aligning the assembled transcripts against the Nr database. For the top BLAST hit species distribution of aligned transcripts in the Nr database, 47.53% were matched with sequences from G. max, followed by Cicer arietinum (15.41%), Phaseolus vulgaris (12.49%), Medicago truncatula (10.35%), and Lotus japonicas (2.78%) (Fig. 6). As expected, more than 88.57% of the distinct sequences in the assembled transcripts had top matches (first hit) with the protein sequences from the plants in the family Leguminosae.

Species distribution of the assembled transcripts. The species distribution of the assembled transcripts was determined by aligning against the Nr database and the top BLAST hit species were recorded and used for species distribution statistics
Prediction of CDS, transcription factor, and miRNA precursors
We utilized Genscan to identify the CDS for each assembled transcript, and 45,677 CDS (65.26% of 69,990 assembled transcripts) were predicted. Of these, 16,055 (35.15%) CDS were longer than 200 amino acids, and the 1755 longest unigenes had lengths over 1000 amino acids. All CDS were translated into peptide sequences according to the standard codon table.
Transcription factors (TFs) play important functions in gene expression regulation during plant growth, development and responses to environmental factors. We identified 3899 (8.54% of the 45,677 CDS) G. max TF analogs. The top four abundant TF families were bHLH (570 numbers), WRKY (513 numbers), MYB_related (272 numbers), and C3H (272 numbers).
Some of the assembled transcripts may represent the stem-loop precursor sequences of miRNAs. By aligning to the stem-loop precursors of A. mongolicus and G. max, a total of 71 predicted miRNA precursors were identified (Supplementary Data 2) most of which may be the orthologs of miRNA genes of A. mongolicus. Among these, 24 aligned to the precursors of conserved miRNAs identified in A. mongolicus, 16 aligned to that of the non-conserved miRNA identified with high confidence, and 31 aligned to that of the non-conserved miRNA candidates identified in A. mongolicus. Homologs of many stem-loop precursors of A. mongolicus were not identified from the assembled transcripts of A. nanus possibly due to the spatio-temporal expression of miRNA genes.
Orthologous contigs and substitution rates between the two Ammopiptanthus species
Orthologous gene identification is a necessary step in comparative genomics analyses. We identified 29,490 pairs of putative orthologous transcripts between A. nanus and A. mongolicus. After incorporating the G. max peptide sequences, 6,606 pairs of putative orthologs with full-length CDS were obtained. This reduction in ortholog number was caused mainly by the exclusion of the relatively young orthologs specific to genus Ammopiptanthus, which were discarded as having low similarity to G. max genes.
Among the 6606 pairs of orthologs between A. nanus and A. mongolicus, 430 pairs were identical, 1583 pairs had only synonymous or nonsynonymous substitutions, and 4593 pairs had both types of substitutions, for which the Ka/Ks ratio were calculated. The mean values of Ka (the number of nonsynonymous substitutions per non synonymous site), Ks (the number of synonymous substitutions per synonymous site), and the Ka/Ks ratio of all orthologous pairs were 0.0065 ± 0.0078, 0.0209 ± 0.0185, and 0.4023 ± 0.3867, respectively.
The peak Ks for orthologous transcript pairs can be used to estimate the times of divergence between closely related species. According to previously described methods, the age of the speciation event between A. nanus and A. mongolicus was calculated to be approximately 0.70 ± 0.62 Mya, which falls in the later stage of middle Pleistocene.
Identification of the rapidly evolved genes that might be under positive selection between the two Ammopiptanthus species
The Ka/Ks ratio in the protein-coding gene was used to estimate the selective pressure. Of the 4593 ortholog pairs, 15 pairs with a Ka/Ks value > 0.5 and a P value < 0.05 were identified, and these rapidly evolved genes were considered likely to have experienced or be experiencing positive selection (candidate positively selected genes, PSGs) (Table 4). For validation, the coding regions of five randomly selected PSGs across two species were isolated by PCR, and the mutations were confirmed by sequencing. The PSGs were aligned against Arabidopsis peptide database (TAIR10, https://www.arabidopsis.org) to reveal the associated biological processes, and the corresponding Arabidopsis homologs were determined. As shown in Table 4, the rapidly evolved genes are involved in multiple biological processes such as plant defense response, epigenetic regulation, development, intracellular transport, and protein folding.
Identification and comparison of the EST-SSRs in the transcriptome sequences of A. nanus and A. mongolicus
After screening for EST-SSRs from the 69,990 assembled transcripts of A. nanus obtained in the present study and the transcriptome dataset of A. mongolicus assembled in a previous study (Gao et al. 2016), 12,064 and 15,034 SSRs distributed in 10,021 and 12,418 sequences were identified from the transcriptome sequences of A. nanus and A. mongolicus, respectively (Table 5). The EST-SSR frequency was 17.24 and 9.58%, and the distribution density was 0.12 and 0.14 per kb in the transcriptome sequences of A. nanus and A. mongolicus, respectively. Based on the repeat motifs, all SSR loci were divided into mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide. For the both species, the most abundant repeat motif was trinucleotide, followed by dinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeat units (Table 5). The top three classes of sequence repeat in the transcriptome sequences of A. nanus were AG/CT, AAG/CTT, and AC/GT, while the top three categories of sequence repeat in the A. mongolicus transcriptome dataset were AG/CT, AAG/CTT, and AT/AT (Fig. 7).

Comparison of the distribution of repeat motifs of SSRs between two Ammopiptanthus species. Grey bar, A. nanus; black bar, A. mongolicus
We further search for SSRs in the putative orthologous transcripts, and found more than 4600 SSRs distributed among 3779 pairs of orthologous transcripts (Table 5, Supplementary Data 3). The distribution of repeat motif of SSRs in orthologous transcripts was similar in the two Ammopiptanthus species (Fig. 8), indicating that a great deal of EST-SSRs identified in orthologous transcripts can used for the two Ammopiptanthus species. At the same time, some species-specific SSRs were also found. For example, Hexanucleotide repeats were only identified in A. mongolicus, thus the 24 Hexanucleotide repeats may be species-specific SSRs (Table 5, Supplementary Data 4). In addition, we found different SSRs in the same pair of orthologous transcripts, and these SSR markers may also be species-specific (Supplementary Data 5). Although the validity of the putative species-specific SSRs is still to be confirmed by further analysis and experiments, our results provided important data which might contribute to marker development for further population-level studies between the two Ammopiptanthus species.

Comparison of the distribution of repeat motifs of SSRs in the putative orthologous transcripts between two Ammopiptanthus species. Grey bar, A. nanus; black bar, A. mongolicus
Discussion
Transcriptome sequencing, De novo assembly, and annotation for A. nanus
Illumina-based transcriptome sequencing has been demonstrated to be an efficient and cost-effective method to obtain transcriptome data and identify genetic markers. In the present study, a general transcriptome dataset of A. nanus was established for the first time via Illumina next generation sequencing and assembly. The N50 length of unigenes was 1662 bp and the average length was 1013 bp. These results are comparable to those of recently published xerophyte transcriptome studies, such as for Haloxylon ammodendron (N50 = 1345 bp, average length = 728 bp) (Long et al. 2014), Reaumuria soongorica (N50 = 1109 bp, average length = 677 bp) (Shi et al. 2013), and Cynanchum komarovii (N50 = 862 bp, average length = 604 bp) (Ma et al. 2015).
The quality and completeness of transcriptome assembly are very important to further transcriptome analysis. Assembly evaluation measures can be classified into two categories: reference-based and reference-free. Besides the two commonly used reference-free measures, the median unigene length and N50 length of contigs, three reference-based assembly evaluations were also adopted to assessed the quality and completeness of our assembled transcripts. We identified all 357 (100%) UCOs and 931 (97.1%) of the 959 shared single copy tribes represented in the PlantTribes database. As an emerging tool for estimating the completeness of genome sequences, BUSCO has been used for the assessment of plant transcriptome completeness. In the present study, although less than 89% BUSCO groups (“complete” and “fragmented”) were found in the assembled transcriptome sequences in A. nanus, the overall BUSCO percentages are greater than that of Spinacia tetrandra and Arundo donax, which were reported in two recent transcriptome studies (Evangelistella et al. 2017; Xu et al. 2015). Together, the results of the functional annotation and assessment of transcriptome coverage demonstrated that our transcriptome sequences represented a substantial portion of A. nanus protein-coding genes. Considering that A. nanus genomic information is not yet available in the public databases, the large batch transcriptome sequences obtained in the present study not only provide a good start for the elucidation of the molecular mechanism underlying the stress tolerance of A. nanus, but also enable the comparative transcriptome analysis between A. nanus and A. mongolicus.
Divergence time between A. nanus and A. mongolicus
Plant species in genus Ammopiptanthus are relic trees of the Tertiary period. It has been hypothesized that the ancestral species of Ammopiptanthus was widely distributed from the eastern border of the Pamir Plateau to the Gobi desert during the Tertiary period (Liu et al. 1995). The aridification and formation of deserts in Central Asia from the early Miocene (24–16 Mya) resulted in the fragmentation of the continuous distribution of the ancestral Ammopiptanthus (Ge et al. 2005). Genetic differentiation between A. nanus and A. mongolicus likely occurred after a geographic barrier formed from the early Miocene. Long-term reproductive isolation led to significant genetic differences between A. nanus and A. mongolicus. In the present study, we estimated that the divergence time between A. nanus and A. mongolicus is approximately 0.70 ± 0.62 Mya before, which falls in the later stage of middle Pleistocene. This estimated time range is roughly consistent with other available evidence. However, considering the disputes about the significance of the substitution rate (Lynch and Conery 2000), this divergence time is only a rough estimate based on the coding regions of orthologous genes; further proof is still needed to determine the accurate splitting time.
Positively selected genes between A. nanus and A. mongolicus
Ka/Ks values have been widely used to identify protein-coding genes under positive or purifying selection (Hurst 2002) and Ka/Ks > 1 is widely accepted as a sign of positive selection. In addition, a Ka/Ks ratio of 0.5 was also interpreted as a useful cut-off to identify genes under positive selection (Swanson et al. 2004). In the present study, 6606 pairs of putative orthologs with full-length CDS were obtained and 15 PSGs that were considered likely to have experienced or be experiencing positive selection were found. These orthologs were involved in multiple biological processes such as plant defense, epigenetic regulation, and plant development regulation, indicating that these biological processes were under evolutionary pressure during the speciation of the two Ammopiptanthus species. Four PSGs are involved in defense response of plant, This result is consistent with those reported in previous studies on two closely related primrose species (Zhang et al. 2013a) and two related Dipteronia species (Zhou et al. 2016). Of the four PSGs, No. 13 encodes the protein SCARECROW-like 14 (SCL14), which is a member of the GRAS family of transcription factors. Arabidopsis SCL14 is involved in the activation of many stress-responsive genes that contribute to the protection of plants against xenobiotic stress (Fode et al. 2008). PSG No. 1 is a homolog of Arabidopsis Constitutive Expresser of PR Genes 1 (CPR1). CPR1 is an F-box protein and functions as a key component in fine control of plant immunity by mediating proteasomal degradation of SNC1, a disease resistance protein (Gou et al. 2012). PSG No. 6 encodes an enhanced downy mildew 2 (EDM2) which mediated disease resistance by functioning as a direct or indirect regulator of Resistance to Peronospora parasitica 7 (RPP7) expression (Eulgem et al. 2007). PSG No. 14 encode SRFR1, which specifically functions as a negative regulator of effector-triggered immunity in Arabidopsis (Kim et al. 2014), and mutations in SRFR1 lead to constitutive expression of SNC1 (Kim et al. 2010).
Of the 15 identified PSGs, two are reported to participate in plant epigenetic regulation. DNA methylation plays an integral role in regulating development and environmental response, and EDM2, Arabidopsis ortholog of PSG No. 6, play a role in regulate genome DNA methylation patterns (Lei et al. 2014). PSG No. 11 encodes an Arabidopsis SWI3C homolog. As a component of the SWI/SNF and RSC chromatin remodeling complexes, SWI3C affects gibberellin biosynthesis and signaling, and regulates plant growth and development including leaf morphogenesis (Sarnowska et al. 2013; Vercruyssen et al. 2014).
It is noteworthy that four of the five PSGs discussed above are also involved in plant development regulation. Among them, PSG No. 1 (Arabidopsis CPR1 ortholog) might regulate leaf pavement cell development via affecting cytoskeleton (Han et al. 2015), PSG No. 6 (Arabidopsis EDM2 ortholog) might regulate vegetative growth and development of leaf epidermal cells (Tsuchiya and Eulgem 2010), PSG No. 11 (Arabidopsis SWI3C ortholog) is likely involved in the transition from cell proliferation to cell differentiation in a developing leaf and affects leaf size (Vercruyssen et al. 2014), and PSG No. 14 (Arabidopsis SRFR1 ortholog) probably affects plant architectures via interaction with TCP transcription factors (Kim et al. 2014). These results indicate that the rapidly evolved genes may coordinate development with the environmental conditions, partially via epigenetic regulation such as DNA methylation. The other PSGs are involved in various biological processes including mRNA export from the nucleus (PSG No. 4), nucleus organization (PSG No. 2), protein targeting to mitochondrion (PSG No. 7), protein folding (PSG No. 5), and photomorphogenesis (PSG No. 3) and associated with peroxisome (PSG No. 10) and cytoskeleton (PSG No. 12).
Taken together, in the present study, we characterized the transcriptomes of A. nanus and conducted a comparative transcriptomic analysis between A. nanus and A. mongolicus. The comparative analysis not only identify large number of orthologous transcripts and a batch of putative positively selected genes between A. nanus and A. mongolicus, estimated the divergence times, but also advance understanding on the evolutionary adaptation of the two species to their individual habitats. In addition, large amount of SSRs were predicted and compared in the two Ammopiptanthus species. These results will serve as a comprehensive genomic resource for functional genomics, population genomics, and association mapping studies in the two Ammopiptanthus species in future.
Author contribution statement
YZ, JF and FG conceived and designed the experiments. CW. and ZX performed the experiments. FG and HL analyzed the data. YZ and FG wrote the paper. All authors read and approved the final manuscript.
References
Altenhoff AM, Dessimoz C (2009) Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol 5:e1000262
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29
Burge C, Karlin S (1997) Prediction of complete gene structures in human genomic DNA. J Mol Biol 268:78–94
Chen J, Sun Y, Sun F, Xia X, Yin W (2011) Tobacco plants ectopically expressing the Ammopiptanthus mongolicus AmCBL1 gene display enhanced tolerance to multiple abiotic stresses. Plant Growth Regul 63:259–269
Cheng T, Fu B, Wu Y, Long R, Liu C, Xia Q (2015) Transcriptome sequencing and positive selected genes analysis of Bombyx mandarina. PLoS One 10:e0122837
Duarte JM, Wall PK, Edger PP et al (2010) Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol Biol 10:61
Elmer KR, Fan S, Gunter HM et al (2010) Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mol Ecol 19:197–211
Eulgem T, Tsuchiya T, Wang XJ et al (2007) EDM2 is required for RPP7-dependent disease resistance in Arabidopsis and affects RPP7 transcript levels. Plant J 49:829–839
Evangelistella C, Valentini A, Ludovisi R et al (2017) De novo assembly, functional annotation, and analysis of the giant reed (Arundo donax L.) leaf transcriptome provide tools for the development of a biofuel feedstock. Biotechnol Biofuels 10:138
Fode B, Siemsen T, Thurow C, Weigel R, Gatz C (2008) The Arabidopsis GRAS protein SCL14 interacts with class II TGA transcription factors and is essential for the activation of stress-inducible promoters. Plant Cell 20:3122–3135
Gao F, Wang J, Wei S et al (2015) Transcriptomic analysis of drought stress responses in Ammopiptanthus mongolicus leaves using the RNA-Seq technique. PLoS One 10:e0124382
Gao F, Wang N, Li H et al (2016) Identification of drought-responsive microRNAs and their targets in Ammopiptanthus mongolicus by using high-throughput sequencing. Sci Rep 6:34601
Ge XJ, Yu Y, Yuan YM, Huang HW, Yan C (2005) Genetic diversity and geographic differentiation in endangered Ammopiptanthus (Leguminosae) populations in desert regions of northwest China as revealed by ISSR analysis. Ann Bot 95:843–851
Gou M, Shi Z, Zhu Y, Bao Z, Wang G, Hua J (2012) The F-box protein CPR1/CPR30 negatively regulates R protein SNC1 accumulation. Plant J 69:411–420
Grabherr MG, Haas BJ, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Graur D, Li WH (2000) Fundamentals of molecular evolution, 2nd edn. Sinauer Associates, Tel Aviv
Han B, Chen L, Wang J, Wu Z, Yan L, Hou S (2015) Constitutive expresser of pathogenesis related genes 1 is required for pavement cell morphogenesis in Arabidopsis. PLoS One 10:e0133249
Hurst LD (2002) The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet 18:486
Kim SH, Gao F, Bhattacharjee S, Adiasor JA, Nam JC, Gassmann W (2010) The Arabidopsis resistance-like gene SNC1 is activated by mutations in SRFR1 and contributes to resistance to the bacterial effector AvrRps4. PLoS Pathog 6:e1001172
Kim SH, Son GH, Bhattacharjee S et al (2014) The Arabidopsis immune adaptor SRFR1 interacts with TCP transcription factors that redundantly contribute to effector-triggered immunity. Plant J 78:978–989
Kozik A, Matvienko M, Kozik I, Van Leeuwen H, Van Deynze A, Michelmore R (2008) Eukaryotic ultra conserved orthologs and estimation of gene capture in EST libraries. Plant and Animal Genome Conference XVI, p P6
Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39(Database issue):D152–D157
Lei M, La H, Lu K et al (2014) Arabidopsis EDM2 promotes IBM1 distal polyadenylation and regulates genome DNA methylation patterns. Proc Natl Acad Sci USA 111:527–532
Liu J, Qiu M (1982) Ecological, physiological and anatomical traits of Ammopiptanthus mongolicus grown in desert of China. Acta Bot Sin 24:568–573
Liu JQ, Qiu MX, Yang K, Shi QH (1995) Studies on the plant community of Ammopiptanthus mongolicus. J Desert Res 15:109–115
Long Y, Zhang J, Tian X et al (2014) De novo assembly of the desert tree Haloxylon ammodendron (C. A. Mey.) based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom 15:1111
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155
Ma X, Wang P, Zhou (2015) De novo transcriptome sequencing and comprehensive analysis of the drought-responsive genes in the desert plant Cynanchum komarovii. BMC Genom 16:753
Necsulea A, Kaessmann H (2014) Evolutionary dynamics of coding and non-coding transcriptomes. Nat Rev Genet 15:734–748
Pang T, Ye CY, Xia X, Yin W (2013) De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa. BMC Genom 14:488
Sarnowska EA, Rolicka AT, Bucior E et al (2013) DELLA-interacting SWI3C core subunit of SWI/SNF chromatin remodeling complex modulates gibberellin responses and hormonal crosstalk in Arabidopsis. Plant Physiol 163:305–317
Shi Y, Yan X, Zhao P et al (2013) Transcriptomic analysis of a tertiary relict plant, extreme xerophyte Reaumuria soongorica to identify genes related to drought adaptation. PLoS One 8:e63993
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212
Swanson WJ, Wong A, Wolfner MF, Aquadro CF (2004) Evolutionary expressed sequence tag analysis of Drosophila female reproductive tracts identifies genes subjected to positive selection. Genetics 168:1457–1465
Tsuchiya T, Eulgem T (2010) Co-option of EDM2 to distinct regulatory modules in Arabidopsis thaliana development. BMC Plant Biol 10:203
Vercruyssen L, Verkest A, Gonzalez N et al (2014) ANGUSTIFOLIA3 binds to SWI/SNF chromatin remodeling complexes to regulate transcription during Arabidopsis leaf development. Plant Cell 26:210–229
Wall PK, Leebens-Mack J, Müller KF, Field D, Altman NS, de Pamphilis CW (2008) PlantTribes: a gene and gene family resource for comparative genomics in plants. Nucleic Acids Res 36:D970–D976
Wang T, Liu SL (2013) Regionalization for regulating oasification and desertification in the arid regions of China: a program. J Desert Res 33:959–966
Wang W, Chen J, Li J, Zhang Y, Shao Z, Kuai B (2007) Extraordinary accumulations of antioxidants in Ammopiptanthus mongolicus (Leguminosae) and Tetraena mongolica (Zygophyllaceae) distributed in extremely stressful environments. Bot Stud 48:55–61
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wei Q, Guo Y, Cao H, Kuai B (2011) Cloning and characterization of an AtNHX2-likeNa+/H+ antiporter gene from Ammopiptanthus mongolicus (Leguminosae) and its ectopic expression enhanced drought and salt tolerance in Arabidopsis thaliana. Plant Cell Tissue Organ 105:309–316
Wei Q, Hu P, Kuai B (2012a) Ectopic expression of an Ammopiptanthus mongolicus H+-pyrophosphatase gene enhances drought and salt tolerance in Arabidopsis. Plant Cell Tissue Organ 110:359–369
Wei Q, Kuai B, Hu P, Ding Y (2012b) Ectopic-overexpression of an HD-Zip IV transcription factor from Ammopiptanthus mongolicus (Leguminosae) promoted upward leaf curvature and non-dehiscent anthers in Arabidopsis thaliana. Plant Cell Tissue Organ 110:299–306
Wu Y, Wei W, Pang X et al (2014) Comparative transcriptome profiling of a desert evergreen shrub, Ammopiptanthus mongolicus, in response to drought and cold stresses. BMC Genom 15:671
Xu S, An L, Feng H, Wang X, Li X (2002) The seasonal effects of water stress on Ammopiptanthus mongolicus in a desert environment. J Arid Environ 51:437–447
Xu C, Jiao C, Zheng Y et al (2015) De novo and comparative transcriptome analysis of cultivated and wild spinach. Sci Rep 5:17706
Yang W, Qi Y, Bi K, Fu J (2012) Toward understanding the genetic basis of adaptation to high-elevation life in poikilothermic species: a comparative transcriptomic analysis of two ranid frogs, Rana chensinensis and R. kukunoris. BMC Genom 13:588
Yang W, Qi Y, Fu J (2014) Exploring the genetic basis of adaptation to high elevations in reptiles: a comparative transcriptome analysis of two toad-headed agamas (Genus Phrynocephalus). PLoS One 9:e112218
Yang Y, Wang L, Han J et al (2015a) Comparative transcriptomic analysis revealed adaptation mechanism of Phrynocephalus erythrurus, the highest altitude lizard living in the Qinghai–Tibet Plateau. BMC Evol Biol 15:101
Yang Y, Yang S, Li J et al (2015b) Transcriptome analysis of the Holly mangrove Acanthus ilicifolius and its terrestrial relative, Acanthus leucostachyus, provides insights into adaptation to intertidal zones. BMC Genom 16:605
Zhang Z, Li J, Zhao XQ, Wang J, Wong GK, Yu J (2006) KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom Proteom Bioinform 4:259–263
Zhang L, Yan HF, Wu W, Yu H, Ge XJ (2013a) Comparative transcriptome analysis and marker development of two closely related Primrose species (Primula poissonii and Primula wilsonii). BMC Genom 14:329
Zhang J, Xie P, Lascoux M, Meagher TR, Liu J (2013b) Rapidly evolving genes and stress adaptation of two desert poplars, Populus euphratica and P. pruinosa. PLoS One 8:e66370
Zhou Y, Gao F, Liu R, Feng J, Li H (2012) De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus. BMC Genom 13:266
Zhou T, Chen C, Wei Y et al (2016) Comparative Transcriptome and chloroplast genome analyses of two related Dipteronia species. Front Plant Sci 7:1512
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (31370356 and 31670335) and the Ministry of Education of China through 111 and 985 projects (B08044, YLDX01013). The authors greatly appreciate Miss Merhaba Abla for collecting the A. nanus seeds.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by R. Alia.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gao, F., Li, H., Xiao, Z. et al. De novo transcriptome analysis of Ammopiptanthus nanus and its comparative analysis with A. mongolicus . Trees 32, 287–300 (2018). https://doi.org/10.1007/s00468-017-1631-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-017-1631-6