
Abstract
Neottopteris nidus is an economically important foliage plant. However, the EST sequence resources available for N. nidus are still very limited. In this study, we present the first transcriptome analysis of N. nidus using the Illumina sequencing technology. More than 53 million reads were generated in transcriptome analysis, and de novo assembly generated 41,173 unigenes with an average length of 969 bp. The similarity search indicated that 24,801 unigenes (60.23 %) had significant similarity with proteins in the NR and Swiss-Prot protein databases. Among these annotated unigenes, 15,683 and 7942 unigenes were assigned to gene ontology categories and Clusters of Orthologous Groups, respectively. A total of 5548 unigenes were mapped into 116 pathways by the Kyoto Encyclopedia of Genes and Genomes Pathway database. Some abundant transcripts related to photomorphogenesis and development, including chlorophyll a–b-binding protein, phototropin, amino acid transporter, ATP-binding protein, expansin, and ribosomal protein, were identified in the N. nidus transcriptome. In total, 5792 potential EST-SSRs were identified among the unigenes. The most abundant type of repeat was dinucleotide (3836, 66.24 %), followed by trinucleotide (1575, 27.19 %) and tetranucleotide (207, 3.57 %) repeats. The dominant repeat motif was CT/GA (1229, 21.22 %), followed by AG/TC (905, 15.63 %), CA/GT (504, 11.32 %), and AC/TG (339, 8.70 %). In addition, a total of 100 potential marker sites were selected to validate the assembly quality and develop EST-SSR markers. This study generated a substantial fraction of N. nidus transcriptome sequences, which were very useful resources for gene discovery and molecular marker development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neottopteris nidus L., a plant of the Aspleniaceae family and native of tropical Asia, is one of the most economically important foliage plants (Ellwood and Foster 2004). N. nidus is an epiphytic fern with erect, simple, and wavy bright green leaves (Ozanne et al. 2003). It was widely cultivated in temperate regions as a houseplant, which can help in cleaning our air naturally (Fernandez and Revilla 2003). Due to its unique shiny beautiful leaves, it has achieved great importance in the ornamental industry and in landscaping especially in areas where it is uncommon. In addition, N. nidus supports a high abundance and wide diversity of taxa, and it has an important ecological role in structuring rain forest arthropod communities (Karasawa and Hijii 2006; Ellwood et al. 2009).
In recent years, next-generation sequencing (NGS) technology has developed rapidly for life sciences research. It not only provides cost-effective, high-throughput, and comprehensive analyses for model organisms, but also can provide opportunities to analyze non-model organisms whose genomes have not been sequenced (Wang et al. 2009; González-Ballester et al. 2010; Li et al. 2010). Transcriptome sequencing is an efficient way to generate functional genomic-level data for non-model plants such as N. nidus, and a large number of expressed sequence tag (EST) sequences are valuable for gene annotation and discovery, expression profiling, comparative genomics, and development of molecular markers (Emrich et al. 2006; Feng et al. 2012; Xiao et al. 2013; Wu et al. 2015; Zhang et al. 2015). Initially, a traditional sequencing technology has made significant contributions to current genomics research and cDNA library construction, but this method is costly, time-consuming, and sensitive to cloning biases (Wei et al. 2011). The newly developed NGS technology provides abundant resources for research on novel gene discovery, molecular marker development, genes mapping, and so on. Recently, an increasing number of EST sequences have become available for model and non-model plants, but very limited EST sequences are currently available for N. nidus. At present, only 76 nucleotide sequences of N. nidus have been deposited in the National Center for Biotechnology Information (NCBI) nucleotide database (as of November 2015).
Simple sequence repeats (SSRs) are 1–6 bp long repeat sequences that occur ubiquitously in plant genomes and serve as important molecular tools for genetic and genomic research (Gur-Arie et al. 2000). SSR markers are useful for a variety of applications in plant genetics and breeding because of their genetic codominance, abundance, multi-allelic variation, high reproducibility, and high level of polymorphism (Powell et al. 1996; Aggarwal et al. 2007). SSRs can be divided into genomic SSRs and EST-SSRs on the basis of the original sequences used to identify simple repeats. However, traditional methods to isolate and identify genomic SSRs are labor-intensive, costly, and time-consuming (Zane et al. 2002). EST-SSRs designed from expressed sequence tag data are more evolutionarily conserved in comparison to markers that are generated from genomic sequences and show more transferability between species (Varshney et al. 2005; Portis et al. 2007). With the increasing number of EST sequences deposited in public databases, a large number of EST-SSRs have been developed, and the polymorphism of EST-SSRs has been evaluated in many plant species (Triwitayakorn et al. 2011; Asadi and Monfared 2014; Kumar et al. 2014; Jia et al. 2015; Chen et al. 2015).
In the present study, we used the Illumina HiSeq™ 2000 platform to characterize the N. nidus transcriptome and to develop a set of EST-SSRs. To our knowledge, this study is the first to characterize the complete transcriptome of N. nidus by analyzing large-scale transcript sequences by the Illumina paired-end sequencing strategy. These EST data sets of N. nidus will serve as a useful sequence resource for novel gene discovery and molecular marker development.
Materials and methods
Plant materials and RNA extraction
Neottopteris nidus was grown in the chamber under a 25/18 °C (day/night temperature), 60 % relative humidity, photoperiod of 16 h light/8 h dark at Jiangsu Academy of Agricultural Sciences, Nanjing, China. Young roots, shoots, and leaves of plants were collected, immediately frozen in liquid nitrogen, and stored at 70 °C for RNA extraction. For Illumina sequencing, total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The extracted RNA was treated with DNase I (Takara Biotechnology, China) for 45 min at 37 °C to remove residual DNA. RNA integrity was verified using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA) with a minimum RNA integrated number (RIN) value of 8. Subsequently, equal amounts of total RNA from three N. nidus tissues were pooled to prepare a cDNA library.
cDNA library construction and Illumina sequencing
The cDNA library was constructed following the Illumina manufacturer’s instructions. First, total RNA was treated by DNA polymerase I, and Poly-A RNA was extracted from total RNA using Oligo (dT) magnetic beads. Fragmentation buffer was added to break the mRNA into short fragments. Using these short fragments for templates, random hexamer primers were used for first-strand cDNA synthesis. The second-strand cDNA was synthesized using buffer, dNTPs, RNase H and DNA polymerase I (Invitrogen, Grand Island, NY, USA). The resulting short fragments were purified using the PCR extraction kit and suspended in EB buffer for end repair and poly (A) addition. The short fragments were then joined to sequencing adaptors of both ends to form a tag library. After PCR amplification, suitable fragments were purified by agarose gel electrophoresis. In the end, the cDNA library was sequenced on the Illumina HiSeq™ 2000 platform.
De novo assembly of sequencing data
Before the transcriptome assembly, we carried out a stringent filtering process of raw sequencing reads. The reads were obtained from raw data by filtering out adaptor-only reads, low-quality reads, and reads with more than 10 % Q < 20 bases. All the clean reads were then used for transcriptome de novo assembly using the short read assembling program Trinity with the default settings (Grabherr et al. 2011). The Trinity first combines reads with an identity value of 95 % and a coverage length of 100 bp to form longer fragments without N, and these are called contigs. Next, Trinity connects the contigs by using N to represent unknown sequences between each two contigs and then forms transcripts. Finally, paired-end reads were performed to fill the gap between different transcripts in order to obtain unigenes. The short reads data sets were deposited into the NCBI Short Read Archive (SRA) with the accession number: SRX269217.
Gene annotation and analysis
All unigenes were analyzed for homology searches against protein databases, including the NR protein database (http://www.ncbi.nlm.nih.gov), Swiss-Prot protein database (http://www.expasy.ch/sprot), GO database (http://www.geneontology.org), COG database (http://www.ncbi.nlm.nih.gov/cog), and KEGG pathway database (http://www.genome.jp/kegg) with BLAST alignment (E value ≤10−5). The best aligning results were used to determine the sequence direction of the unigenes. To identify the best BLAST hits from the alignments, putative gene names and predicted proteins of the corresponding assembled sequences were produced. Functional annotations of all unigenes by gene ontology (GO) terms were performed based on the best BLAST hits using the Blast2GO software. WEGO was used for GO functional classification of unigenes and to view the distribution of gene functions at the macro level (Ye et al. 2006). For pathway-enrichment analysis, unigenes were mapped to terms in the KEGG database (Kanehisa et al. 2008).
SSR identification and primer design
To further evaluate the assembly quality and develop molecular markers, all unigenes were used to mine potential SSRs with the Simple Sequence Repeat Identification Tool (SSRIT, http://www.gramene.org/db/markers/ssrtool). The parameters were adjusted for identification of perfect di-, tri-, tetra-, penta-, and hexanucleotide motifs with a minimum of 6, 5, 5, 4, and 4 repeats, respectively. Mononucleotide repeats were ignored since distinguishing genuine mononucleotide repeats from polyadenylation products and single nucleotide stretch errors generated by sequencing was difficult. Primer pairs were designed for the flanking regions of SSRs using the BatchPrimer3 software. The primer design parameters were set as follows: primer length of 18–23 bases, GC content of 40–60 %, PCR product size of 150–500 bp, and annealing temperatures of 50–60 °C. In total, 100 pairs of primers were designed and validated by PCR reactions. PCR amplifications using DNA templates extracted from the ten fern germplasms were performed in 10 µl reaction mixtures containing template DNA 30 ng, 10× PCR buffer 1.0 µl, dNTPs (10 mM) 0.8 µl, primer (10 µM) 0.8 µl, and Taq DNA polymerase 0.5 U. The PCR reaction cycling profile was 94 °C for 5 min, followed by 33 cycles of 94 °C for 40 s, 50–60 °C for 50 s, 72 °C for 1 min, and a final step of 72 °C for 10 min. PCR products were analyzed by electrophoresis on 8 % non-denaturing polyacrylamide gels and visualized by silver staining.
Results
Illumina paired-end sequencing and de novo assembly
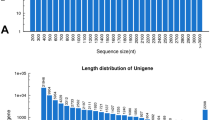
With the purpose of understanding the transcriptome of N. nidus, we constructed a cDNA library of pooled RNA samples to generate the transcriptome and sequenced using the Illumina sequencing platform. After a stringent quality check and data filtering, 53,313,049 paired-end reads with a total of 10.66 Gbp were generated and assembled into 41,173 unigenes. Among the clean reads, more than 87 % had Phred-like quality scores at the Q20 level. As a result, a total of 2,104,392 contigs with a N50 length of 316 bp and an average length of 223 bp, 63,529 transcripts (>200 bp) with a N50 length of 1895 bp and an average length of 1139 bp, and 41,173 unigenes (>200 bp) with a N50 length of 1737 bp and an average length of 969 bp were assembled, respectively (Supplementary Table S1). The length distributions of the contigs, transcripts, and unigenes are listed in Fig. 1. Taking unigenes as an example, the lengths of unigenes ranged from 201 to 14,471 bp. Of the 41,173 unigenes, 19,953 (48.46 %) were between 201 and 500 bp; 7793 (18.93 %) ranged from 501 to 1000 bp; 7815 (18.98 %) ranged from 1001 to 2000 bp; and 5612 (13.63 %) were larger than 2000 bp.

Overview of the N. nidus transcriptome assembly. a Size distribution of contigs, b size distribution of transcripts, and c size distribution of unigenes
Functional annotation of assembled unigenes
For the validation and annotation of the assembled unigenes, all unigenes were searched against the NCBI non-redundant (NR) protein database and Swiss-Prot protein database. Of the 41,173 unigenes, 24,657 (59.89 %) could be annotated based on sequences in the NR protein database, and 17,436 unigenes (42.35 %) had significant similarity to annotated proteins in the Swiss-Prot protein database (Supplementary Table S2). Altogether, 24,801 unigenes (60.23 %) were successfully annotated in the NR and Swiss-Prot protein databases, suggesting that this Illumina sequencing project generated a substantial fraction of N. nidus genes in this research. The proportion of unigenes with homologous matches decreased with the decrease of the length of the unigene. About 95.37 % of the assembled unigenes over 1000 bp in length showed homologous matches in the NR protein database, whereas only 29.34 % of the assembled unigenes shorter than 300 bp showed homologous matches (Fig. 2). The E value distribution of the top matches showed that 50.55 % of the aligned sequences had significant homology in the NR protein database (E value <1E−50), and almost 33.80 % of the aligned sequences had alignment identities greater than 60 % (Supplementary Fig. S1A, C). As expected, a comparable pattern of E value and similarity distributions of the top matches was found in the Swiss-Prot protein database. The result indicated that 42.70 and 26.89 % of the aligned sequences had significant homologies and alignment identities higher than 60 % in the Swiss-Prot protein database, respectively (Supplementary Fig. S1B, D).

Comparison of unigene length with or without hits. Longer unigenes were more likely to have BLAST hits in protein databases
Functional classification by GO, COG, and KEGG
Based on the NR annotation, 15,683 unigenes with BLAST matches to known proteins were categorized into 55 functional groups within the GO database, which could be classified into the three main categories: biological process, cellular components, and molecular function clusters (Supplementary Fig. S2). Among the cellular component category, cell part (9942, 24.80 %) was the most dominant group, followed by cell (9699, 24.19 %) and organelle (8421, 21.01 %). With the classification of molecular function, catalytic activity (7626, 45.58 %) and binding (6716, 40.13 %) were separately the first and second largest groups, whereas other categories such as metallochaperone activity, protein tag, receptor activity, nutrient reservoir activity, and antioxidant activity contained 223 unigenes only representing 1.32 %. As for the category of biological process, metabolic process (10,132, 22.66 %) and cellular process (9543, 21.35 %) were the dominant groups, followed by response to stimulus (4407, 9.86 %), biological regulation (3732, 8.35 %), and cellular component organization or biogenesis (2749, 6.15 %).
To further evaluate the integrity of the N. nidus transcriptome and the effectiveness of the annotation process, unigene sequences were subjected to a search against the Clusters of Orthologous Groups (COG) database for functional prediction and classification. Out of 24,657 unigenes with significant similarity to the NR protein database in our study, 7942 unigenes were assigned to the COG classifications (Supplementary Fig. S3). COG-annotated putative proteins were functionally classified into 25 categories. Among the 25 COG categories, the cluster for general function prediction only (2175, 19.39 %) represented the largest group, followed by replication, recombination and repair (1097, 9.78 %), transcription (1018, 9.08 %), signal transduction mechanisms (894, 7.97 %), and translation, ribosomal structure, and biogenesis (730, 6.51 %), whereas the percentages of five groups were less than 1.00 %, such as RNA processing and modification, chromatin structure and dynamics, cell motility, extracellular structures, and nuclear structure.
To identify biological pathways activated in N. nidus, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was conducted with an E value cutoff less than 10−5. According to the KEGG database, 5548 unigenes were assigned to five main categories including 116 KEGG functional pathways (Supplementary Table S3). Among the five categories, metabolism was the biggest category (3263, 58.81 %), followed by genetic information processing (1629, 29.36 %), cellular processes (312, 5.62 %), environmental information processing (204, 3.68 %), and organismal systems (140, 2.53 %). The specific pathways, including ribosome, RNA transport, spliceosome, purine metabolism, plant hormone signal transduction, and the other 20 top mapped pathways, were shown (Supplementary Fig. S4). In addition, the KEGG metabolic pathways contained many amino acid synthesis and degradation pathways, among which arginine and proline metabolism, cysteine and methionine metabolism, phenylalanine metabolism, and alanine, aspartate, and glutamate metabolism occupied the majority of positions. The functional classification of KEGG provided a valuable resource for investigating specific processes, functions, and pathways involved in the transcriptome of N. nidus.
The most abundant transcripts in the N. nidus transcriptome
Of all the 41,173 unigenes, 23 contained more than 50 reads, which represented the most abundant transcripts in the N. nidus transcriptome (Table 1). In this study, two transcripts encoding chlorophyll a–b-binding protein and phototropin were predominantly expressed, which are known to be involved in photomorphogenesis. Four other abundant transcripts encode cytochrome P450, serine/threonine-protein kinase, E3-ubiquitin protein ligase, and non-specific lipid-transfer protein, which are required for plant defense responses. Besides the stress response proteins, two transcripts encoding phenylalanine ammonia lyase and chalcone synthase were predominantly expressed, which were associated with the biosynthesis of flavonoid and performed a variety of functions in plants. Moreover, the transcripts encoding DNA-binding protein, ribosomal protein, amino acid transporter, expansin, ATP-binding protein, zinc finger protein, and RNA-binding protein, which are considered to play roles in amino acid metabolism, energy metabolism, cell proliferation, development, and growth in plants, suggest that these genes might be very important in N. nidus growth and development. There were also some highly frequent transcripts that had matches to unknown or predicted proteins. These genes may play important roles in N. nidus growth, development, defense responses, and secondary metabolite production and are worthy of further functional investigation.
Development and characterization of EST-SSR markers
To further evaluate the assembly quality and develop new molecular markers, the 41,173 unigenes assembled were used to mine potential microsatellites that were defined as di- to hexanucleotide motifs. Using the SSRIT tool, a total of 5792 potential EST-SSRs were identified from 4823 unigenes (Supplementary Table S4). Among the 4823 unigenes, 3665 and 1158 unigenes contained one and more than one SSR, respectively. For these unigenes, the EST-SSR frequency was 11.71 %, and an average of one SSR was found every 4.89 kb in the unigenes.
The frequency, type, and distribution of the potential EST-SSRs were also analyzed. Dinucleotide repeats were the most abundant type (3836, 66.23 %), followed by trinucleotide (1575, 27.19 %), tetranucleotide (207, 3.57 %), hexanucleotide (116, 2.00 %), and pentanucleotide (58, 1.01 %) repeats (Table 2). The frequencies of EST-SSRs with different numbers of tandem repeats were further evaluated. As shown in Table 2, EST-SSRs with six repeat motifs (1481, 25.57 %) were the most common, followed by seven repeat motifs (1003, 17.32 %), five repeat motifs (981, 16.94 %), eight repeat motifs (739, 12.78 %), and nine repeat motifs (576, 9.93 %). The EST-SSRs length was mostly distributed from 15 to 24 bp, accounting for 86.54 % of the total EST-SSRs. The dominant repeat motif in EST-SSRs was CT/GA (1229, 21.22 %), followed by AG/TC (905, 15.63 %), CA/GT (504, 11.32 %), and AC/TG (339, 8.70 %) (Fig. 3). The four types of repeat motifs mentioned above represented about 56.87 %, whereas the remaining types of repeat motifs only accounted for 43.13 %.

Frequency distribution of EST-SSRs based on motif sequence types. The frequency of main motif types was shown
Using the EST-SSR-containing sequences as a source, a total of 100 potential marker sites were randomly selected to validate the assembly quality and develop EST-SSR markers. Of the 100 primer pairs, 79 primer pairs were successfully PCR amplified with the N. nidus genomic DNA (Supplementary Table S5). The remaining 21 primers failed to generate PCR products at various annealing temperatures and Mg2+ concentrations and were therefore excluded from further analysis. Among the 79 successful primer pairs, 65 primer pairs generated PCR products at the expected size, 10 generated PCR products larger than expected, and 4 generated PCR products smaller than expected. A total of 306 amplifying bands were detected with 79 primer pairs, and the number of amplifying bands per primer pair ranged from one to eight, with an average of 3.87 per marker. The 79 primer pairs were further examined with 10 fern germplasms as PCR templates; 68 and 11 primer pairs could amplify polymorphic (alleles of different size) and monomorphic products, respectively. Supplementary Figure S5 shows the polymorphic bands amplified by three primer pairs. The polymorphic EST-SSR markers are important for research, including genetic diversity, cultivar identification, genetic diversity analysis, and marker-assisted selection breeding in N. nidus. These results clearly demonstrated that developing SSR markers based on assembled unigenes derived from Illumina paired-end sequencing in N. nidus is an effective and feasible approach.
Discussion
Illumina sequencing technology is an extremely high-throughput and effective method to obtain large amounts of transcriptome data, which is essential for identifying novel genes and developing molecular markers. Recent algorithmic and experimental advances are likely to increase the applicability of Illumina sequencing and de novo assembly, which has been successfully used in model and non-model plants (Libault et al. 2010; Xie et al. 2012; Verma et al. 2013; Jia et al. 2015). Consistent with these reports, the results from our study also suggested that short reads from Illumina sequencing can be effectively assembled and used for gene identification and EST-SSR marker development in non-model plants. About 10.66 Gbp of data were generated and assembled into a reference transcriptome, which was the first library in N. nidus to be reported. The assembly result showed that the average length of all unigenes was 969 bp, which was longer than that documented in previous studies, such as tea (402 bp), sweet potato (581 bp), and sesame (629 bp) (Wang et al. 2010; Wei et al. 2011; Tan et al. 2013). There were two possible reasons for obtaining longer unigenes in our study. First, longer paired-end reads (100 bp) were obtained than those of a previous study (75 or 90 bp). Second, the novel assembly program Trinity was utilized, which can recover more full-length transcripts across a broad range of expression levels and provide a unified solution for transcriptome reconstruction in species without a reference genome (Grabherr et al. 2011). These results suggested that the transcriptome data of N. nidus were effectively assembled, which was further validated by the high proportion of unigenes searched with public protein databases.
Further, assembly validation may be done by comparison with protein databases. In this study, 60.23 % unigenes had homologs in the NCBI NR or Swiss-Prot protein databases, suggesting their relatively conserved functions. The lengths of the assembled unigenes are crucial in determining the level of significance of a BLAST match (Shi et al. 2011). We found that the proportion of the assembled unigenes with BLAST matches decreased with the decrease of the length of the assembled unigenes in our study. Close to 40 % of the N. nidus unigenes had no BLAST matches in protein databases, which might be due to the relatively short length or poor alignment of the assembled unigenes. The shorter sequences may lack conserved functional domains, or they may contain a known protein domain but not show sequence matches because of the short query sequence, resulting in false-negative results (Hou et al. 2011). However, 345 unigenes with sequence length longer than 1000 bp had no BLAST matches. The lack of BLAST matches for these unigenes was not because of a shorter sequence length but because of a genuine lack of hits to sequences in database.
In this study, many unigenes matched to known proteins were assigned to a wide range of GO categories and COG classifications (Supplementary Fig. S2, S3), suggesting that our sequencing data represented a wide diversity of transcripts in N. nidus. Of the GO category, cell part and binding activity were the largest subcategories in the cellular component and molecular function categories, respectively. This result was consistent with that in litchi, sweet potato, and alfalfa (Wang et al. 2010; Li et al. 2013). As for the COG classification, the second and third largest classifications were separately replication, recombination and repair, and transcription, which was consistent with other studies (Wang et al. 2010; Wei et al. 2011; Li et al. 2013). Based on the KEGG pathway database, most representative unigenes were mapped to specific pathways, such as metabolism pathways, biosynthesis of secondary metabolites, transport and catabolism, translation, and plant hormone signal transduction. GO, COG, and KEGG classifications of the N. nidus data set revealed that the assembled unigenes had diverse molecular functions and were involved in many metabolic pathways, indicating the diversity of the assembled unigenes while reflecting the global landscape of the transcriptome. Many of these novel unigenes might represent potential N. nidus-specific genes. In summary, the large number of genes we detected should provide sufficient transcriptomic sequence information to increase our understanding of the processes regulating growth and development in N. nidus.
It is well known that SSR markers are important tools for analysis of genetic diversity, comparative genetics, phylogenetic relationship, linkage mapping, QTL analysis, and marker-assisted selection (Saha et al. 2006; Cavagnaro et al. 2010). Therefore, the development of SSR markers from the N. nidus transcriptome could be more useful for genetic studies and breeding applications. In our study, a total of 5792 potential EST-SSRs were identified based on assembled sequences generated from Illumina paired-end sequencing, and 11.71 % unigene sequences possessed SSRs. The EST-SSR frequency was 1 per 4.89 kb (5792 SSRs in 28.29 Mb), which was similar to that in poplar (1/4 kb) and radish (1/3.4 kb) (Tuskan et al. 2004; Wang et al. 2012a, b), much higher than that in rice (1/40 kb) and mungbean (1/67 kb) (Temnykh et al. 2001; Tangphatsornruang et al. 2009), and lower than those in cucumber (1/1.8 kb) and tea (1/2.4 kb) (Cavagnaro et al. 2010; Tan et al. 2013). The EST-SSR frequency is dependent on several factors, such as the genome structure, size of sequence data analysis, the mining tool used, and the parameters for exploration of SSRs (Gupta et al. 2003).
Dinucleotide repeats were the most abundant motif type, followed by trinucleotide and tetranucleotide repeats in our study, which was consistent with previous reports (Wei et al. 2011; Li et al. 2012; Tan et al. 2013). The most abundant di- and trinucleotide motifs were CT/GA and ACC/TGG, respectively (Fig. 3). This finding is consistent with the results reported for other plant species (Kantety et al. 2002; Kumpatla and Mukhopadhyay 2005; La et al. 2005; Wang et al. 2010; Li et al. 2012; Kumar et al. 2014). It was speculated that CT repeats are typically found in transcribed regions and occur at a high frequency in 5′-UTRs, and they may be involved in antisense transcription and play a role in gene regulation (Martienssen and Colot 2001; Wang et al. 2012a, b). The repeat GA-rich was the most abundant dinuleotide motif, whereas TA-rich was the most frequent motif in poplar, sorghum, and mungbean sequences (Tuskan et al. 2004; Tangphatsornruang et al. 2009; Yonemaru et al. 2009). The proportion of GC-rich motifs was the smallest in the dinucleotide repeat, which is in agreement with pervious studies (Wei et al. 2011; Yu et al. 2011; Zhu et al. 2012). The other motif repeats appeared to be evenly distributed with a frequency lower than 1 % except AAG/TTC, ACC/TGG, ACT/TGA, AGG/TCC, AGT/TCA, ATC/TAG, CAG/GTC, CCT/GGA, CGA/GCT. and CTC/GAG in trinucleotide repeats. Differences in SSRs abundance in different studies from different plant species are often seen. However, some motifs showed a greater abundance in most plants, such as AT/TA and AG/TC (Morgante et al. 2002).
The majority of EST-SSR primers generated high-quality amplicons, suggesting that EST-SSRs are suitable for specific primer designs. In this study, 100 pairs of PCR primers were designed and used to assess assembly quality, and 79 primer pairs (79 %) successfully amplified PCR fragments. The failure of 21 primer pairs to produce amplification may have been caused by the large introns, the location of the primer across splice sites, chimeric primers, or poor-quality sequences (Varshney et al. 2005). Of these 79 working primer pairs, 65 amplified PCR products of the expected size. The deviation of 14 primer pairs from the expected size may be due to the presence of introns in the amplicons, large insertions or repeat number variations, or a lack of specificity (Saha et al. 2006). The possibility of assembly errors also cannot be ruled out. These PCR results validate the quality of our assembled unigenes and demonstrate the possible utility of the EST-SSRs produced in this study. The large number of EST-SSRs identified from our transcriptome data will be used for population genetics, genetic diversity, linkage mapping, comparative genomics, and other genetic studies of N. nidus. The results suggest that the unigenes assembled were of high quality and that the EST-SSRs identified in our data set will provide a wealth of resources for developing EST-SSR markers in N. nidus.
References
Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, Singh L (2007) Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet 114:359–372
Asadi AA, Monfared SR (2014) Characterization of EST–SSR markers in durum wheat EST library and functional analysis of SSR-containing EST fragments. Mol Genet Genomics 289:625–640
Cavagnaro PF, Senalik DA, Yang L, Simon PW, Harkins TT, Kodria CD, Huang S (2010) Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom 11:569–586
Chen LY, Cao YN, Yuan N, Nakamura K, Wang GM, Qiu YX (2015) Characterization of transcriptome and development of novel EST–SSR makers based on next-generation sequencing technology in Neolitsea sericea (Lauraceae) endemic to East Asian land-bridge islands. Mol Breed 35:187
Ellwood MDF, Foster WA (2004) Doubling the estimate of invertebrate biomass in a rainforest canopy. Nature 429:549–551
Ellwood MDF, Manica A, Foster WA (2009) Stochastic and deterministic processes jointly structure tropical arthropod communities. Ecol Lett 12:277–284
Emrich SJ, Barbazuk WB, Li L, Schnable PS (2006) Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res 16:1–5
Feng C, Chen M, Xu CJ, Bai L, Yin XR, Li X, Allan AC, Ferguson IB, Chen KS (2012) Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genom 13:19
Fernandez H, Revilla MA (2003) In-vitro culture of ornamental ferns. Plant Cell Tissue Org 73:1–13
González-Ballester D, Casero D, Cokus S, Pellegrini M, Merchant SS, Grossman AR (2010) RNA-Seq analysis of sulfur-deprived chlamydomonas cells reveals aspects of acclimation critical for cell survival. Plant Cell 22:2058–2084
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech 29:644–652
Gupta PK, Rustgi S, Sharma S, Singh R, Kumar N, Balyan HS (2003) Transferable EST–SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol Genet Genomics 270:315–323
Gur-Arie R, Cohen CJ, Eitan Y, Shelef L, Hallerman EM, Kashi Y (2000) Simple sequence repeats in Escherichia coli: abundance, distribution, composition, and polymorphism. Genome Res 10:62–71
Hou R, Bao ZM, Wang S et al (2011) Transcriptome sequencing and de novo analysis for yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PLoS ONE 6:e21560
Jia XP, Deng YM, Sun XB, Liang LJ, Ye XQ (2015) Characterization of the global transcriptome using Illumina sequencing and novel microsatellite marker information in seashore paspalum. Genes Genomics 37:77–86
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:480–484
Kantety RV, La Rota M, Matthews DE, Sorrells ME (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol 48:501–510
Karasawa S, Hijii N (2006) Does the existence of bird’s nest ferns enhance the diversity of oribatid (Acari: Oribatida) communities in a subtropical forest? Biodiversity Conserv 15:4533–4553
Kumar S, Shah N, Garg V, Bhatia S (2014) Large scale in silico identification and characterization of simple sequence repeats (SSRs) from de novo assembled transcriptome of Catharanthus roseus (L.) G. Don. Plant Cell Rep 33:905–918
Kumpatla SP, Mukhopadhyay S (2005) Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 48:985–998
La RM, Kantety R, Yu JK, Sorrells M (2005) Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley. BMC Genom 6:23
Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y (2010) The sequence and de novo assembly of the giant panda genome. Nature 463:311–317
Li D, Deng Z, Qin B, Liu XH, Men ZH (2012) De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST–SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom 13:192
Li CQ, Wang Y, Huang XM, Li J, Wang HC, Li JG (2013) De novo assembly and characterization of fruit transcriptome in Litchi chinensis Sonn and analysis of differentially regulated genes in fruit in response to shading. BMC Genom 14:552
Libault M, Farmer A, Joshi T, Takahashi K, Langley R, Franklin LD, He J, Xu D, May G, Stacey G (2010) An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J 63:86–99
Martienssen RA, Colot V (2001) DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293:1070–1074
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet 30:194–200
Ozanne CMP, Anhuf D, Boulter SL, Keller M, Kitching RL, Kőrner C, Meinzer FC, Mitchell AW, Nakashizuka T, Silva Dias PL, Stork NE, Wright SJ, Oshimura MY (2003) Biodiversity meets the atmosphere: a global view of forest canopies. Science 301:183–186
Portis E, Nagy I, Sasva Z, Stagelri A, Barchi L, Lanteri S (2007) The design of Capsicum spp. SSR assays via analysis of in silico DNA sequence, and their potential utility for genetic mapping. Plant Sci 172:640–648
Powell W, Machray GC, Provan J (1996) Polymorphism revealed by simple sequence repeats. Trends Plant Sci 1:215–222
Saha MC, Cooper JD, Mian MAR, Chekhovskiy K, May GD (2006) Tall fescue genomic SSR markers: development and transferability across multiple grass species. Theor Appl Genet 113:1449–1458
Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Sun J, Li YY, Xia T, Wan XH (2011) Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom 12:131
Tan LQ, Wang LY, Wang K, Cheng CZ, Li YW, Gui NQ, Hao C, Qiang Z, Cui QM, Liang JB (2013) Floral transcriptome sequencing for SSR marker development and linkage map construction in the tea plant (Camellia sinensis). PLoS One 8:e81611
Tangphatsornruang S, Somta P, Uthaipaisanwong P, Chanprasert J, Sangsrakru D, Seehalk W, Sommanas W, Tragoonrung S, Srinives P (2009) Characterization of microsatellites and gene contents from genome shotgun sequences of mungbean (Vigna radiata (L.) Wilczek). BMC Plant Biol 9:137–148
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations and genetic marker potential. Genome Res 11:1441–1452
Triwitayakorn K, Chatkulkawin P, Kanjanawattanawong S, Sraphet S, Yoocha T, Sangsrakru D, Chanprasert J, Ngamphiw C, Jomchai N, Therawattanasuk K, Tangphatsornruang S (2011) Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res 18:471–482
Tuskan GA, Gunter LE, Yang ZK, Yin T, Sewell MM, DiFazio SP (2004) Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa. Can J For Res 34:85–93
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Verma P, Shah N, Bhatia S (2013) Development of an expressed gene catalogue and molecular markers from the de novo assembly of short sequence reads of the lentil (Lens culinaris Medik.) transcriptome. Plant Biotechnol J 11:894–905
Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L (2009) Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res 38:5075–5087
Wang ZY, Fang BP, Chen JY, Zhang XJ, Luo ZX, Huang L, Chen X, Li Y (2010) De novo assembly and characterization of root transcriptome using Illumina paired–end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genom 11:726
Wang HX, Walla JA, Zhong SB, Huang DQ, Dai WH (2012a) Development and cross-species/genera transferability of microsatellite markers discovered using 454 genome sequencing in chokecherry (Prunus virginiana L.). Plant Cell Rep 31:2047–2055
Wang S, Wang X, He Q, Liu X, Xu W, Li L, Gao J, Wang F (2012b) Transcriptome analysis of the roots at early and late seedling stages using Illumina paired-end sequencing and development of EST–SSR markers in radish. Plant Cell Rep 31:1437–1447
Wei WL, Qi XQ, Wang LH, Zhang YX, Hua W, Li D, Lv H, Zhang X (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST–SSR markers. BMC Genom 12:451
Wu TQ, Luo SB, Wang R, Zhong YJ, Xu XM, Lin YE, He XM, Sun BJ, Huang HX (2015) The first Illumina-based de novo transcriptome sequencing and analysis of pumpkin (Cucurbita moschata Duch.) and SSR marker development. Mol Breeding 34:1437–1447
Xiao J, Jin XH, Jia XP, Wang HY, Cao AZ, Zhao WP, Pei HY, Xue ZK, He LQ, Chen QG, Wang XE (2013) Transcriptome-based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC Genom 14:197
Xie F, Burklew CE, Yang Y, Liu M, Xiao P, Zhang B, Qiu D (2012) De novo sequencing and a comprehensive analysis of purple sweet potato (Ipomoea batatas L.) transcriptome. Planta 236:101–113
Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:293–297
Yonemaru J, Ando T, Mizubayashi T, Kasugo S, Matsumoto T, Yano M (2009) Development of genome-wide simple sequence repeat markers using whole-genome shotgun sequences of sorghum (Sorghum bicolor (L.) Moench). DNA Res 16:187–193
Yu F, Wang BH, Feng SP, Wang JY, Li WG, Wu YT (2011) Development, characterization, and cross-species/genera trans-ferability of SSR markers for rubber tree (Hevea brasiliensis). Plant Cell Rep 30:335–344
Zane L, Bargelloni L, Patarnello T (2002) Strategies for microsatellite isolation: a review. Mol Ecol 11:1–16
Zhang LW, Wan XB, Xu JT, Lin LH, Qi JM (2015) De novo assembly of kenaf (Hibiscus cannabinus) transcriptome using Illumina sequencing for gene discovery and marker identification. Mol Breeding 35:192
Zhu H, Senalik D, McCown BH, Zeldin EL, Speers J, Hyman J, Bassil N, Hummer K, Simon PW, Zalapa JE (2012) Mining and validation of pyrosequenced simple sequence repeats (SSRs) from American cranberry (Vaccinium macrocarpon Ait.). Theor Appl Genet 124:87–96
Acknowledgments
This research was supported by the Jiangsu Provincial Agricultural Science and Technology Innovation Project (CX-13-5057).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have read and approved the manuscript. It is not being submitted to any other journal. The authors have declared that no competing interests exist.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
Characterization of the assembled unigenes against the NR and Swiss-Prot protein databases. (A) E-value distribution of the top BLAST hits for unigenes in the NR database. (B) E-value distribution of BLAST hits for unigenes in the Swiss-Prot database. (C) Similarity distribution of the top BLAST hits for unigenes in the NR database. (D) Similarity distribution of the top BLAST hits for unigenes in the Swiss-Prot database (JPEG 73 kb)
Figure S2
Gene ontology (GO) classification of the assembled unigenes; 15,683 unigenes with BLAST matches were assigned to three main GO categories: cellular component, molecular function, and biological process. The x-axis indicates the subcategories, and the y-axis indicates the numbers related to the total GO terms present (JPEG 371 kb)
Figure S3
Clusters of Orthologous Groups (COG) classification. All unigenes were aligned to the COG database to predict and classify possible functions; 7942 unigenes were assigned to 25 COG classifications (JPEG 136 kb)
Figure S4
The top 25 mapped pathways annotated by the KEGG database (JPEG 220 kb)
Figure S5
Polyacrylamide gel electrophorogram of amplification results using three EST-SSR markers. A, B, and C show the polymorphic of Primer ES_19, ES_46, and ES_67. Lines 1~10 represent the amplification products across ten fern germplasms (line 1~10 stands for Neottopteris nidus, Neottopteris phyllitidis, Neottopteris latipes, Davallia bullata, Asplenium trichomanes, Adiantum capillus-veneris, Pteris cretica, Nephrolepis auriculata, Nephralepis exaltata, and Platycerium wallichii, respectively). (JPEG 95 kb)
Table S1
Characteristics of assembled contigs, transcripts, and unigenes (DOC 39 kb)
Table S2
Summary of functional annotation of assembled unigenes (DOC 39 kb)
Table S3
Summary of the unigenes annotated to the reference canonical pathways in the KEGG database (DOCX 21 kb)
Table S4
Summary of SSRs identified in the N. nidus transcriptome (DOC 42 kb)
Table S5
The characterizations of EST-SSR markers (DOCX 24 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Jia, X., Deng, Y., Sun, X. et al. De novo assembly of the transcriptome of Neottopteris nidus using Illumina paired-end sequencing and development of EST-SSR markers. Mol Breeding 36, 94 (2016). https://doi.org/10.1007/s11032-016-0519-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0519-2