
Abstract
Background
We recently showed that a 3-year growth hormone (GH) treatment improves linear growth in severely short children with X-linked hypophosphatemic rickets (XLH). It is unknown if GH therapy increases adult height in XLH patients.
Methods
We carried out a follow-up analysis of a randomized controlled open-label GH study in short prepubertal children with XLH on phosphate and active vitamin D treatment. The changes in SD scores (SDS) of height, sitting height, leg and arm length, and sitting height index (i.e., the ratio between sitting height and height) were analyzed in 11 out of 16 patients followed-up until adult height.
Results
At baseline, XLH patients showed disproportionately short stature with reduced standardized height (−3.2 ± 0.6), sitting height (−1.7 ± 0.6), leg (−3.7 ± 0.7) and arm (−2.5 ± 0.8) length, and markedly elevated sitting height index (3.3 ± 0.6; each p < 0.01 versus healthy children). In GH-treated patients, adult height, sitting height, leg length, and arm length exceeded baseline values by 0.7 SDS, 1.7 SDS, 0.7 SDS, and 1.2 SDS respectively, although this was only significant for sitting height. In controls, no significant changes in linear body dimensions were noted. Adult height did not statistically differ between groups (−2.4 ± 0.7 vs −3.3 ± 1.2, p = 0.082). GH did not exaggerate body disproportion.
Conclusions
Growth hormone treatment did not significantly increase adult height in this group of short children with XLH, which may be at least partly due to the small number of patients included in our study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
X-linked hypophosphatemic rickets (XLH, MIM #307800) is the most frequent inherited phosphate-wasting disorder, with a prevalence of 1/20,000. Patients suffering from XLH have a loss-of-function mutation in the phosphate-regulating gene with homologies to endopeptidase on the X chromosome (PHEX) gene, leading to overexpression of the phosphaturic fibroblast growth factor 23 (FGF23) hormone in bone [1, 2]. In XLH patients, an excess of FGF23 results in renal phosphate-wasting and consecutive hypophosphatemia via down-regulation of the sodium-dependent phosphate transporter in the proximal tubule and reduced 1,25(OH)2D3 levels by inhibition of renal 1-α hydroxylase and stimulation of the catabolic 24-hydroxylase [3, 4]. The characteristic features of XLH are disproportionately short stature, rickets, and osteomalacia starting early in life [5,6,7].
Initiation of early treatment, i.e., within the first year of life, with active vitamin D (calcitriol or 1alpha (OH)D3) and oral doses of phosphate, is of crucial importance in improving growth and hindering skeletal deformities in XLH patients [8,9,10]. Unfortunately, especially in isolated cases, diagnosis is often made after infancy resulting in delayed treatment and thus the majority of XLH patients show poor long-term growth outcome (height < 3rd percentile) despite phosphate and active vitamin D treatment [7, 11,12,13]. In a recent report from France. the mean adult height in female and male XLH patients was 153 cm and 160 cm respectively [13]. Conventional treatment with phosphate and active vitamin D is limited by its renal (hypercalciuria, nephrocalcinosis) and gastrointestinal (abdominal pain, diarrhea) side effects. In addition, both phosphate and active vitamin D trigger further FGF23 expression in bone. resulting in a vicious circle, which may limit the efficacy and safety of the current standard treatment [14].
Several uncontrolled and two controlled studies have shown that treatment with recombinant human growth hormone (GH) is able to improve growth in short children with XLH for treatment periods of up to 3 years [15,16,17,18,19,20,21]. Alongside its direct effects on growth cartilage, GH is thought to stimulate growth in XLH patients by increasing renal phosphate reabsorption and thereby serum phosphate levels via induced circulating insulin-like growth factor 1 (IGF1) [20,21,22].
We recently performed a randomized controlled open-label GH study in 16 severely short prepubertal children with XLH (height z-score < −2.5) on phosphate and active vitamin D treatment [21]. A 3-year GH treatment significantly improved linear growth in these patients, without progression of body disproportion. However, it remains unclear whether GH treatment ultimately increases adult height in XLH patients, as suggested in two uncontrolled trials of GH treatment, including 9 XLH patients in total [22, 23].
We therefore analyzed linear growth, i.e., height, sitting height, leg and arm length, and sitting height index (i.e., ratio between sitting height and height) as a measure of body disproportion, in 11 out of 16 XLH patients, who were initially enrolled in our 3-year randomized controlled study and treated with GH or followed up as controls until attainment of adult height.
Patients and methods
Study design and patients
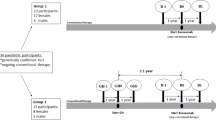
This is a follow-up analysis of a multicenter, prospective, open-label, randomized trial (registered at http://ClinicalTrials.gov, Identifier: NCT00473187) of 3-year GH treatment in severely short prepubertal children with XLH. Details of the study and the 3-year results have been published previously [21]. In brief, 16 patients (8 female) with XLH were enrolled between February 2003 and May 2005 fulfilling the following inclusion criteria:
-
1.
Clinical diagnosis of XLH, i.e., family history, symptoms, physical evaluation consistent with XLH, and the presence of hypophosphatemia due to selective phosphate-wasting in the absence of kidney malformations
-
2.
Age: 3–9 years (girls), 3–10 years (boys)
-
3.
Tanner stage 1 (B1 in girls, G1 and testicular volume < 3 ml in boys)
-
4.
Height z-score below −2.5
-
5.
Height velocity below 75th percentile
-
6.
Complete growth monitoring during the last 12 months
-
7.
Treatment with oral phosphate and calcitriol for at least 1 year
Exclusion criteria were as follows:
-
1.
GH deficiency (i.e., IGF1 < −2 SDS)
-
2.
Untreated hypothyroidism
-
3.
Severe rickets (i.e., serum alkaline phosphatase >800 IU/L)
-
4.
Severe leg deformities (e.g. bowing index >0.15)
-
5.
Uncontrolled secondary hyperparathyroidism (i.e., intact parathyroid hormone (PTH) > 120 pg/ml)
-
6.
Estimated glomerular filtration rate (eGFR) < 60 ml/min per 1.73 m2)
-
7.
Hereditary hypophosphatemic rickets with hypercalciuria
-
8.
Severe organ involvement possibly contributing to growth failure
-
9.
Glucose intolerance
-
10.
History of malignancy
-
11.
Previous treatment with GH, anabolic steroids or glucocorticoids
Fifteen of 16 patients completed the 3-year trial and agreed to continue in this study and written informed consent was obtained from all patients and their parents. During follow-up, 3 patients (2 in the GH and 1 in the control group) were lost to follow-up for social reasons after a median period of 4 years (range 3–7 years) and were therefore excluded from the statistical analysis. In addition, one girl suffered from an idiopathic juvenile thoracolumbar scoliosis, diagnosed at the age of 3 years, which was treated by a full-time anti-gravity brace. In this patient, standardized leg length markedly improved (+1.2 SD score [SDS] within 9 years of GH treatment). However, this only resulted in a slight increase in height (+0.4 SDS), because of progressive scoliosis (Cobb angle: at start of GH, 60°, 2 years, 21°; 3 years, 40°, 5 years, 40°; 6 years, 74°), and GH treatment was stopped before attainment of adult height at the age of 13.6 years. Therefore, this patient was excluded from the adult height analysis. Baseline clinical characteristics of the remaining 11 patients who could be followed up until attaining adult height are given in Table 1. Patients originated from 8 different family trees, and 7 families had one affected child each. Four patients had one or more affected siblings, but only one of the affected siblings was enrolled from each of these two families. In all but one patient the clinical diagnosis was confirmed genetically, as previously published [21].
Treatment protocol
Patients were randomized into a GH-treated group (Genotropin™, Pfizer, Berlin, Germany, dosage: 0.40 mg per kilogram body weight per week given daily by subcutaneous injections; n = 5) and a control group (no additional treatment; n = 6). Patients were evaluated every 3 months until attainment of adult height (i.e., epiphyseal closure on hand X-rays and/or a height velocity < 1 cm/year). At every clinical visit, GH dosage was adjusted according to weight by the responsible physician and a complete interval history was recorded and a physical examination, including Tanner pubertal staging and blood pressure measurement, was performed. Patients were monitored at 3-month intervals with respect to:
-
1.
Complete blood cell count, serum creatinine, sodium, calcium, potassium, chloride, bicarbonate, phosphate, cholesterol, liver enzymes, alkaline phosphatase activity, PTH, and glycosylated hemoglobin
-
2.
Second morning spot urine to determine phosphate, calcium, and creatinine levels
Serum thyroid function was measured at yearly intervals. Finally, renal ultrasound (assessment of nephrocalcinosis according to Hoyer et al. [24]) and optional determination of bone age (according to Greulich and Pyle [25]) was performed at yearly intervals. Tubular maximum for the reabsorption of phosphate (TmP/GFR), and estimated glomerular filtration reate (eGFR) were assessed according to Brodehl et al. [26] and the revised Schwartz formula [27] respectively.
Anthropometry
The same investigator (M.Ž) performed all measurements, as recommended by the International Biological Program [28] using standardized equipment (Stadiometer Dr Keller I, Längen-Messtechnik, Limbach-Oberfrohna, Germany; Siber Hegner Anthropometer Zürich, Switzerland). Height and length of the three longitudinal body segments (i.e., sitting height, arm, and leg length) were measured yearly and height and sitting height data were used to calculate the sitting height index [21]. A detailed description of the underlying procedures and their definition was previously published [29, 30]. The distance between the medial condyles of the right and left femur and the ratio between this distance and leg lengths (i.e., the bowing index) were assessed as indicators for the degree of leg bowing (i.e., genu varum) [7]. Age- and gender-related z scores (SD scores, SDS) were calculated with reference limits derived from healthy children [29, 31]. Target height was calculated as mid-parental height + 10 cm for boys and −2.6 cm for girls [32]. Predicted adult height was calculated according to Greulich and Pyle [33].
Statistics
Data are given as mean ± SD unless otherwise indicated. The normality of distribution was evaluated using the Kolmogorov–Smirnov test for each observed variable. SDS values were calculated according to the equation SDS = (xi – x s)/SD, with xi representing actual patient data, and xs and SD representing corresponding mean and standard deviation from age- and sex-matched healthy peers. Sitting height index was calculated according to the formula: sitting height index = (sitting height/stature)*100. Comparison between groups was performed using Student’s t test in cases of normal data distribution of data and otherwise the Mann–Whitney U test was employed. The longitudinal changes in anthropometric and biochemical data were evaluated using repeated-measures ANOVA, including a within-subject factor (time) and a between-subject factor (i.e., GH group vs controls). SPSS software, version 24 (SPSS, Chicago, IL, USA) was used for all statistical calculations. All p values resulted from two-sided statistical tests and p < 0.05 was considered significant.
Results
Effect of GH on longitudinal growth and adult height
Longitudinal body dimensions were significantly reduced in XLH patients (height SDS, −3.2 ± 0.6; sitting height SDS, −1.7 ± 0.6; leg length SDS, −3.7 ± 0.7, arm length SDS, −2.5 ± 0.8; each p < 0.001 vs healthy children; Table 1.). Leg length was more impaired than trunk length (p < 0.01) resulting in a markedly elevated sitting height index SDS (3.3 ± 0.6; p < 0.001 vs healthy children). Baseline anthropometric data did not differ significantly between groups. The median duration of GH treatment was 7.8 years (range 6.0–11.3 years), and the mean age at attainment of adult height was 17.4 ± 1.3 years and 17.5 ± 1.6 years in GH-treated patients and controls, respectively (p = 0.662).
In GH-treated patients, a significant increase in all linear body dimensions was noted during the first 3 years compared with baseline (3-year increase: height, +1.1 SDS; sitting height, +1.3 SDS; leg length, +0.7 SDS; arm length, +1.3 SDS; each p < 0.05 vs baseline). Sitting height remained significantly increased compared with baseline values until attainment of adult height (total increase, +1.7 SDS; p < 0.05), whereas no significant changes were noted for height (+0.7 SDS, p = 0.167), leg length (+0.7 SDS, p = 0.268), and arm length (+1.2 SDS, p = 0.055; Figs. 1, 2a). By contrast, controls showed no significant changes. Consequently, mean standardized height was significantly higher in GH-treated patients at 2 and 3 years, and mean standardized sitting height and arm length were significantly higher at 2 to 6 years compared with controls (each p < 0.05). Mean adult height was significantly higher compared with predicted adult height in GH-treated patients (−2.39 ± 1.02 SDS vs −4.21 ± 0.36 SDS, p < 0.01), and in controls (−3.25 ± 1.17 SDS vs −4.49 ± 0.72 SDS, p = 0.019). Mean adult height did not differ statistically between groups (GH, −2.39 ± 1.02 SDS; non-GH, −3.25 ± 1.17 SDS; p = 0.082). In both GH-treated patients and controls, mean sitting height index did not change significantly during the study (Fig. 2b).

a–d Mean values for standard deviation scores (SDS) of height, sitting height, arm length, and leg length in patients with growth hormone-treated X-linked hypophosphatemic rickets (XLH) patients (3 boys, 2 girls, black circles) and controls (3 boys, 3 girls, white circles) during the study period. Only patients with complete follow-up data are shown. Data are given as mean ± SEM. *p < 0.05 vs baseline; # p < 0.05 GH-treated patients vs controls. AH adult height, GH growth hormone

a Cumulative changes in standard deviation scores (SDS) of height, sitting height, arm length, and leg length during the study period until attainment of adult height (AH) in growth hormone (GH)-treated patients (3 boys, 2 girls, black circles) and controls (3 boys, 3 girls, white circles). There were no significant differences between groups. Only patients with complete follow-up data are shown. b Change in mean standardized sitting height index during the observation period in GH-treated patients (3 boys, 2 girls, black circles) and controls (3 boys, 3 girls, white circles). No significant change was observed in GH-treated patients and controls. c Change in mean bowing index (i.e., the ratio of the distance between the medial condyles of the right and left femurs and leg lengths) during the observation period in GH-treated patients (3 boys, 2 girls, black circles) and controls (3 boys, 3 girls, white circles). No significant change was observed in GH-treated patients and controls. Data are given as mean ± SEM
Effect of GH on leg bowing
At the time of enrolment, the median distance between the medial condyles of the right and left femur was 31.2 mm (range 10.0–67.2 mm) and 30.6 mm (range 10.0–94.8 mm) in GH-treated patients and controls (p = 0.931). The respective values of the bowing index amounted to 0.045 (range 0.01–0.09) and 0.050 (range 0.01–0.16) in GH-treated patients and controls (p = 1.000; Fig. 2c). Seven patients (4 on GH treatment and 3 controls) underwent a total of 10 orthopedic surgeries for the correction of leg deformities (genu valgum/varum) after a median period of 6.4 years (range 4.5–9.7 years; Table 2). In both groups no significant changes in standardized height or bowing index were noted after surgery at next annual measurement after a median period of 6 months (range 3–9 months) compared with the last measurement before surgery. During the whole study period, no significant changes in the mean bowing index were noted in either patient group (Fig. 2c).
Effect of GH treatment on pubertal development
The median age at start of puberty in GH-treated and non-GH-treated boys was 11.1 years (range 10.9–11.2 years) and 11.4 years (range 11.0–12.2 years) respectively (p = 0.800). The median age at start of puberty in GH-treated and non-GH-treated girls was 10.3 years (range 10.0–10.8 years) and 10.5 years (range 9.9–10.9 years) respectively (p = 1.000). The median age at menarche was 12.8 years (range 12.2–13.6 years) and 14.0 years (11.8–15.9 years) in GH-treated and non-GH-treated girls respectively (p = 0.700).
Effect of GH on serum and urine biochemistry and safety parameters
The mean weight-related dosage of calcitriol and phosphate remained constant throughout the study (data not shown) and did not differ statistically between groups (Table 1). Alkaline phosphatase serum levels and TmP/GFR were elevated compared with baseline during 1–6 years of GH treatment, although this reached statistical significance for alkaline phosphatase only (Table 3). Mean serum PTH levels were significantly elevated after 3 years of GH treatment compared with baseline, whereas no significant changes in serum phosphate, calcium, eGFR, and blood pressure values were noted in either group. However, blood pressure values were above the 95th percentile in one patient of each group. Urinary calcium excretion was not affected by GH and none of the patients showed hypercalciuria before or after beginning GH treatment (Table 1). Low-grade nephrocalcinosis (grades 1 and 2), which was present at the time of enrolment in 4 and 5 patients in the GH and control groups respectively, was resolved at the end of the study in one patient from each group and remained constant in the others. In general, GH was well tolerated and no severe side effects were observed.
Discussion
Our randomized trial showed that GH treatment resulted in a sustained increase in all linear body dimensions in severely short children with XLH, with no worsening of body disproportion. However, adult height did not significantly differ between GH-treated patients and controls in this small patient cohort (p = 0.082). Nevertheless, increased sitting height (+1.7 SDS, p < 0.05) and arm length (+1.2 SDS, p = 0.055) may improve function of daily activities and/or self-esteem in XLH patients, although this remains to be proven.
Catch-up growth during GH treatment was most pronounced during early years, which was followed by non-significant decreases in height z-scores during puberty, suggesting that the benefit of GH treatment might be obtained mainly during the prepubertal growth period. The GH-induced cumulative increases in standardized height amounted to 1.1 SDS within 3 years, and 0.7 SDS until attainment of adult height, although the latter failed to be significant (p = 0.167). Nevertheless, individual patients with severe growth retardation showed a clear benefit of the addition of GH treatment, and final height significantly exceeded predicted adult height at baseline by 1.82 SDS. It is important to note that adult height exceeded predicted adult height at baseline in controls by 1.24 SDS, underlining the importance of initiating medical treatment with active vitamin D and phosphate supplements as soon as possible [8,9,10].
The cumulative increase at adult height was comparable with two previously reported non-controlled studies involving a total of 9 XLH patients treated with GH for periods of 3.1 to 10.6 years (median, 1.0 SDS; range, 0.3–1.3) [22, 23]. In addition, in one study, adult height exceeded predicted adult height to a similar extent (0.8 SDS) as in the present study [22]. However, as most patients in the above-mentioned studies had already entered puberty during the first or second year of GH treatment, and in view of the small number of patients included in all studies, comparisons must be made cautiously.
Concern has been raised that GH treatment may induce the early onset of puberty in XLH patients [23]. Therefore, it is important to note that all patients experienced a normal onset of puberty irrespective of GH treatment in the present study. Similarly, adult height was reached at the expected ages in boys and girls.
At the time of enrolment, XLH patients presented with considerable body disproportion, with a preferential impairment in leg length and preserved trunk length resulting in a markedly elevated sitting height index (3.3 SDS). Similar findings on body disproportion have been seen in XLH patients previously and, as a rule of thumb, the shortest patients present with the highest degree of body disproportion [7]. However, because our study was initiated in 2001, our case series may not represent the average patient with XLH who is nowadays diagnosed early in life.
Concern was previously raised that GH may preferentially stimulate trunk growth, thereby worsening body disproportion [18]. In our study, the GH-induced cumulative increase in standardized sitting height appeared somewhat higher than that of leg length (1.7 vs 0.7 SDS, p = 0.225). However, sitting height index as a measure of body disproportion, in addition to the bowing index indicating the degree of leg deformities, remained stable throughout the study period, irrespective of GH treatment, which confirms the results of our previous 3-year GH trial [21]. Moreover, in non-GH-treated patients, an increase in seating height index was noted during puberty [7]. It is important to note that 8 of the 11 patients underwent orthopedic surgery for correction of leg deformities at puberty between the 5th and 10th years of the study. This is in line with the literature, where the need for orthopedic surgery for leg deformities was reported in up to 89% of XLH patients, although its need apparently dropped in more recent years because of earlier diagnosis and more vigorous medical treatment [13, 34]. Interestingly, the mean standardized height at the first follow-up visit after leg surgery did not significantly differ from the last measurement before surgery. However, one has to bear in mind, that correction of leg deformity by transient hemiepiphysiodesis takes considerable time and that in pubertal XLH patients (age 12–15 years) a mean loss of height of approximately 0.3 SDS can be expected within a period of 6 months [7].
In GH-treated patients, a transient increase in PTH levels was noted but not in controls. A tendency toward exacerbation of secondary hyperparathyroidism in GH-treated XLH patients was also noted in a previous study [18] and is thought to be due to the phosphate-raising properties of GH [21]. This highlights the need for careful monitoring of PTH and adaptation of vitamin D dosage in XLH patients treated with GH.
Standard treatment with active vitamin D and phosphate supplementation is known to further stimulate FGF23 serum levels and thereby promote renal phosphate-wasting in XLH patients [14, 35]. A recent study in GH-deficient children showed that GH treatment resulted in an up-regulation of the FGF23/Klotho system, suggesting that the phosphate-raising properties of GH are mediated by insulin-like growth factor-1 rather than by down-regulation of the FGF23/Klotho system [36]. Therefore, it would be interesting to know whether GH treatment also stimulated circulating FGF23 in our patients. Unfortunately, determination of FGF23 was not available when our study was initiated in 2001 and thus serum sampling was not performed.
Growth hormone was well tolerated and not associated with hypercalciuria or nephrocalcinosis. Instead, ultrasound examination revealed resolution of nephrocalcinosis in several of our patients with and without GH treatment and at a rate similar to previous observations in non-GH-treated pediatric XLH patients [37]. It is important to note that in one girl, who was not included in this analysis, as GH was stopped before attainment of adult height, progressive preexisting scoliosis was noted during GH treatment, which substantially limited height benefit in this patient (+0.4 SDS). Growth hormone treatment was not associated with an increased risk of scoliosis in children with idiopathic short stature or Prader–Willi syndrome [38, 39]. By contrast, worsening of preexisting scoliosis was suggested in a recent study in 35 patients with Ullrich–Turner syndrome followed up over 4 years on GH treatment [40]. Thus, GH-induced stimulation of spine growth may also have worsened preexisting idiopathic scoliosis in the above-mentioned patient. However, no other potentially GH-induced adverse events were noted in the present study, and there was no negative impact from orthopedic surgery in the careful follow-up of GH-treated patients.
Several limitations of this study must be noted: the total number of patients enrolled was low; several patients underwent surgical correction of leg bowing during puberty; and 5 out of 16 patients were lost to follow-up during the study period of 16 years. This substantially limited statistical power and may have biased the results of this study.
In conclusion, in this randomized- controlled trial, GH treatment resulted in a sustained increase in all linear body dimensions in severely short children with XLH, without a worsening of body disproportion. However, GH treatment did not significantly increase adult height compared with controls in this small patient cohort. Further adequately powered studies are needed to evaluate the effect of GH on adult height in XLH patients.
References
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S (2002) Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87(11):4957–4960
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348(17):1656–1663
Goetz R, Nakada Y, MC H, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M (2010) Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-klotho complex formation. Proc Natl Acad Sci U S A 107(1):407–412
Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD (2006) Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 17(5):1305–1315
Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL (2011) A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res 26(7):1381–1388
Pavone V, Testa G, Gioitta Iachino S, Evola FR, Avondo S, Sessa G (2015) Hypophosphatemic rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol 25(2):221–226
Zivicnjak M, Schnabel D, Billing H, Staude H, Filler G, Querfeld U, Schumacher M, Pyper A, Schroder C, Bramswig J, Haffner D (2011) Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr Nephrol 26(2):2232–2231
Kruse K, Hinkel GK, Griefahn B (1998) B calcium metabolism and growth during early treatment of children with X-linked hypophosphataemic rickets. Eur J Pediatr 157(11):894–900
Makitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E (2003) Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 88(8):3591–3597
Quinlan C, Guegan K, Offiah A, Neill RO, Hiorns MP, Ellard S, Bockenhauer D, Hoff WV, Waters AM (2012) Growth in PHEX-associated X-linked hypophosphatemic rickets: the importance of early treatment. Pediatr Nephrol 27(4):581–588
Santos F, Fuente R, Mejia N, Mantecon L, Gil-Pena H, Ordonez FA (2013) Hypophosphatemia and growth. Pediatr Nephrol 28(4):595–603
Ariceta G, Langman CB (2006) Growth in X-linked hypophosphatemic rickets. Eur J Pediatr 166(4):303–309
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prie D, Rothenbuhler A, Wicart P, Harvengt P (2014) Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect 3(1):R13–R30
Carpenter TO, Insogna KL, Zhang JH, Ellis B, Nieman S, Simpson C, Olear E, Gundberg CM (2010) Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status. J Clin Endocrinol Metab 95(11):E352–E357
Lanes R, Harrison HE (1990) Growth hormone therapy in a poorly growing child with hypophosphatemic rickets. J Endocrinol Investig 13(10):833–837
Wilson DM, Lee PD, Morris AH, Reiter EO, Gertner JM, Marcus R, Quarmby VE, Rosenfeld RG (1991) Growth hormone therapy in hypophosphatemic rickets. Am J Dis Child 145(10):1165–1170
Saggese G, Baroncelli GI, Bertelloni S, Perri G (1995) Long-term growth hormone treatment in children with renal hypophosphatemic rickets: effects on growth, mineral metabolism, and bone density. J Pediatr 127(3):395–402
Haffner D, Wuhl E, Blum WF, Schaefer F, Mehls O (1995) Disproportionate growth following long-term growth hormone treatment in short children with X-linked hypophosphataemia. Eur J Pediatr 1154(8):610–613
Reusz GS, Miltenyi G, Stubnya G, Szabo A, Horvath C, Byrd DJ, Peter F, Tulassay T (1997) X-linked hypophosphatemia: effects of treatment with recombinant human growth hormone. Pediatr Nephrol 11(5):573–577
Seikaly MG, Brown R, Baum M (1997) The effect of recombinant human growth hormone in children with X-linked hypophosphatemia. Pediatrics 100(5):879–884
Zivicnjak M, Schnabel D, Staude H, Even G, Marx M, Beetz R, Holder M, Billing H, Fischer DC, Rabl W, Schumacher M, Hiort O, Haffner D (2011) Three-year growth hormone treatment in short children with X-linked hypophosphatemic rickets: effects on linear growth and body disproportion. J Clin Metab 96(12):2097–2105
Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G (2001) Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr 138(2):236–243
Haffner D, Nissel R, Wuhl E, Mehls O (2004) Effects of growth hormone treatment on body proportions and final height among small children with X-linked hypophosphatemic rickets. Pediatrics 113(6):E593–E596
Hoyer PF (2004) Nephrokalzinose. In: Hofmann V, Deeg KH, Hoyer PF (eds) Ultraschall-diagnostik in Pädiatrie und Kinderchirurgie. Thieme, Stuttgart, pp 467–470
Greulich WW, Pyle SI (1959) Radiographic atlas of skeletal development of the hand and wrist. Stanford University Press, Stanford
Brodehl J, Krause A, Hoyer PF (1988) Assessment of maximal tubular phosphate reabsorption: comparison of direct measurement with the nomogram of Bijvoet. Pediatr Nephrol 2(2):183–189
De Souza VC, Rabilloud M, Cochat P, Selistre L, Hadj-Aissa A, Kassai B, Ranchin B, Berg U, Herthelius M, Dubourg L (2012) Schwartz formula: is one k-coefficient adequate for all children? PLoS One 7(12):E53439
Weiner JS, Lourie JA (1981) Practical human biology. Academic Press, London
Zivicnjak M, Narancic NS, Szirovicza L, Franke D, Hrenovic J, Bisof V (2003) Gender-specific growth patterns for stature, sitting height and limbs length in Croatian children and youth (3 to 18 years of age). Coll Antropol 27(1):321–334
Zivicnjak M, Franke D, Filler G, Haffner D, Froede K, Nissel R, Haase S, Offner G, Ehrich JH, Querfeld U (2007) Growth impairment shows an age-dependent pattern in boys with chronic kidney disease. Pediatr Nephrol 22(3):420–429
Zivicnjak M, Smolej Narancic N, Szirovicza L, Franke D, Hrenovic J, Bisof V, Tomas Z, Skaric-Juric T (2008) Gender-specific growth patterns of transversal body dimensions in Croatian children and youth (2 to 18 years of age). Coll Antropol 32(2):419–431
Molinari L, Largo RH, Prader A (1985) Target height and secular trend in the Swiss population. In: Borms J, Hauspie R, Sand A, Susanne C, Habbelinck M (eds) Human growth and development. Plenum, New York, pp 193–200
Bayley N, Pinneau S (1952) Tables for predicting adult height from skeletal age: revised for use with the Greulich-Pyle hand standards. J Pediatr 40(4):423–441
Nielsen LH, Rahbek ET, Beck-Nielsen SS, Christesen HT (2014) Treatment of hypophosphataemic rickets in children remains a challenge. Dan Med J 61(7):A4874
Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ (2010) Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab 95(4):1846–1850
Efthymiadou A, Kritikou D, Mantagos S, Chrysis D (2016) The effect of GH treatment on serum FGF23 and klotho in GH-deficient children. Eur J Endocrinol 174(4):473–479
Auron A, Alon US (2005) Resolution of medullary nephrocalcinosis in children with metabolic bone disorders. Pediatr Nephrol 20(8):1143–1145
Allen DB (2011) Safety of growth hormone treatment of children with idiopathic short stature: the US experience. Horm Res Paediatr 76 [Suppl 3]:45–47
Wolfgram PM, Carrel AL, Allen DB (2013) Long-term effects of recombinant human growth hormone therapy in children with Prader-Willi syndrome. Curr Opin Pediatr 25(4):509–514
Ricotti S, Petrucci L, Carenzio G, Klersy C, Calcaterra V, Larizza D, Dalla Toffola E (2011) Prevalence and incidence of scoliosis in Turner syndrome: a study in 49 girls followed-up for 4 years. Eur J Phys Rehabil Med 47(3):447–453
Acknowledgements
We very much appreciate the willingness of our patients and their families to participate in this trial. We thank all the members of the Hypophosphatemic Rickets Study Group of the “Deutsche Gesellschaft für Kinderendokrinologie und -diabetologie” (German Society for Pediatric Endocrinology and Diabetology) and the “Gesellschaft für Pädiatrische Nephrologie” (German Society for Pediatric Nephrology) for their help in the design and realization of this project. We especially thank E. Schmid (Kinderpraxen Hirschaid, Germany) and D. Wölfel (Children’s Hospital Göppingen, Germany, who recruited patients into this study. This project was supported by a research grant from Pfizer Pharma GmbH, who also provided Genotropin™ for treatment.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
The study received full approval from the Ethics Committee of the Charité Hospital, Berlin and each participating center. Assent and written informed consent for participation was obtained from all patients and/or their parents.
Conflicts of interest
DH has served as a consultant for Sandoz and has received honoraria and/or research support from Amgen Inc., Horizon Pharma, Kyowa Kirin, Merck Serono, Pfizer, and Sandoz. DS has served as a consultant for Sandoz and Novo Nordisk and has received honoraria from Kyowa Kirin, Alexion, Merck Serono, Pfizer, and Ipsen. OH has served as a consultant for Novo Nordisk and has received honoraria from Sandoz, Ipsen, Merck Serono, and Ultragenyx. MŽ, NM, HS, EW, MM, RB, UQ, MH, HB, WR, CS, JB, and AR declare that they have no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Meyerhoff, N., Haffner, D., Staude, H. et al. Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol 33, 447–456 (2018). https://doi.org/10.1007/s00467-017-3820-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-017-3820-3