
Abstract
Objective
In the present study, we sought to identify causal relationships between obesity and other complex traits and conditions using a data-driven hypothesis-free approach that uses genetic data to infer causal associations.
Methods
We leveraged available summary-based genetic data from genome-wide association studies on 1498 phenotypes and applied the latent causal variable method (LCV) between obesity and all traits.
Results
We identified 110 traits causally associated with obesity. Of those, 109 were causal outcomes of obesity, while only leg pain in calves was a causal determinant of obesity. Causal outcomes of obesity included 26 phenotypes associated with cardiovascular diseases, 22 anthropometric measurements, nine with the musculoskeletal system, nine with behavioural or lifestyle factors including loneliness or isolation, six with respiratory diseases, five with body bioelectric impedances, four with psychiatric phenotypes, four related to the nervous system, four with disabilities or long-standing illness, three with the gastrointestinal system, three with use of analgesics, two with metabolic diseases, one with inflammatory response and one with the neurodevelopmental disorder ADHD, among others. In particular, some causal outcomes of obesity included hypertension, stroke, ever having a period of extreme irritability, low forced vital capacity and forced expiratory volume, diseases of the musculoskeletal system, diabetes, carpal tunnel syndrome, loneliness or isolation, high leukocyte count, and ADHD.
Conclusions
Our results indicate that obesity causally affects a wide range of traits and comorbid diseases, thus providing an overview of the metabolic, physiological, and neuropsychiatric impact of obesity on human health.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity is a complex, multifactorial and preventable disease in which an imbalance between daily caloric energy intake and expenditure leads to unwanted and atypical accumulation of fat or adipose tissue, which in turn results in the impairment of human health (Hruby and Hu 2015; Purnell 2018; Panuganti et al. 2020). Obesity is the second most common cause of preventable death after smoking (Hurt et al. 2010; Mitchell et al. 2011; Ng et al. 2014; Panuganti et al. 2020), making it an essential target of public health interventions. Globally, its prevalence has increased by 27.5% for adults and 47.1% for children in the last three decades (Ng et al. 2014; Apovian 2016), affecting over 500 million adults (Panuganti et al. 2020).
Obesity is typically defined according to body mass index (BMI), which is estimated as the ratio of weight in kilograms and height in meters squared (De Lorenzo et al. 2016; Panuganti et al. 2020). Typically, an individual with obesity has a BMI higher than 30 kg/m2 (De Lorenzo et al. 2016; Panuganti et al. 2020). The BMI-based classification includes underweight (BMI < 18.5), normal range (18.5 < BMI < 24.9), overweight (25 < BMI < 29.9), obesity class I (30 < BMI < 34.9), obesity class II (35 < BMI < 39.9) and obesity class III (BMI > 40) (De Lorenzo et al. 2016; Panuganti et al. 2020).
Genetic epidemiological studies have made considerable advances in the understanding of the genetic propensity to obesity. Genome-wide association studies (GWAS) have identified ~ 950 genomic loci associated with an obesity measure (Yengo et al. 2018; Tam et al. 2019) and genetic overlap with other anthropometric measurements, coronary artery disease, blood pressure and type 2 diabetes, among others, has been reported (Locke et al. 2015). A genetic correlation (rG) between two traits could be explained by horizontal pleiotropy (i.e. genetic variants have a direct effect on both traits, not via each other) or by vertical pleiotropy (i.e. the effect of a genetic variant on a trait is mediated by its effect on another trait) (O’Connor and Price 2018; Haworth et al. 2020).
Horizontal pleiotropy represents a challenge for statistical methods seeking to determine causality between two traits. For example, traditional Mendelian randomisation (MR) methods can be biased due to horizontal pleiotropy, which increases the likelihood of false-positive findings (O’Connor and Price 2018; Koellinger and de Vlaming 2019; García-Marín et al. 2021). The latent causal variable (LCV) is a recently developed statistical approach developed to investigate whether a genetic correlation between traits is explained by causal effects or by horizontal pleiotropic effects (O’Connor and Price 2018; Haworth et al. 2020; García-Marín et al. 2021).
Understanding the extent to which obesity is causally associated with other conditions is a fundamental question in obesity research. Here, we conduct a genetic screening using GWAS summary data to identify potential causal associations between obesity and other phenotypes. Specifically, we apply the LCV method to perform a hypothesis-free phenome-wide screening to the extensive collection of phenotypes with GWAS summary data (N = 1498) compiled in the Complex Trait Genetics Virtual Lab (CTG-VL) web platform.
Methods
Data
The present study used summary statistics from GWAS for obesity and 1498 other phenotypes. Summary statistics summarise relevant parameters such as allele frequency, effect size, standard error and the p value of genetic variants tested on the trait of interest. Several published GWAS have made available their summary statistics to the scientific community to enable researchers to advance understanding of the genetic components of several phenotypes. The CTG-VL web platform (https://genoma.io/) (Cuéllar-Partida et al. 2019) has compiled a set of 1610 GWAS summary statistics, and the inclusion criteria was a nominally significant heritability derived from LD-score regression. The CTG-VL web platform includes GWAS summary statistics from the UK Biobank released by Neale’s Lab (www.nealelab.is/uk-biobank/) (Neale’s Lab 2018) and from GWAS consortia. For this study, we only used GWAS derived from European populations to avoid potential biases due to population differences in linkage disequilibrium and allele frequencies.
Obesity dataset
The obesity GWAS summary statistics used here correspond to a sample (N = 361,194) of European ancestry from the second wave of GWAS results released by Neale’s Lab (ICD10 code E66) (Neale’s Lab 2018; Cuéllar-Partida et al. 2019) available in the CTG-VL web platform. Obesity was assessed as a binary trait based on BMI classification (BMI > 30), and GWAS summary statistics were adjusted for age, age2, inferred_sex, age * inferred_sex, age2 * inferred_sex, and 20 genetic ancestry principal components (Neale’s Lab 2018; Cuéllar-Partida et al. 2019).
LCV analysis
Genetic causal proportion (GCP) between the obesity GWAS and 1498 GWAS was estimated using the phenome-wide LCV pipeline implemented in the CTG-VL web platform as described previously (Haworth et al. 2020; García-Marín et al. 2021). Briefly, GWAS summary statistics for obesity were formatted and uploaded onto the CTG-VL web platform. Then, we used the phenome-wide analysis pipeline (Haworth et al. 2020) that is available at the CTG-VL web platform, which includes LD-score regression (Bulik-Sullivan et al. 2015b) as well as LCV analysis (O’Connor and Price 2018). Lastly, causal architecture plots were used to visualise the results. In particular, as part of the phenome-wide analysis pipeline, the LCV method was applied to all traits that showed a genetic correlation with obesity based on bivariate LD-score regression (Bulik-Sullivan et al. 2015b) at Benjamini–Hochberg’s False Discovery Rate (FDR < 5%). Then, to account for multiple testing on LCV estimates, we applied an FDR < 5% to the GCP estimates.
The phenome-wide analysis pipeline (Haworth et al. 2020), which is publicly available in the CTG-VL web platform, is performed in R 4.00 based on the R script that the original authors of the LCV method (O’Connor and Price 2018) have made available (https://github.com/lukejoconnor/LCV). Within this pipeline, to ensure consistency of alleles and variants across GWAS summary statistics, data are formatted using munge_sumstats.py made available by the LD-score software and HapMap SNPs are extracted using the list of SNPs w_hm3.snplist (https://github.com/bulik/ldsc/wiki). Full details about the phenome-wide analysis pipeline in the CTG-VL web platform are described and illustrated in previous studies (Haworth et al. 2020; García-Marín et al. 2021).
The LCV method does not distinguish between the ‘exposure’ and the ‘outcome’ (O’Connor and Price 2018). These are exchangeable labels that do not affect the degree of causality; specifically, the sign of the result denotes which trait is the determinant and which trait is the outcome (O’Connor and Price 2018). Further, the LCV method estimates GCP by assuming a latent variable L that mediates the genetic correlation between two traits, which is assumed to be a causal component mediating the genetic correlation between the phenotypes (O’Connor and Price 2018; Haworth et al. 2020; García-Marín et al. 2021). The GCP ranges from -1 (full genetic causality of Trait 2 on Trait 1) to 1 (full genetic causality of Trait 1 in Trait 2). A |GCP| of 1 indicates that a genetic correlation between traits may be explained by vertical pleiotropy, whereas a GCP of 0 indicates that a genetic correlation between traits may be explained by horizontal pleiotropy. (O’Connor and Price 2018; Haworth et al. 2020; García-Marín et al. 2021). Notably, a |GCP|< 0.60 is considered low and indicates limited partial genetic causality (O’Connor and Price 2018).
Sensitivity analysis
As a sensitivity analysis, we applied Bonferroni correction for multiple testing comparisons to identify traits with statistically significant genetic correlations and with evidence of a causal relationship with obesity based on their genetic correlation and GCP estimates. Bonferroni is a stricter, more conservative approach than FDR to determine statistical significance.
Results
We conducted a phenome-wide LCV analysis between obesity and 1498 other phenotypes to estimate their genetic correlation and GCP. We identified 266 traits genetically correlated with obesity at FDR < 5%. Of those, 105 were causally associated (|GCP|> 0.6; FDR < 5%; Online Resource 1) and five showed evidence of a limited partial genetic causality (|GCP|< 0.6; FDR < 5%; Online Resource 1). Putative outcomes of obesity included cardiovascular diseases, anthropometric measurements, the health of the musculoskeletal system, behavioural or lifestyle factors, respiratory diseases, body bioelectric impedances, psychiatric disorders, diseases of the nervous system, disabilities or long-standing illness, health of the gastrointestinal system, use of analgesics, metabolic diseases, inflammatory response and neurodevelopmental disorders, among others (Table 1 and Fig. 1).

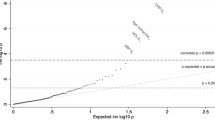
Causal associations for obesity (FDR < 5%). Causal architecture plots showing the latent causal variable exposome-wide analysis results. Each dot represents a trait with a significant genetic correlation with obesity. The x-axis shows the GCP estimate, whilst the y-axis shows the genetic causality proportion (GCP) absolute Z-score (as a measure of statistical significance). The statistical significance threshold (FDR < 5%) is represented by the red dashed lines, while the division for causal determinants of obesity (on the left) and causal outcomes of obesity (on the right) is represented by the grey dashed lines. Results are shown separately for traits with a positive genetic correlation with obesity (a) and with a negative genetic correlation with obesity (b). *Phenotypes causally associated with obesity in the present study but not with BMI in Haworth et al. (2020). Phenotypes without an asterisk (*) were also identified for BMI in Haworth et al. (2020)
Our results show 26 cardiovascular phenotypes as potential consequences of obesity. For instance, we identified obesity as a causal determinant of self-reports in hypertension (GCP = 0.94, p valueGCP = 7.71 × 10−76) and heart attack (GCP = 0.39, p valueGCP = 1.68 × 10−04). Conditions diagnosed by a doctor such as high blood pressure (GCP = 0.93, p valueGCP = 4.34 × 10−55), heart attack (GCP = 0.32, p valueGCP = 1.58 × 10−03) and angina problems (GCP = 0.63, p valueGCP = 1.82 × 10−02) were causal outcomes of obesity. A similar pattern was observed for chronic ischaemic heart disease (GCP = 0.72, p valueGCP = 1.26 × 10−04) and diastolic blood pressure. In contrast, obesity was found to cause a decline in high-density lipoprotein (HDL) cholesterol (GCP = 0.68, p valueGCP = 1.68 × 10−02; Fig. 1 and Table 2).
Our results show a potential causal effect of obesity on 22 anthropometric measurements. In particular, obesity caused increments of 18 traits, including fat percentages throughout the body and ankle spacing width, while a decline in height (GCP = 0.72, p valueGCP = 1.22 × 10−04) and handgrip strength were caused by obesity. Similarly, we found evidence of obesity causing the decrease of five body bioelectrical impedance measures, including in both arms and legs.
Obesity was a causal determinant of eight phenotypes involving diseases of the musculoskeletal and connective tissue (Table 2). Pain-related phenotypes such as knee (GCP = 0.87, p valueGCP = 5.81 × 10−14) and hip (GCP = 0.79, p valueGCP = 2.44 × 10−07) pain in the last month along with self-reported osteoarthritis, arthrosis and gonarthrosis (ICD10), were caused by obesity. In contrast, leg pain in calves (GCP = − 0.80, p valueGCP = 4.18 × 10−06) was a causal determinant of obesity.
Six causal relationships were observed between obesity and respiratory-related phenotypes. For instance, obesity was observed to increase risk of shortness of breath (GCP = 0.85, p valueGCP = 1.43 × 10−10) and whistling in the chest (GCP = 0.69, p valueGCP = 8.09 × 10−04). Consistently, obesity caused the decline of forced vital capacity (FVC; GCP = 0.89, p valueGCP = 1.48 × 10−23) and forced expiratory volume in one second (FEV1; GCP = 0.83, p valueGCP = 3.69 × 10−12) (Fig. 1 and Table 2).
Obesity was a causal determinant of four psychiatric-related traits (Fig. 1 and Table 2), including gaining weight during the worst period of depression (GCP = 0.63, p valueGCP = 1.82 × 10−02). Also, obesity was observed to increase irritability through traits such as ever having a period of extreme irritability (GCP = 0.85, p valueGCP = 5.67 × 10−13), experiencing manifestations of mania or irritability (GCP = 0.68, p valueGCP = 1.77 × 10−03) and ever highly irritable for 2 days. Similarly, an increase in the behavioural trait loneliness or isolation (GCP = 0.93, p valueGCP = 6.11 × 10−52) was found to be caused by obesity (Fig. 1 and Table 2).
Obesity was a causal determinant of four diseases of the nervous system (GCP = 0.76, p valueGCP = 8.77 × 10−06; Table 2) including mononeuropathies of upper limb (ICD10; GCP = 0.72, p valueGCP = 1.37 × 10−04), carpal tunnel syndrome (GCP = 0.68, p valueGCP = 1.27 × 10−03) and nerve, nerve root and plexus disorders (GCP = 0.66, p valueGCP = 3.7 × 10−03; Fig. 1).
Three phenotypes related to the gastrointestinal system, including diverticular disease of the intestine (ICD10; GCP = 0.67, p valueGCP = 3.43 × 1 −03) and self-reported gastro-oesophageal reflux (GCP = 0.62, p valueGCP = 1.22 × 10−02) were identified as potential outcomes of obesity (Table 2).
Diabetes, both self-reported (GCP = 0.72, p valueGCP = 1.90 × 10−03) and diagnosed by a doctor (GCP = 0.75, p valueGCP = 3.49 × 10−04), showed evidence of being causal outcomes of obesity. Also, obesity was also found to pose a causal effect on high leukocyte levels (GCP = 0.66, p valueGCP = 2.00 × 10−03) and attention-deficit/hyperactivity disorder (ADHD; GCP = 0.64, p valueGCP = 1.84 × 10−02).
Sensitivity analysis
As a sensitivity analysis, we applied a Bonferroni correction to our results instead of FDR to account for multiple testing (Bonferroni < 0.05; Fig. 2). Bonferroni is well known to be a much more conservative approach than FDR (Noble 2009). Using this approach, we identified 86 genetic correlations. Of those, 52 were causal outcomes of obesity (GCP > 0.6; Online Resource 2); these included anthropometric and bioelectrical impedance measurements, poor health of the musculoskeletal system, hypertension, diabetes, and ADHD (Table 3). We did not find any causal determinants of obesity with this approach.

Causal associations for obesity (Bonferroni < 5%). Causal architecture plots showing the latent causal variable exposome-wide sensitivity analysis results. Each dot represents a trait with a significant genetic correlation with obesity. The x-axis shows the GCP estimate, whilst the y-axis shows the genetic causality proportion (GCP) absolute Z-score (as a measure of statistical significance). The statistical significance threshold (Bonferroni < 5%) is represented by the red dashed lines, while the division for causal determinants of obesity (on the left) and causal outcomes of obesity (on the right) is represented by the grey dashed lines. Results are shown separately for traits with a positive genetic correlation with obesity (a) and with a negative genetic correlation with obesity (b)
Discussion
In this work, we performed a phenome-wide screening of potential causes and effects of obesity on a number of health conditions. Although previous research has made extensive efforts to describe how obesity and metabolic syndrome affect several body systems, their relationships with inflammatory response, hypertension, cardiovascular disease, neurodevelopmental disorders, and the musculoskeletal and nervous system has not been fully elucidated. In this study, we found that obesity was causally associated with increased leukocyte count, self-reported hypertension, high blood pressure and diabetes diagnosed by a doctor. Leukocytes are white blood cells involved in both local and general inflammatory response (Langer and Chavakis 2009; Leick et al. 2014). Further, it has previously been reported that obesity increases adipose tissue dysfunction, leading to a pro-inflammatory state, which in turn can result in vascular dysfunction impairing endothelium vasodilation with an impact on hypertension and affecting the responsiveness of the insulin-vasodilator mechanism (Campia et al. 2012; Swarup et al. 2020). Also, obesity is considered the main cause of metabolic syndrome components such as high blood pressure and triglycerides, while the increased risk of diabetes is attributed to a decrease in insulin secretion as a consequence of obesity-related effects (Goodarzi 2018).
Obesity and metabolic syndrome are major risk factors for cardiovascular disease (Goodarzi 2018; Swarup et al. 2020; Panuganti et al. 2020). In the present study, several cardiovascular diseases were causal outcomes of obesity. Specifically, our results indicate that obesity is a causal determinant of major coronary heart disease events, heart attacks, myocardial infarctions, chronic ischaemic heart disease (ICD10) and angina problems. Consistently, an increase in Aspirin’s intake, which is commonly prescribed for secondary prevention of cardiovascular diseases (Ansa et al. 2019), was also identified as an outcome of obesity. Further, it has been previously shown that inflammation is a significant risk factor for heart disease events (Hoffman et al. 2004; Kim et al. 2017). Thus, our findings are consistent with previous studies reporting a causal effect of obesity on cardiovascular disease and support the hypothesis in which obesity-related inflammation (i.e. high leukocyte levels) could lead to high blood pressure and hypertension, which in turn are known risk factors for cardiovascular disease.
Obesity causes an increase in the mechanical load across weight-bearing joints, which has been associated with musculoskeletal deterioration and neuropathic pain (Anandacoomarasamy et al. 2008; Hozumi et al. 2016). Further, previous studies have suggested that an increase in fat mass may result in a decrease in bone mass (Anandacoomarasamy et al. 2008). Our findings uncovered causal associations in which obesity is the putative causal determinant of musculoskeletal pain and diseases such as osteoarthritis. A similar pattern was observed for diseases of the nervous system, including mononeuropathies and nerve, nerve root and plexus disorders. Consistently, obesity caused an increase in the use of analgesics such as Aspirin, Codamol and Paracetamol. Our results suggest that an increase in analgesics use may be explained by the development of musculoskeletal pain and damage to the nervous system. It is possible that the inflammatory state induced by obesity may also result in poorer musculoskeletal and nervous system health. However, more research is needed to disentangle the complex relationships between specific-tissue inflammation, pain and obesity.
Previous studies have described a higher incidence of disability among people with obesity (Anandacoomarasamy et al. 2008; Queirós et al. 2015). Our results identified causal associations between obesity and disability-related phenotypes such as disability living allowance and long-standing illness or disability. Thus, it is possible that the development of disabling conditions among people with obesity is explained by poor musculoskeletal and nervous system health.
Despite extensive efforts to advance our understanding of the relationship between obesity and lung function, the effects of obesity on the respiratory system have not been fully elucidated. Previous findings suggest that obesity may be associated with complex respiratory diseases such as chronic obstructive pulmonary disease (COPD) and its severity (Zammit et al. 2010; Mafort et al. 2016; Dixon and Peters 2018). Consistently, our results suggest that obesity causes increased risk of shortness of breath and whistling in the chest, as well as decreases in FVC and FEV1. Moreover, traits such as COPD onset and other obstructive pulmonary disease (ICD10) were identified as causal outcomes of obesity. Previous studies have also reported leukocyte accumulation in lung tissue in individuals with a chronic obstructive pulmonary disease, which in turn increases the expression of adhesion molecules in bronchial blood vessels (Davis et al. 2012; Koo et al. 2017). Our results would suggest that obesity poses a deteriorative effect on the respiratory system, which could contribute to the development of complex respiratory diseases. We speculate that this relationship could be explained by the accumulation of adipose tissue and inflammation arising from an increase in trunk fat mass, which in turn could lead to physiological changes decreasing lung capacity and weakening the respiratory muscles. However, more research is needed to disentangle the intricate relationship between obesity, inflammation, lung function and disease.
Observational studies have reported an association between obesity and gastrointestinal diseases such as diverticular diseases and gastro-oesophageal reflux (Camilleri et al. 2017). Similarly, increased adipose tissue has been inversely linked with adiponectin levels, which are a protective factor of gastro-oesophageal reflux complications (Chang and Friedenberg 2014). Consistently, our findings show causal associations in which obesity is a causal determinant of diverticular diseases of the intestine (ICD10) and self-reported gastro-oesophageal reflux.
Previous studies have sought to describe the extent to which obesity could be involved in the development of depression, leading to an unclear set of conclusions (Mannan et al. 2016; Day et al. 2018; Chauvet-Gelinier et al. 2019; Speed et al. 2019). For instance, some studies suggest a bidirectional relationship between obesity and depression (Mannan et al. 2016; Chauvet-Gelinier et al. 2019), while others report that anthropometric measurements such as BMI, fat mass and height are only risk factors for depression (Speed et al. 2019). Also, a Mendelian randomisation analysis reports a one-way causal association for BMI causing loneliness and a bidirectional causal association between BMI and depressive symptoms, suggesting that the relationship between these traits is complex and perhaps a consequence of shared biological mechanisms (Day et al. 2018) or lifestyle factors such as sleep quality, diet and physical inactivity (Hawkley and Cacioppo 2010). In the present study, we provide evidence for obesity as a causal determinant of psychiatric-related phenotypes, such as gaining weight during the worst period of depression and loneliness or isolation. However, we did not find evidence for a causal association between obesity and depression (Supplementary File 1). It is most likely that obesity causes weight gain due to the effect of obesity’s genetic variants regardless if an individual is depressed or not. Nonetheless, as previous studies have noted, there may be bidirectional effects between obesity and depression (Luppino et al. 2010; Day et al. 2018). As we discuss in the limitations below, LCV is not able to estimate bidirectional causality, and null findings from LCV analyses could be due to bidirectional causal effects or the absence of causality.
Previous research has pointed out an association between ADHD and obesity. However, cause–effect links and the underpinning molecular mechanisms of this association remain unclear (Cortese and Tessari 2017; Cortese 2019). Observational studies have examined the relationship between ADHD and obesity with both traits as exposure and outcome, suggesting that this association is independent of potential confounding factors (Cortese and Tessari 2017). Also, some genetic studies show a one-way causal relationship in which high BMI is a causal determinant of ADHD (Martins-Silva et al. 2019), while others suggest that a plausible bidirectional causal association may exist between obesity and ADHD (Liu et al. 2020). In the present study, results show a causal association in which obesity is a causal determinant of ADHD. However, to fully understand the effect of obesity on ADHD, additional research should seek to elucidate potential mechanisms underlying this association.
Pleiotropic effects, both horizontal and vertical, among obesity-related phenotypes have become a focal point of interest in genetic epidemiological studies, and pleiotropic effects have been identified between abdominal obesity and immune response (Kaur et al. 2019). However, Kaur et al. (2019) were unable to delineate vertical from horizontal pleiotropic effects in the association between abdominal obesity and immune response. Other studies have reported that abdominal obesity increases the risk to develop autoimmune diseases due to a chronic inflammatory state (Gustafsson et al. 2013; Rosenberg et al. 2013). Our results add up to the evidence (Lumeng and Saltiel 2011; Andersen et al. 2016; Kaur et al. 2019) suggesting that obesity prompts an immune response and a chronic inflammatory state with detrimental effects on the overall health.
The main strengths of the LCV method as compared to traditional MR methods include (O’Connor and Price 2018; Koellinger and de Vlaming 2019; Haworth et al. 2020; García-Marín et al. 2021): (i) it is less prone to bias due to horizontal pleiotropy; (ii) it is robust to sample overlap, and (iii) it uses aggregated information across the entire genome, increasing statistical power and enabling analyses between pairs of phenotypes that would be considered “underpowered” for other statistical methods.
Our results are consistent with previous studies reporting that LCV is a meaningful tool to detect potential causal associations in underpowered phenotypes for which MR methods have not been able to determine potential causation (O’Connor and Price 2018; Haworth et al. 2020; García-Marín et al. 2021). We suggest that the causal relationships pointed out through LCV could be used as a testable hypothesis for future epidemiological observational and genetic studies.
Methods and findings in the present study must be compared to those in Haworth et al. (2020). The main difference between the work by Haworth et al. (2020) and ours is the phenotypes that were investigated. For instance, the GWAS summary statistics used by Haworth et al. (2020) represent BMI as a continuous variable; whereas, in the present study, GWAS summary statistics correspond to obesity defined as the dichotomisation of BMI (De Lorenzo et al. 2016; Panuganti et al. 2020). This difference between phenotypes is directly reflected in the genetic correlation between the GWAS of obesity used here and GWAS of BMI used by Haworth et al., 2020 (rG = 0.67, SE = 0.11, p value = 7.87 × 10–10), showing that although BMI is a common screening tool for obesity (Hall and Cole 2006), a continuous BMI measurement does not entirely reflect obesity as a disease. Furthermore, the study by Haworth et al. (2020) assessed BMI against 1340 other phenotypes. In contrast, in the present study, we have expanded the panel of traits and tested obesity against 1498 other phenotypes.
Regarding the results, one of the principal differences is that in our study, we only identified one potential causal association in which obesity was the outcome of another phenotype (leg pain in calves), which is most likely explained by lack of physical activity. In contrast, the study by Haworth et al. (2020) identified 23 traits potentially affecting BMI; however, none of them included leg pain in calves. Most of the causal determinants of BMI in the study by Haworth et al. included occupational-related phenotypes, which are most likely explained by lack of physical activity and socioeconomic variables such as assortative mating and educational attainment. As we discuss in the limitations below, it is possible that we did not observe as many causal determinants for obesity as Haworth et al. did for BMI due to a reduction of statistical power. In addition, Haworth et al. (2020) identified 110 traits as outcomes of BMI, whereas our study identified 109 traits as outcomes of obesity. A total of 40 causal outcomes were identified in both studies (Fig. 1, Table 2 and Online Resource 1); these included self-reported hypertension, high blood pressure diagnosed by a doctor, gonarthrosis (ICD10), diastolic blood pressure, heart attack diagnosed by a doctor, knee pain in the last month, hip pain in the last month, FEV1, carpal tunnel syndrome, waist circumference, bioelectrical impedance measurements and fat percentages throughout the body.
Sixty-eight potential causal outcomes were only observed in the present study, but not by Haworth et al. (2020). These include those between obesity and leukocyte levels, ADHD, loneliness, long-standing illness or disability, self-reported and diagnosed by a doctor diabetes, cardiovascular diseases, diseases of the nervous system, and diseases of the musculoskeletal system and connective tissue, among others (Fig. 1, Table 2 and Online Resource 1). We attribute these substantial differences among the findings of both studies to the differences in phenotype definition. Also, we note that the following 11 phenotypes causally associated with obesity in the present study were not tested in Haworth et al. (2020): Any ischemic stroke (Europeans only), any ischaemic stroke (trans-ethnic meta-analysis), any stroke (trans-ethnic meta-analysis), any stroke (European only), coronary artery disease, HDL Cholesterol (both sexes, males only and females only), age at menarche, ADHD, and leukocyte count.
Establishing causal associations should always arise only after convergent evidence from studies with multiple designs. Ideally, at least one should be an interventional design (e.g. a randomised controlled trial). However, interventional studies are not only expensive and time-consuming, but also, in many instances, are unfeasible or unethical to conduct (i.e. when an exposure known to harm participants is evaluated). In these cases, assessing causality using genetics can be informative. Nonetheless, some limitations of the present study must be acknowledged. Although our data included GWAS summary statistics derived from consortia, which include only individuals of European ancestry but are on participants from a number of countries, most of our data were retrieved primarily from the UK Biobank, which predominantly consists of participants of European ancestry, and previous studies have highlighted ethnic differences in obesity (Higgins et al. 2019). Thus, our results’ generalisability is limited to European ancestry individuals until tested in other ethnicities. Also, by dichotomising BMI to define obesity, statistical power is reduced compared to the continuous BMI measurement used in the study by Haworth et al. (2020). It is possible that the results identified in Haworth et al. (2020) but not in the present study are due to the difference of statistical power between studies.
Differences between methods used to correct for multiple comparisons should be noted. FDR has been used in previous studies describing LCV analyses results (O’Connor and Price 2018; Haworth et al. 2020; García-Marín et al. 2021). A main advantage of FDR is that it does not require tests to be independent of each other. Thus, it is useful when assessing several hypotheses that are simultaneously tested (Chen et al. 2017), like in the present study. However, FDR is less stringent than other multiple testing correction methods, such as Bonferroni (Chen et al. 2017). Although a Bonferroni correction for multiple tests is much stricter and less prone to false-positive findings, it assumes that all tests must be independent of each other (Stevens et al. 2017). This condition is not met in the present study because some traits in the phenome-wide scan are correlated (i.e. cardiovascular phenotypes, anthropometric traits, psychiatric phenotypes, among others). Here, we included results for potential causal associations between obesity and 1 498 other phenotypes using FDR < 5% correction and, as a sensitivity analysis, we have included the results for the phenome-wide analysis pipeline using a Bonferroni < 5% correction; however, we note that our tests are not entirely independent from one another.
Regarding potential bias in our analyses due to sample overlap, the LCV method and genetic correlations estimated with LD-score regression can handle sample overlap (Bulik-Sullivan et al. 2015a; O’Connor and Price 2018). For potential bias due to sample ascertainment, we note that the UK Biobank cohort is selected for older adults aged between 40 and 69. Around 9 million individuals were invited to participate (Munafò et al. 2018). However, the UK Biobank only achieved a 5% response rate (Munafò et al. 2018); thus, the resulting sample is not entirely representative of the UK population. For instance, in the first release of genetic data, current smokers were underrepresented (19% in the general population vs 11% in UK Biobank) (Munafò et al. 2018). However, this was no longer the case for the full release of genome-wide association data (Munafò et al. 2018), which is the one available at the CTG-VL web platform. Ascertainment bias within the UK Biobank can induce spurious correlations and genetic effects. For example, recent analyses have shown autosomal heritability of sex in big cohorts is likely explained by participation bias (Pirastu et al. 2020).
Our analyses included more than 1400 phenotypes; however, causal associations with other traits not tested here may exist. Related to this is the interpretability of some of the phenotypes used here, such as taking medication: Aspirin which could be considered a proxy trait for pain or cardiovascular disease, taking medication: Candesartan cilexetil and lisinopril which could be considered a proxy trait for hypertension. Unfortunately, other traits for which GWAS summary statistics are available lack such a straightforward proxy interpretation.
Similarly, diabetes phenotypes, both self-reported and diagnosed by a doctor, included gestational diabetes, type 1 diabetes, type 2 diabetes, diabetes insipidus and unclassified diabetes. Considering that the physiopathology of these phenotypes is substantially different (Skyler et al. 2017), future studies should aim to further assess the associations between obesity and specific types of diabetes. In addition, even though the LCV method uses genetic information aggregated across the entire genome, the GCP estimates are still tied to the statistical power of the GWAS. Thus, the ability to causal associations for some phenotypes is limited, particularly for those with small sample sizes. Also, the LCV may estimate spurious associations when the genetic correlation between traits is mediated by multiple latent factors (O’Connor and Price 2018). However, the presence of multiple latent factors would reduce statistical power and lower GCP estimates biasing results towards the null (O’Connor and Price 2018). Lastly, the LCV method seeks to detect the predominant causal direction between two phenotypes (O’Connor and Price 2018; Haworth et al. 2020), and therefore, bidirectional causality cannot be tested between traits. This limitation is intrinsic to the nature of the LCV method in which a bidirectional causal association would mimic horizontal pleiotropy biasing the GCP towards the null. Therefore, null findings may have occurred due to a lack of power, because there is truly no causal relationship or because the relationship is bidirectional. The possibility of bidirectional effects should be further explored in future studies using methods that can test for bidirectionality.
In summary, we assessed potential causal relationships between obesity and 1498 phenotypes, and identified 110 traits with significant causal associations with obesity. Our findings uncovered the effect of obesity on leukocyte-related inflammation, which may incur in a chronic pro-inflammatory state and several metabolic syndrome components. Further, we provide evidence for the impact of obesity on cardiovascular disease, poor health of the respiratory and musculoskeletal systems and its potential damage to the nervous system. We observe a causal effect of obesity on gastrointestinal disorders, psychiatric phenotypes and the neurodevelopmental disorder ADHD. Also, we identified causal associations of obesity on bioelectrical impedances and physical disability. Altogether, our results confirm some previously reported associations and identify some new testable hypotheses that could contribute to advance our understanding of the effects of obesity on metabolic inflammation in specific tissues and organs, which in turn may provide novel perspectives on the metabolic implications of obesity and the development of anti-inflammatory therapeutics.
Data availability
Summary-level data used in the present study are publicly available at the Complex Traits Genomics Virtual Lab (https://genoma.io/) web platform.
Code availability
Code used as part of the present manuscript is web platform-based work, which is available at the Complex Traits Genomics Virtual Lab (https://genoma.io/) web platform. Links and details to access the pipeline used in the present study are provided in the Methods section of this manuscript.
References
Anandacoomarasamy A, Caterson I, Sambrook P et al (2008) The impact of obesity on the musculoskeletal system. Int J Obes 32:211–222. https://doi.org/10.1038/sj.ijo.0803715
Andersen CJ, Murphy KE, Fernandez ML (2016) Impact of obesity and metabolic syndrome on immunity. Adv Nutr 7:66–75. https://doi.org/10.3945/an.115.010207
Ansa BE, Hoffman Z, Lewis N et al (2019) Aspirin use among adults with cardiovascular disease in the United States: implications for an intervention approach. J Clin Med Res. https://doi.org/10.3390/jcm8020264
Apovian CM (2016) Obesity: definition, comorbidities, causes, and burden. Am J Manag Care 22:c176–c185
Bulik-Sullivan B, Finucane HK, Anttila V et al (2015a) An atlas of genetic correlations across human diseases and traits. Nat Genet 47:1236–1241. https://doi.org/10.1038/ng.3406
Bulik-Sullivan BK, Loh P-R, Finucane HK et al (2015b) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47:291–295. https://doi.org/10.1038/ng.3211
Camilleri M, Malhi H, Acosta A (2017) Gastrointestinal complications of obesity. Gastroenterology 152:1656–1670. https://doi.org/10.1053/j.gastro.2016.12.052
Campia U, Tesauro M, Cardillo C (2012) Human obesity and endothelium-dependent responsiveness. Br J Pharmacol 165:561–573. https://doi.org/10.1111/j.1476-5381.2011.01661.x
Chang P, Friedenberg F (2014) Obesity and GERD. Gastroenterol Clin N Am 43:161–173. https://doi.org/10.1016/j.gtc.2013.11.009
Chauvet-Gelinier J-C, Roussot A, Cottenet J et al (2019) Depression and obesity, data from a national administrative database study: geographic evidence for an epidemiological overlap. PLoS ONE 14:e0210507. https://doi.org/10.1371/journal.pone.0210507
Chen S-Y, Feng Z, Yi X (2017) A general introduction to adjustment for multiple comparisons. J Thorac Dis 9:1725–1729. https://doi.org/10.21037/jtd.2017.05.34
Cortese S (2019) The association between ADHD and obesity: intriguing, progressively more investigated, but still puzzling. Brain Sci. https://doi.org/10.3390/brainsci9100256
Cortese S, Tessari L (2017) Attention-deficit/hyperactivity disorder (ADHD) and obesity: update 2016. Curr Psychiatry Rep 19:4. https://doi.org/10.1007/s11920-017-0754-1
Cuéllar-Partida G, Lundberg M, Kho PF et al (2019) Complex-traits genetics virtual lab: a community-driven web platform for post-GWAS analyses. bioRxiv 518027
Davis BB, Shen Y-H, Tancredi DJ et al (2012) Leukocytes are recruited through the bronchial circulation to the lung in a spontaneously hypertensive rat model of COPD. PLoS ONE 7:e33304. https://doi.org/10.1371/journal.pone.0033304
Day FR, Ong KK, Perry JRB (2018) Elucidating the genetic basis of social interaction and isolation. Nat Commun 9:2457. https://doi.org/10.1038/s41467-018-04930-1
De Lorenzo A, Soldati L, Sarlo F et al (2016) New obesity classification criteria as a tool for bariatric surgery indication. World J Gastroenterol 22:681–703. https://doi.org/10.3748/wjg.v22.i2.681
Dixon AE, Peters U (2018) The effect of obesity on lung function. Expert Rev Respir Med 12:755–767. https://doi.org/10.1080/17476348.2018.1506331
García-Marín LM, Campos AI, Martin NG et al (2021) Inference of causal relationships between sleep-related traits and 1527 phenotypes using genetic data. Sleep. https://doi.org/10.1093/sleep/zsaa154
Goodarzi MO (2018) Genetics of obesity: what genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol 6:223–236. https://doi.org/10.1016/S2213-8587(17)30200-0
Gustafsson S, Lind L, Söderberg S et al (2013) Oxidative stress and inflammatory markers in relation to circulating levels of adiponectin. Obesity 21:1467–1473. https://doi.org/10.1002/oby.20097
Hall DMB, Cole TJ (2006) What use is the BMI? Arch Dis Child 91:283–286. https://doi.org/10.1136/adc.2005.077339
Hawkley LC, Cacioppo JT (2010) Loneliness matters: a theoretical and empirical review of consequences and mechanisms. Ann Behav Med 40:218–227. https://doi.org/10.1007/s12160-010-9210-8
Haworth S, Kho PF, Holgerson PL et al (2020) Assessment and visualization of phenome-wide causal relationships using genetic data: an application to dental caries and periodontitis. Eur J Hum Genet. https://doi.org/10.1038/s41431-020-00734-4
Higgins V, Nazroo J, Brown M (2019) Pathways to ethnic differences in obesity: the role of migration, culture and socio-economic position in the UK. SSM Popul Health 7:100394. https://doi.org/10.1016/j.ssmph.2019.100394
Hoffman M, Blum A, Baruch R et al (2004) Leukocytes and coronary heart disease. Atherosclerosis 172:1–6. https://doi.org/10.1016/S0021-9150(03)00164-3
Hozumi J, Sumitani M, Matsubayashi Y et al (2016) Relationship between neuropathic pain and obesity. Pain Res Manag 2016:2487924. https://doi.org/10.1155/2016/2487924
Hruby A, Hu FB (2015) The epidemiology of obesity: a big picture. Pharmacoeconomics 33:673–689. https://doi.org/10.1007/s40273-014-0243-x
Hurt RT, Kulisek C, Buchanan LA, McClave SA (2010) The obesity epidemic: challenges, health initiatives, and implications for gastroenterologists. Gastroenterol Hepatol 6:780–792
Kaur Y, Wang DX, Liu H-Y, Meyre D (2019) Comprehensive identification of pleiotropic loci for body fat distribution using the NHGRI-EBI Catalog of published genome-wide association studies. Obes Rev 20:385–406. https://doi.org/10.1111/obr.12806
Kim JH, Lim S, Park KS et al (2017) Total and differential WBC counts are related with coronary artery atherosclerosis and increase the risk for cardiovascular disease in Koreans. PLoS ONE 12:e0180332. https://doi.org/10.1371/journal.pone.0180332
Koellinger PD, de Vlaming R (2019) Mendelian randomization: the challenge of unobserved environmental confounds. Int J Epidemiol 48:665–671. https://doi.org/10.1093/ije/dyz138
Koo H-K, Kang HK, Song P et al (2017) Systemic white blood cell count as a biomarker associated with severity of chronic obstructive lung disease. Tuberc Respir Dis 80:304–310. https://doi.org/10.4046/trd.2017.80.3.304
Langer HF, Chavakis T (2009) Leukocyte-endothelial interactions in inflammation. J Cell Mol Med 13:1211–1220. https://doi.org/10.1111/j.1582-4934.2009.00811.x
Leick M, Azcutia V, Newton G, Luscinskas FW (2014) Leukocyte recruitment in inflammation: basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res 355:647–656. https://doi.org/10.1007/s00441-014-1809-9
Liu C-Y, Schoeler T, Davies NM et al (2020) Are there causal relationships between attention-deficit/hyperactivity disorder and body mass index? Evidence from multiple genetically informed designs. Int J Epidemiol. https://doi.org/10.1093/ije/dyaa214
Locke AE, Kahali B, Berndt SI et al (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518:197–206. https://doi.org/10.1038/nature14177
Lumeng CN, Saltiel AR (2011) Inflammatory links between obesity and metabolic disease. J Clin Invest 121:2111–2117. https://doi.org/10.1172/JCI57132
Luppino FS, de Wit LM, Bouvy PF et al (2010) Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry 67:220–229. https://doi.org/10.1001/archgenpsychiatry.2010.2
Mafort TT, Rufino R, Costa CH, Lopes AJ (2016) Obesity: systemic and pulmonary complications, biochemical abnormalities, and impairment of lung function. Multidiscip Respir Med 11:28. https://doi.org/10.1186/s40248-016-0066-z
Mannan M, Mamun A, Doi S, Clavarino A (2016) Prospective associations between depression and obesity for adolescent males and females—a systematic review and meta-analysis of longitudinal studies. PLoS ONE 11:e0157240. https://doi.org/10.1371/journal.pone.0157240
Martins-Silva T, dos Santos VJ, Hutz MH et al (2019) Assessing causality in the association between attention-deficit/hyperactivity disorder and obesity: a Mendelian randomization study. Int J Obes 43:2500–2508. https://doi.org/10.1038/s41366-019-0346-8
Mitchell NS, Catenacci VA, Wyatt HR, Hill JO (2011) Obesity: overview of an epidemic. Psychiatr Clin N Am 34:717–732. https://doi.org/10.1016/j.psc.2011.08.005
Munafò MR, Tilling K, Taylor AE et al (2018) Collider scope: when selection bias can substantially influence observed associations. Int J Epidemiol 47:226–235. https://doi.org/10.1093/ije/dyx206
Neale’s Lab (2018) GWAS Results. In: UK Biobank—Neale Lab. http://www.nealelab.is/uk-biobank. Accessed 29 Jul 2020
Ng M, Fleming T, Robinson M et al (2014) Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384:766–781. https://doi.org/10.1016/S0140-6736(14)60460-8
Noble WS (2009) How does multiple testing correction work? Nat Biotechnol 27:1135–1137. https://doi.org/10.1038/nbt1209-1135
O’Connor LJ, Price AL (2018) Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet 50:1728–1734. https://doi.org/10.1038/s41588-018-0255-0
Panuganti KK, Nguyen M, Kshirsagar RK (2020) Obesity. In: StatPearls. StatPearls Publishing
Pirastu N, Cordioli M, Nandakumar P et al (2020) Genetic analyses identify widespread sex-differential participation bias. bioRxiv
Purnell JQ (2018) Definitions, classification, and epidemiology of obesity. In: Endotext [Internet]. MDText.com, Inc.
Queirós FC, Wehby GL, Halpern CT (2015) Developmental disabilities and socioeconomic outcomes in young adulthood. Public Health Rep 130:213–221. https://doi.org/10.1177/003335491513000308
Rosenberg L, Nanda KS, Zenlea T et al (2013) Histologic markers of inflammation in patients with ulcerative colitis in clinical remission. Clin Gastroenterol Hepatol 11:991–996. https://doi.org/10.1016/j.cgh.2013.02.030
Skyler JS, Bakris GL, Bonifacio E et al (2017) Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 66:241–255. https://doi.org/10.2337/db16-0806
Speed MS, Jefsen OH, Børglum AD et al (2019) Investigating the association between body fat and depression via Mendelian randomization. Transl Psychiatry 9:184. https://doi.org/10.1038/s41398-019-0516-4
Stevens JR, Al Masud A, Suyundikov A (2017) A comparison of multiple testing adjustment methods with block-correlation positively-dependent tests. PLoS ONE 12:e0176124. https://doi.org/10.1371/journal.pone.0176124
Swarup S, Goyal A, Grigorova Y, Zeltser R (2020) Metabolic Syndrome. In: StatPearls. StatPearls Publishing
Tam V, Turcotte M, Meyre D (2019) Established and emerging strategies to crack the genetic code of obesity. Obes Rev 20:212–240. https://doi.org/10.1111/obr.12770
Yengo L, Sidorenko J, Kemper KE et al (2018) Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet 27:3641–3649. https://doi.org/10.1093/hmg/ddy271
Zammit C, Liddicoat H, Moonsie I, Makker H (2010) Obesity and respiratory diseases. Int J Gen Med 3:335–343. https://doi.org/10.2147/IJGM.S11926
Acknowledgements
AIC and LMGM are supported by UQ Research Training Scholarships from The University of Queensland (UQ). MER thanks the National Health and Medical Research Council and Australian Research Council’s support through a Research Fellowship (APP1102821).
Funding
L.M.G.M and A.I.C. are supported by UQ Research Training Scholarships from The University of Queensland (UQ). M.E.R. thanks support of the NHMRC and Australian Research Council (ARC) through a Research Fellowship (GNT1102821). P.F.K. is supported by an Australian Government Research Training Program Scholarship from Queensland University of Technology (QUT).
Author information
Authors and Affiliations
Contributions
MER and GC-P conceived and directed the study. LMG-M performed the statistical and bioinformatics analyses, with support and input from AIC, P-FK, NGM, GC-P and MER. LMG-M wrote the first draft of the paper and integrated input and feedback from all co-authors.
Corresponding authors
Ethics declarations
Conflict of interest
GC-P contributed to this study while employed at The University of Queensland. He is now an employee of 23andMe Inc., and he may hold stock or stock options. All other authors declare having no conflicts of interest.
Ethics approval
This study was approved by the Human Research Ethics Committee of the QIMR Berghofer Medical Research Institute.
Consent to participate (ethics)
Informed consent was obtained from all individual participants included in the study.
Consent to publish (ethics)
All participants provided informed consent for the publication of study results.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
García-Marín, L.M., Campos, A.I., Kho, PF. et al. Phenome-wide screening of GWAS data reveals the complex causal architecture of obesity. Hum Genet 140, 1253–1265 (2021). https://doi.org/10.1007/s00439-021-02298-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02298-9