
Abstract
The immune cell system is a critical component of host defense. Recruitment of immune cells to sites of infection, immune reaction, or injury is complex and involves coordinated adhesive interactions between the leukocyte and the endothelial cell monolayer that lines blood vessels. This article reviews basic mechanisms in the recruitment of leukocytes to tissues and then selectively reviews new concepts that are emerging based on advances in live cell imaging microscopy and mouse strains. These emerging concepts are altering the conventional paradigms of inflammatory leukocyte recruitment established in the early 1990s. Indeed, recent publications have identified previously unrecognized contributions from pericytes and interstitial leukocytes and their secreted products that guide leukocytes to their targets. Investigators have also begun to design organs on a chip. Recent reports indicate that this avenue of research holds much promise.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The conventional multistep adhesion cascade paradigm of leukocyte recruitment
The inflammatory response is the cornerstone of the body’s defense mechanism against bacterial and viral pathogens and against physical-, chemical- and environmental-mediated tissue and organ damage. Acute inflammation occurs within minutes to hours and the telltale signs are readily observed microscopically: the recruitment of blood leukocytes and an increase in vascular permeability (Kumar et al. 2010). Successful leukocyte transmigration across postcapillary venules is the culmination of sequential and overlapping steps mediated by multiple classes of adhesion molecules expressed on the surface of endothelial cells and their interactions with cognate ligands expressed by leukocytes. Drs. von Andrian and Butcher were the first to describe a molecular model of leukocyte recruitment in vivo: a two-step model consisting in leukocyte rolling via L-selectin (formerly called LECAM-1) followed by firm adhesion via β2 integrins (von Andrian et al. 1991). As with most other molecules discovered later to play a role in leukocyte transmigration, critical information was acquired by using function-blocking monoclonal antibodies to these adhesion molecules and direct visualization in real time of rabbit postcapillary venules by intravital microscopy (IVM; for a review, see Butcher 1991). Another paradigm shift in the field was the demonstration that the vascular endothelium plays an active and essential role in recruitment of circulating leukocytes. Upon treatment with tumor necrosis factor-α (TNF-α) or interleukin-1β (IL-1β) cytokines or Escherichia coli Gram-negative lipopolysaccharide treatment, the normally non-adhesive and non-thrombogenic surface of the endothelium becomes pro-adhesive or “activated” (Bevilacqua and Gimbrone 1987). Reports from several laboratories during this time revealed that this transformation in the endothelial cell phenotype was mediated by a transcriptionally regulated program involving the nuclear factor kappa-B (NFκB)-dependent pathway triggered by pro-inflammatory cytokines or bacterial endotoxins (for a review, see Collins et al. 1995).
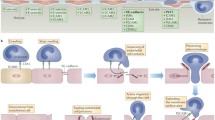
The paradigm for neutrophil recruitment in postcapillary venules has since been further expanded by investigators, based primarily on the study of neutrophils (Fig. 1; for a review, see Kolaczkowska and Kubes 2013) and one can broadly generalize that most leukocytes follow a similar multi-step cascade in the peripheral (non-lymphoid) vasculature with some exceptions. Accordingly, an updated adhesion cascade in postcapillary venules involves the initial attachment or tethering of free-flowing leukocytes and then their slow velocity rolling (step 1), stable adhesion (arrest) on endothelial cells (step 2), flattening (step 3) and subsequent crawling on the vascular endothelium, followed by transendothelial cell migration (TEM) between (paracellular route) or through (transcellular) the vascular endothelium (step 4) and uropod elongation to complete the transmigration of postcapillary venules (step 5). The initial attachment and rolling steps are initiated by interactions of endothelial E- and P-selectins and their counter-receptors on leukocytes, namely L-selectin, P-selectin glycoprotein ligand-1, CD44, CD43 and E-selectin ligand-(ESL)1. The rolling step is reversible, unless followed by endothelial-presented chemoattractants and/or chemokines that activate leukocyte α4β1 (also called VLA4) and two members of the β2 integrin family, namely lymphocyte function-associated antigen 1 (LFA-1) and macrophage-1 antigen (Mac-1), to cause leukocyte arrest by binding to their cognate ligands, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), respectively. Neutrophils from patients with leukocyte adhesion deficiencies (LAD), which lack selectin ligands (LAD type II), are unable to roll on endothelium, whereas leukocytes from patients lacking β2 integrins or expressing mutant β2 integrins (LAD type I) are unable to arrest stably. LAD type 1 and 2 mutations are rare and result in defects in leukocyte recruitment and severe recurrent infections in these patients (for a review, see van de Vijver et al. 2012). Leukocytes from patients that have mutations in kindlin-3 (FERMT3 gene), a cytosolic protein that binds to the cytoplasmic domains of β1, β2 and β3 integrins, fail to activate these three integrins and, as a result, emigrate poorly into tissues (Svensson et al. 2009). This disorder is called LAD type III. Since kindlin-3 binds to αIIbβ3 integrin (the fibrinogen receptor) expressed in platelets, LAD type III patients also have defects in platelet adhesion and coagulation, in addition to defects in leukocyte adhesion and present with severe and recurrent infections and hemostasis defects (for a review, see van de Vijver et al. 2012). Once stably arrested on the endothelial surface, leukocytes flatten, probably to reduce their exposure to the shear stress force of flowing blood and collisions with circulating blood cells and crawl variable distances before initiating transendothelial migration. These events rely on leukocyte β1 and β2 integrins binding to their endothelial-expressed cognate ligands, ICAM-1 and VCAM-1, respectively. In particular, recent studies in neutrophils and monocytes indicate that the Mac-1 integrins (αMβ2) mediate apical leukocyte crawling (Schenkel et al. 2004; Phillipson et al. 2006).

Steps of the conventional paradigm for leukocyte transmigration and ventral lamellipodia recovery of transmigration gaps. Inflammation from an abscess activates the endothelium to express adhesion molecules E-selectin, intercellular cell adhesion molecule (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) and chemokines that mediate the multistep adhesion cascade. 1 Rolling of leukocytes via expression of selectin ligands (P-selectin glycoprotein ligand-1, L-selectin). 2 Arrest and firm adhesion is induced when β2 integrins on the leukocyte (lymphocyte-function-associated antigen 1 [LFA-1]) bind to ICAM-1 on the activated endothelium under laminar shear flow conditions. Further integrin-mediated adhesion strengthening and spreading (3) is LFA-1/ICAM-1-dependent; VCAM-1 is also involved in adhesion of monocytes, T cells, basophils and eosinophils. 4 Transmigration occurs primarily by the paracellular pathway, as shown here and in some cases by a transcellular transendothelial route (for a review, see Alcaide et al. 2009). The leukocyte crosses the basement membrane preferentially at areas of lower collagen and laminin densities and migrates (5) toward the inflammatory focus. A schematic model of transmigration pore closure with ventral lamellipodia is shown enlarged in the inset (upper right). Loss of tension in the endothelial cell actin filaments (black lines in the cell) is proposed to be a trigger initiating ventral lamellipodia formation
Dissecting molecular mechanisms of transendothelial migration: contributions of both endothelium and leukocytes are necessary
Recent studies suggest that crawling T lymphocytes probe the apical surface for sites to transmigrate by generating membrane protrusions, called podoprints or filopodia, into the endothelial surface; however, whether neutrophils or other leukocyte types produce these membrane protrusions is as yet unknown (Carman et al. 2007; Shulman et al. 2009). Colocalizing with the T cell membrane protrusions are clusters of endothelial molecules including ICAM-1; VCAM-1 does not appear to exhibit this behavior (Shulman et al. 2009). The adhesion of many leukocyte types to ICAM-1, E-selectin, VCAM-1, CD47 and platelet endothelial cell adhesion molecule-1 (PECAM-1) has been documented to trigger multiple intracellular signals in the endothelium preceding leukocyte TEM (for reviews, see Alcaide et al. 2009; van Buul and Hordijk 2009). The proximal signaling events detected include transient intracellular Ca2+ mobilization, the activation of many tyrosine kinases including Src and Pyk-2 and phosphotyrosine phosphatases and the subsequent association of many of these adhesion molecules to the cytoskeleton through binding to cytoskeletal adaptor molecules such as cortactin, α-actinin and filamin. Intracellular vesicles, termed the lateral border recycling compartment (LBRC), contain leukocyte adhesion molecules that are essential for leukocyte TEM. LBRC are actively transported to locations at cell-cell borders where transmigration of leukocytes occurs (see below for details of the LBRC). In addition, VCAM-1, ICAM-1 and E-selectin have been shown to interact with one another and are organized in the plasma membrane into endothelial adhesion platforms (EAPs) by tetraspanins. EAPs are envisioned to facilitate leukocyte attachment and signaling during transmigration (Barreiro et al. 2008) and to contribute to the activation of small GTPases that initiate cytoskeletal reorganization. Ultimately, the signals generated by adherent leukocytes trigger the formation of gaps in the protein complexes that comprise the adherens junctions at endothelial cell-cell borders. The major complex includes the vascular endothelial (VE)-cadherin and its associated cytosolic binding partners, namely α-, β-, γ- and p120-catenins and vascular endothelial-receptor protein tyrosine phosphatase (VE-PTP; Vestweber 2012). The mechanisms underlying the dissociation of VE-cadherin-catenin-VE-PTP and gap formation have not been completely elucidated but they appear to involve tyrosine phosphorylation of the cytoplasmic tail of VE-cadherin, an event that destabilizes or dissociates VE-cadherin from catenin and VE-PTP, thus enabling VE-cadherin to be either endocytosed or transiently moved aside (for reviews, see Alcaide et al. 2009; Vestweber 2012). Recently, VCAM-1-induced Rac1 activation, reactive oxygen species (ROS) and Pyk2 signaling have been reported to play a key role in triggering the dissociation of the VE-PTP-VE-cadherin complex via a surprising requirement for the binding of an as yet unidentified a substrate for the VE-PTP (Vockel and Vestweber 2013). Whether this is a universal mechanism is unclear, since not all leukocytes express the ligand VLA-4, which binds to VCAM-1, suggesting that other molecule(s) are needed to initiate this process.
The lateral border recycling compartment
A certain amount of membrane movement has been described to occur at endothelial cell borders during leukocyte transendothelial migration, with membrane constitutively being internalized and recycled in what has been termed the “lateral border recycling compartment” (LBRC) by Muller and colleagues (Muller 2011). Homophilic interactions between PECAM-1 (also called CD31) on leukocytes and PECAM-1 at the endothelial border trigger the targeted recycling of PECAM-1-containing membrane vesicles from the LBRC located beneath actively transmigrating leukocytes at the lateral borders (Mamdouh et al. 2003, 2008). Interestingly, although the LBRC contains other junctional molecules involved in transmigration including CD99, junctional adhesion molecule-A and poliovirus receptor (CD155; also called PVR), VE-cadherin is not included. As mentioned above, during transmigration, VE-cadherin moves out of the way, forming characteristic gaps in the otherwise linear VE-cadherin complex (Shaw et al. 2001; Allport et al. 2000). The LBRC membrane surrounds the transmigrating leukocyte and is transported along microtubules to the site of diapedesis by kinesin motors (Mamdouh et al. 2008). Indeed, the blocking of PECAM-1, the depolymerization of microtubules, or the blocking kinesin function all inhibit transmigration without affecting leukocyte adhesion and without being additive, indicating a role in the same pathway. The authors conclude that the LBRC integrates those molecules necessary for leukocyte passage into a “transmigration complex”, with a high number of unligated adhesion molecules available for interactions, while at the same time opening or loosening junctions by excluding VE-cadherin. Recently, the Muller group has also found that CD155, which is involved in transmigration through interactions with its cognate leukocyte ligand DNAM-1 (CD266) expressed on monocytes, can also be found in the LBRC (Reymond et al. 2004; Sullivan et al. 2013). Adhesion of monocytes to CD155 of endothelial cells activates a src-kinase-dependent recruitment of the tyrosine-protein phosphatase non-receptor type 11 (also called Shp-2). The authors, therefore, suggest that the localization of adhesion/signaling molecules to the lateral border recycling compartment and the recruitment of Shp-2 are common mechanisms for the regulation of TEM. Some key aspects of the LBRC remain unanswered. How do the dynamics of the LBRC vesicular compartment synchronize with the dissociation of the VE-cadherin-VE-PTP-catenin complex to create the portal for leukocyte transmigration and by extension, how does the LBRC play a role in the recovery of the VE-cadherin-VE-PTP-catenin complex at cell-cell borders? Each of these processes is also of great relevance to the control of vascular permeability.
New insights into molecules that regulate leukocyte integrin affinity
Integrins are a family of αβ heterodimeric adhesion receptors that mediate attachment between cells and other cells or to the basement membrane and transmit, via signal transduction, information about the environmental status into cells; they are thus vital players in leukocyte recruitment. The β2 integrin family is exclusively expressed in leukocytes and, as mentioned earlier, is essential for leukocyte arrest on the endothelium and for migration across the endothelium (Ley et al. 2007). Integrin activation is required for these adhesive functions. An important aspect of integrin biology is their dual role of sensing and interacting with the surrounding environment through outside-in and inside-out signaling (Hynes 2002). Integrins exist in various states of activation that are in equilibrium with one another and that depend on the activation status of the cell. In unstimulated leukocytes, integrins are usually in a conformation with low binding affinity, until they receive stimulating signals from other receptors, such as chemokine receptors and the T cell receptor complex or in conjunction with ligands including ICAM-1. Upon activation, a dramatic shift occurs in the conformation of the molecule from a bent compact shape to an extended open one, thereby enabling the molecule to display high affinity for ligands (Luo et al. 2007). Ligand binding appears necessary to stabilize the high affinity conformations. Monoclonal antibodies recognizing these conformations have been described for β2 integrins (Schurpf and Springer 2011). In the presence of lateral shear forces from flowing blood, integrin switching to a higher affinity conformation is faster, because the transmembrane subunits are pushed slightly apart, facilitating the fully extended conformation (Kim et al. 2003). This allows for rapid and tight binding to ICAM-1 (Zhu et al. 2008). Dysregulation of integrin affinity can occur through mutations in integrins or cytosolic binding molecules. Therefore, not surprisingly, integrins are also highly regulated molecules.
CD47 regulates the affinity of LFA-1 and VLA-4 in T cells
Recently, we identified CD47 as an important transmembrane protein that regulates the expression of high affinity conformations of leukocyte VLA-4 and LFA-1 and their adhesive functions (Azcutia et al. 2012, 2013; Stefanidakis et al. 2008; Martinelli et al. 2013b). CD47 has the following characteristics: (1) it is a broadly expressed 50-kDa transmembrane glycoprotein (Brown et al. 1990), (2) it interacts in “trans” with signal regulatory protein (SIRP), SIRPα and SIRPγ and with thrombospondin-1, (3) it has also been called an integrin-associated protein because of its ability to interact in “cis” with many integrins including αVβ3, α2β1 and αvβ3 in non-leukocyte cell types (for reviews, see Brown and Frazier 2001; Oldenborg 2013), (4) it is expressed on both endothelial cells and leukocytes in which we have shown that it plays a role during leukocyte transmigration in both in vivo and in vitro models of inflammation (Azcutia et al. 2012, 2013; Stefanidakis et al. 2008; Martinelli et al. 2013b). In the endothelium, CD47 engagement by anti-CD47 antibody crosslinking promotes src-kinase activation, actin stress fiber formation and VE-cadherin tyrosine phosphorylation (Martinelli et al. 2013b; Stefanidakis et al. 2008). However, its ability to interact in “cis” and to regulate endothelial cell integrins has not been reported. Murine T helper type 1 (Th1) cells from CD47−/− mice exhibit defects in tethering to TNF-α-activated endothelium in the cremaster muscle microcirculation and show impaired arrest and transmigration of TNF-α-activated murine heart endothelium in an in vitro flow chamber assay. More recently, CD47 has been shown to associate with human leukocyte β2 integrins, primarily LFA-1, in “cis” by fluorescence lifetime imaging microscopy and Förster resonance energy transfer. The use of reporter monoclonal antibodies that recognize high affinity forms of human β1 and β2 integrins has revealed that the loss of CD47 impairs their ability to express the high affinity “extended and open” conformation. These results support the concept that CD47 associates in “cis” with Th1 cell LFA-1 and VLA-4 integrins, that CD47 is necessary to induce the high affinity conformations of these integrins allowing them to bind to their endothelial cell ligands ICAM-1 and VCAM-1 and that CD47 is involved in leukocyte adhesion and transendothelial migration (Azcutia et al. 2013).
Preservation of barrier function during transmigration
The endothelium, although an obstacle that transmigrating leukocytes have to overcome, is a thin monolayer of cells attached to a basement membrane by integrins in focal adhesion complexes and to each other by gap, tight and adherens junctions (Bazzoni and Dejana 2004). The endothelium is well adapted to respond to biomechanical forces mediated by blood flow and blood pressure (Gimbrone et al. 2000). Transmigrating leukocytes pose a problem to endothelial barrier function by disrupting cell-cell and cell-matrix junctions and by disturbing VE-cadherin connections as mentioned above. Transmigrating leukocytes induce micrometer-scale disruptions in endothelial layer integrity through F-actin protrusions such as pseudopodia and invasive podosomes (for a review, see Ley et al. 2007). Although inflammatory cytokines and neutrophil-secreted substances such as ROS, proteases and other secreted products (DiStasi and Ley 2009) can cause significant barrier loss accompanying transmigration, little loss in barrier function or significant plasma leakage is observed during transmigration per se, because the formed VE-cadherin gaps disappear quickly; moreover, the transmigration pores are rapidly resealed behind the leukocyte, so that endothelial integrity is restored, revealing the strong self-restorative capacity of the endothelium (Shaw et al. 2001; Carman and Springer 2004; Sage and Carman 2009; He 2010). In this respect, Martinelli et al. (2013a) recently described a Rac1-, arp2/3- and ROS-dependent actin remodeling mechanism in the endothelium monitoring local barrier disruptions and effectively closing these micro-wounds. In the case of transcellular pores, these studies reveal the existence of an asymmetric actin-mediated wound-closure mechanism related to membrane protrusions. Total internal reflective microscopy has revealed that such actin structures are ventrally located, with the authors thus coining the term “ventral lamellipodia”. These authors also reported that ventral lamellipodia formation depends on alpha3 and alpha5 integrin association with the basolateral extracellular matrix. Similarly, leukocyte paracellular transmigration pores are rapidly closed by ventral lamellipodia travelling peripherally to fill the gaps and re-establish cell-cell contacts. In live-cell imaging, VE-cadherin has been shown to regain its linear pattern and adherens junctions are reconstituted within 5 min of gap closure (see Fig. 1), a finding that agrees nicely with previous reports that describe VE-cadherin gap formation and closure during leukocyte transmigration (Shaw et al. 2001; Allport et al. 2000). Martinelli et al. (2013a) found that these ventral lamellipodia are initiated by tension loss (after adhesion rupture, for example) that occurs in wounding or is transmitted through the monolayer; this induces local signaling for repair that is initiated from pre-existing actin filaments. These lamellipodia propagate beneath cells and hence, this is a type of actin remodeling different from known lamellipodia or dorsal ruffles (Chhabra and Higgs 2007). The ventral lamellipodia, which have also been observed in epithelia, probably represent a more general mechanism of closing pores and gaps and of cell migration and thus maintain barrier integrity during leukocyte transmigration or during recovery from stimuli that promote vascular leakage (thrombin, histamine, vascular endothelial growth factor). The determination of whether the transmigration-induced dynamics of the LBRC and the VE-cadherin-VE-PTP-catenin complex are mechanistically linked to the formation of endothelial ventral lamellipodia is therefore of interest.
Modification of the “conventional” multistep paradigm for organs such as the liver, lung and kidney
IVM of postcapillary venules in the cremaster muscle, mesentery and hamster cheek pouch microcirculation in combination with either fluorescently labeled leukocytes or the recent novel mouse strains expressing leukocyte-specific fluorescent reporter molecules have been the most commonly studied tissues employed to visualize the behavior of leukocyte subsets and to gain insight into the mechanisms that mediate their recruitment. This “conventional” multistep paradigm has been discussed above (Fig. 1). Reports of new methodologies for in vivo imaging including two photon-IVM (TP-IVM) and spinning disk confocal microscopy-IVM suggest that leukocyte-endothelial interactions in certain organs do not conform to the multi-step model paradigm observed in postcapillary venules. In particular, the liver, lung and kidney are also prone to inflammatory diseases (e.g., hepatitis, acute respiratory distress syndrome, glomerulonephritis) and recent studies have made progress in deciphering the steps and molecules involved in leukocyte-endothelial interactions in these organs. These organs obviously are structurally different from one another and hence are characterized by unique functional specializations reflected in their microvasculature. In these organs, blood leukocytes interact with capillaries (lung), sinusoids (liver) and the glomerulus capillary plexus (kidney), rather than postcapillary and collecting venules. Here, we briefly review recent reports of models of inflammation in these organs.
The liver lobule microvasculature receives an arterial and venous supply and is composed of sinusoid capillaries that are lined with fenestrated endothelium and that drain into post-sinusoidal central veins. The endothelium is separated from hepatocytes by a discontinuous basement membrane and a space called the space of Disse, which contains microvilli of the hepatocytes and Kupffer cells, the resident liver macrophages. Liver sinusoidal endothelium express a different leukocyte adhesion molecule profile from that of the postcapillary venules of peripheral tissue, with little E- and P-selectin or VCAM-1 expression, while exhibiting constitutively high expression of ICAM-1 and vascular adhesion protein-1 (McNab et al. 1996). Whereas leukocytes do indeed adhere to sinusoidal endothelium, rolling interactions have not been reported. In the post-sinusoidal central veins in liver, leukocyte rolling does occur (for a review, see Jenne and Kubes 2013). In knockout mouse models, leukocyte transmigration has been shown to be independent of selectin expression but dependent on ICAM-1 (Wong et al. 1997). Moreover, in a model of neutrophil recruitment induced by lipopolysaccharide, adhesive interactions have been found to be mediated by the endothelial-cell-expressed glycosaminoglycan hyaluronan acid (HA), which binds serum-derived HA-binding protein, which in turn interacts with its ligand CD44 expressed by neutrophils (McDonald et al. 2008). Other leukocyte types are present in the liver, including invariant natural killer T cells and Kupffer cells (for a detailed discussion of the dynamics of both of these cells in various models, see the review by Jenne and Kubes 2013).
The lung vasculature is composed of capillaries that are extremely thin in order to facilitate gas exchange. The capillaries form a complex interconnecting network of short vessels that are continuous and not fenestrated. Previous reports have described leukocyte recruitment as occurring at capillaries, without the conventional multistep cascade or reliance on either the β2 integrins or selectins (Wang et al. 2004). TP-IVM in a recent study revealed significant numbers of extravascular neutrophils in the lung of mechanically ventilated LysM-green-fluorescent-protein (GFP) transgenic mice at baseline (Kreisel et al. 2010). These neutrophils appear to be weakly motile (~2-3 μm/min). Within minutes of Listeria monocytogenes bacterial challenge, however, a rapid and significant influx of neutrophils occurs. Their motility speed increases 3.5-fold (~10 μm/min) and they form clusters containing ~20 cells. In a model of ischemia-reperfusion, induced as a result of wild-type lung transplanted into LysM-GFP recipients, blood monocytes have also been imaged, because of their fortuitous uptake of fluorescent 655-nm Q-dots. With this approach, monocytes are infrequently found to be arrested or extravasated in lung capillaries. Significant neutrophil extravasation has been noted in this model and the cells show high velocity crawling, forming dynamic clusters similar to those seen after bacterial challenge. Monocytes are required for this behavior, because clodronate liposome injection to deplete monocytes markedly reduces extravascular neutrophils and cluster formation. The authors speculate that monocytes secrete chemoattractants that cause the neutrophil clustering behavior in both the bacterial and lung transplantation models. One can also speculate that a chemotaxis relay system is also at work in this lung model. For example, chemotaxing “leader” interstitial neutrophils following the initial monocyte-derived chemoattractant(s) might be stimulated to produce their own chemoattractant, perhaps leukotriene B4 (LTB4), to amplify locally the initial monocyte product in order to recruit additional neutrophils (see below).
Leukocyte interactions with the vascular wall in the kidney glomerulus was reported recently under resting conditions and after induction of acute inflammation by multiphoton-IVM (immune complex deposition; Devi et al. 2013). Again, contrary to the “conventional” paradigm discussed earlier, leukocyte interactions with the glomerulus microvessels of the kidney are distinctly different. Under resting conditions in the kidney, both neutrophils and monocytes interact with the apical endothelial surface of glomerular capillaries for >30 s to periods as long as a few minutes, which the authors term the “dwell time”. These adherent and crawling leukocytes have been localized to the lumen and characterized as performing patrolling activities with migration velocities of ~11 μm/min. Conceptually, intravascular patrolling has also been observed in the liver and lung and is thought to confer intravascular immune surveillance in organs. These investigators envision that such localization enables rapid responses by these leukocytes to infectious and sterile inflammatory stimuli. In the kidney study, this is exactly what occurs. Upon initiating inflammation in the kidney (i.e., glomerulonephritis) by immune complex deposition in the glomerulus basement membrane, leukocyte dwell time in vessel lumen increases dramatically and is greater than 20 min. Many adherent and crawling leukocytes produce toxic ROS that might damage the endothelium and lead to proteinuria in these mice. These types of studies revealed novel concepts for glomerular homeostasis and a clearer understanding of the molecular mechanisms of leukocyte trafficking; instead of undergoing transendothelial migration after adhesion, the leukocytes increase their intravascular retention time and, under pro-inflammatory conditions, produce ROS and other pro-inflammatory mediators that can damage tissues and organs.
Mechanisms of post-diapedesis leukocyte migration: pass through the basement membrane and “turn right” onto pericyte “avenues” for guidance to interstitial targets
Leukocytes that have successfully transmigrated across the vascular endothelium in most tissues encounter the basement membrane and in many tissues, pericytes, which discontinuously wrap around arterioles, capillaries and postcapillary and collecting venules (Fig. 2; for a review, see Armulik et al. 2005). With the improving characterization of pericytes, their importance in leukocyte migration has recently come into sharp focus. Neutrophils and monocytes have been reported to migrate preferentially through specific areas in the basement membrane in which the matrix proteins, namely collagen type IV and laminin 10, are expressed at a lower density (Wang et al. 2006; Voisin et al. 2009). A recent study revealed that emigrated interstitial neutrophils interact extensively with neuron-glial-2-positive (NG2+) pericytes after migrating across the vascular endothelium in a murine model of sterile TNF-α-induced inflammation (Proebstl et al. 2012). Another recent study revealed that pericytes express ICAM-1 and secrete chemoattractants (Stark et al. 2013). Notably, two different populations of pericytes have been described that associate with distinct blood vessel types: NG2+ and α-smooth-muscle-actin-positive (α-SMC+) pericytes are located along arterioles and capillaries, whereas NG2-negative (NG2-) and α-SMC+ pericytes are located along postcapillary venules in a dermal mouse model. Emigrated neutrophils and monocytes first encounter the NG2- population on postcapillary venules and crawl along these and then become attracted to the NG2+ pericytes. This is because NG2+ pericytes are stimulated by tissue-derived damage and pathogen-associated molecular patterns to produce the chemokine macrophage migration-inhibitory factor and other chemokines that attract those neutrophils that have just transmigrated. These studies provide a broader picture of mechanisms that mediate innate cell recruitment and strongly suggest that the endothelium, the basement membrane and pericytes are active participants in the innate immune response. They also beg the question of whether other interstitial cells or physiological systems are involved in regulating and guiding leukocytes to sites of inflammation. A recent report shed light on this (Lammermann et al. 2013).

Transmigration and interactions of neutrophils after transendothelial migration. Neutrophils cross the basement membrane of a postcapillary venule (top endothelial layer) at a region of lower collagen IV and laminin 10 densities and are guided within the interstitium by interactions with pericytes. Neutrophils first crawl along the neuron-glial-2-negative (NG2-) pericytes (green) outside of postcapillary venules. Migration-inhibitory factor (MIF) secretion from activated pericytes on arterioles (red) further guides cells towards the inflammatory focus. We speculate that neutrophils attract a further influx of neutrophils by secreting leukotriene B4 (LTB4) or other chemoattractants that trigger a swarming behavior of neutrophils around a wound (DAMPs damage-associated molecular patterns)
Interstitial neutrophil dance ballad called “The Swarming of Neutrophils”
A deeper understanding of the migration of neutrophils in the interstitial space was obtained from a model of laser-induced sterile skin injury by using TP-IVM (Lammermann et al. 2013); in contrast to the conventional multistep model of neutrophil extravasation, an integrin-independent molecular-relay mechanism was uncovered that occurs in conjunction with coordinated chemotaxis and cluster formation of neutrophils in the extravascular space of damaged dermal tissue. This type of behavior is also known as swarming. It is temporally coordinated and is characterized by initial chemotaxis of the neutrophils closest to the damaged tissue, followed by a strong influx of neutrophils from more distant sites and finally neutrophil clustering as reported previously (Ng et al. 2011). Neutrophils immediately swarm the wound as demonstrated by highly directional chemotaxis at high speeds, whereas chemokine receptor CXCR3+ monocytes migrate at a later time and at slower speeds and do not enter the developing cluster of neutrophils. This phase is followed by a second phase of neutrophil influx from more distant sites, the second phase being catalyzed by the death of only a small number of neutrophils. Studying neutrophils lacking various G-protein-coupled receptors and then narrowing down the respective receptors, the authors (Lammermann et al. 2013) were able to identify the receptor for LTB4 (LTB4R1) as being crucial and the neutrophil-secreted LTB4 chemoattractant as the relaying signal among neutrophils over larger distances. It is this relaying signal that amplifies cell death signals, thus broadening the radius of chemotactic signal. Indeed, neutrophils lacking LTB4R1 (Ltb4r1−/−) still migrate towards the wound but the second phase of the neutrophil response is missing. The clustering step, however, has been found to be integrin-mediated. The authors suggest these events trigger a self-organized neutrophil influx that forms a wound seal ending with the later monocyte recruitment that isolates the injured from the viable tissue (Lammermann et al. 2013).
Neutrophil extracellular traps: their role in host defense and in disease
The ability of neutrophils to swarm, phagocytose and/or neutralize foreign materials, necrotic tissues and bacteria is unparalleled in nature. Moreover, as neutrophils perform their “clean-up” job and once their clean-up is complete, they keep on giving themselves through the formation of neutrophil extracellular traps (NETs; Brinkmann et al. 2004). During an inflammatory response, neutrophils can be stimulated by physiological agonist and bacterial endotoxins to release NETs, a cloud-like mixture of highly negatively charged strands of DNA and associated proteins and granule enzymes. NETs are envisioned to enhance the ability of the cell to trap and inactivate virulence factors and have been demonstrated to kill Gram-positive (Staphylococcus aureus) and Gram-negative (Salmonella typimurium) pathogenic bacteria (Brinkmann and Zychlinsky 2012; Brinkmann et al. 2004). Subsequent studies have corroborated these findings and provided the stepwise pathway for the generation and release of NETS by neutrophils. Other granulocyte leukocytes including mast cells and basophils have been demonstrated to release NETs (Kolaczkowska and Kubes 2013). Interestly, NETs also appear to have a “dark” side. Recent reports have shown that NETs contribute to thrombus formation in an experimental deep vein thrombosis model (Brinkmann and Zychlinsky 2012; Fuchs et al. 2010). Once again, Mother Nature has provided yet another layer of defense in the immune system that was unexpected.
A few words about organs on a chip
The advent of microdevices has provided new approaches for modeling and studying various aspects of organ function. Organs on chips are a class of microengineered tissue models that might be an appropriate conduit to the transition from basic single cell in vitro models to in vivo experimental disease models (van der Meer and van den Berg 2012). Although beyond the scope of this review, recent reports from the Ingber laboratory and other laboratories have advanced this field by providing a fertile environment in which cell biologists and bioengineers can work collaboratively to develop microfluidic devices. These devices use living cells in well-controlled environmental conditions to induce cells to express and maintain tissue-specific differentiated properties that mimic organs including physiological relevant three-dimensional structural units of living organs such as kidney, liver, brain, heart, intestine and lung (Huh et al. 2013). Spatiotemporal chemical gradients or mechanical forces such as breathing movements, fluid flow, or gut peristalsis can be mimicked creating a more physiologically relevant cell culture model, upgrading the classical in vitro experiments and thus representing a possible low-cost alternative to animal models. In such devices, inflammation can be successfully mimicked and studied. The future task is to ascertain the best means of utilizing these organs on chips and of integrating these technologies and to determine whether their “added value” will hold up to expectations and lead to an in-depth understanding of how leukocytes enter and exit during normal and pathological settings and where and when we can intervene successfully.
Concluding remarks
Recent studies have highlighted the role of inflammation in obesity, diabetes, hypertension, autoimmune disease and cardiovascular diseases including stroke and atherosclerosis. A common approach for developing innovative and effective anti-inflammatory therapeutics is to understand basic mechanisms regulating leukocyte entry and exit from tissues and organs and then to translate these observations into a physiological model that mimics human disease. Several important points need to be discussed in the future. How can we harness our knowledge of these key events to build a rational therapeutic strategy with minimal off-target effects? Will the organ on a chip become the intermediate step from basic single cell in vitro assays to murine models? Will the new imaging modalities enable researchers to assemble data for faster drug discoveries?
References
Alcaide P, Auerbach S, Luscinskas FW (2009) Neutrophil recruitment under shear flow: it’s all about endothelial cell rings and gaps. Microcirculation 16:43–57
Allport JR, Muller WA, Luscinskas FW (2000) Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J Cell Biol 148:203–216
Andrian UH von, Chambers JD, McEvoy LM, Bargatze RF, Arfors KE, Butcher EC (1991) Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc Natl Acad Sci U S A 88:7538–7542
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97:512–523
Azcutia V, Stefanidakis M, Tsuboi N, Mayadas T, Croce KJ, Fukuda D, Aikawa M, Newton G, Luscinskas FW (2012) Endothelial CD47 promotes vascular endothelial-cadherin tyrosine phosphorylation and participates in T cell recruitment at sites of inflammation in vivo. J Immunol 189:2553–2562
Azcutia V, Routledge M, Williams MR, Newton G, Frazier WA, Manica A, Croce K, Parkos CA, Schmider AB, Turman MV, Soberman RJ, Luscinskas FW (2013) CD47 plays a critical role in T cell recruitment by regulation of LFA-1 and VLA-4 integrin adhesive functions. Mol Biol Cell 24:3358–3368
Barreiro O, Zamai M, Yanez-Mo M, Tejera E, Lopez-Romero P, Monk PN, Gratton E, Caiolfa VR, Sanchez-Madrid F (2008) Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol 183:527–542
Bazzoni G, Dejana E (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84:869–901
Bevilacqua MP, Gimbrone MA Jr (1987) Inducible endothelial functions in inflammation and coagulation. Semin Thromb Hemost 13:425–433
Brinkmann V, Zychlinsky A (2012) Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol 198:773–783
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Brown EJ, Frazier WA (2001) Integrin-associated protein (CD47) and its ligands. Trends Cell Biol 11:130–135
Brown E, Hooper L, Ho T, Gresham H (1990) Integrin-associated protein: a 50-kD plasma membrane antigen physically and functionally associated with integrins. J Cell Biol 111:2785–2794
Butcher EC (1991) Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 67:1033–1036
Buul JD van, Hordijk PL (2009) Endothelial adapter proteins in leukocyte transmigration. Thromb Haemost 101:649–655
Carman CV, Springer TA (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 167:377–388
Carman CV, Sage PT, Sciuto TE, Fuente MA de la, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA (2007) Transcellular diapedesis is initiated by invasive podosomes. Immunity 26:784–797
Chhabra ES, Higgs HN (2007) The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol 9:1110–1121
Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T (1995) Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J 9:899–909
Devi S, Li A, Westhorpe CL, Lo CY, Abeynaike LD, Snelgrove SL, Hall P, Ooi JD, Sobey CG, Kitching AR, Hickey MJ (2013) Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med 19:107–112
DiStasi MR, Ley K (2009) Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol 30:547–556
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107:15880–15885
Gimbrone MA Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G (2000) Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci 902:230–239
He P (2010) Leucocyte/endothelium interactions and microvessel permeability: coupled or uncoupled? Cardiovasc Res 87:281–290
Huh D, Kim HJ, Fraser JP, Shea DE, Khan M, Bahinski A, Hamilton GA, Ingber DE (2013) Microfabrication of human organs-on-chips. Nat Protoc 8:2135–2157
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687
Jenne CN, Kubes P (2013) Immune surveillance by the liver. Nat Immunol 14:996–1006
Kim M, Carman CV, Springer TA (2003) Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301:1720–1725
Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175
Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, Pless R, Gelman AE, Krupnick AS, Miller MJ (2010) In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A 107:18073–18078
Kumar V, Abbas AK, Fausto N, Aster J (2010) Robbins and Cotran: pathologic basis of disease, 8th edn. Elsevier, Philadelphia
Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, Germain RN (2013) Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498:371–375
Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689
Luo BH, Carman CV, Springer TA (2007) Structural basis of integrin regulation and signaling. Annu Rev Immunol 25:619–647
Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA (2003) Targeted recycling of PECAM from endothelial surface-connected compartments during diapedesis. Nature 421:748–753
Mamdouh Z, Kreitzer GE, Muller WA (2008) Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J Exp Med 205:951–966
Martinelli R, Kamei M, Sage PT, Massol R, Varghese L, Sciuto T, Toporsian M, Dvorak AM, Kirchhausen T, Springer TA, Carman CV (2013a) Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J Cell Biol 201:449–465
Martinelli R, Newton G, Carman CV, Greenwood J, Luscinskas FW (2013b) Novel role of CD47 in rat microvascular endothelium: signaling and regulation of T-cell transendothelial migration. Arterioscler Thromb Vasc Biol 33:2566–2576
McDonald B, McAvoy EF, Lam F, Gill V, Motte C de la, Savani RC, Kubes P (2008) Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med 205:915–927
McNab G, Reeves JL, Salmi M, Hubscher S, Jalkanen S, Adams DH (1996) Vascular adhesion protein 1 mediates binding of T cells to human hepatic endothelium. Gastroenterology 110:522–528
Meer AD van der, Berg A van den (2012) Organs-on-chips: breaking the in vitro impasse. Integr Biol (Camb) 4:461–470
Muller WA (2011) Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 6:323–344
Ng LG, Qin JS, Roediger B, Wang Y, Jain R, Cavanagh LL, Smith AL, Jones CA, Veer M de, Grimbaldeston MA, Meeusen EN, Weninger W (2011) Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. J Invest Dermatol 131:2058–2068
Oldenborg PA (2013) CD47: a cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol 2013:614619
Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P (2006) Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 203:2569–2575
Proebstl D, Voisin MB, Woodfin A, Whiteford J, D’Acquisto F, Jones GE, Rowe D, Nourshargh S (2012) Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med 209:1219–1234
Reymond N, Imbert AM, Devilard E, Fabre S, Chabannon C, Xerri L, Farnarier C, Cantoni C, Bottino C, Moretta A, Dubreuil P, Lopez M (2004) DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med 199:1331–1341
Sage PT, Carman CV (2009) Settings and mechanisms for trans-cellular diapedesis. Front Biosci (Landmark Ed) 14:5066–5083
Schenkel AR, Mamdouh Z, Muller WA (2004) Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol 5:393–400
Schurpf T, Springer TA (2011) Regulation of integrin affinity on cell surfaces. EMBO J 30:4712–4727
Shaw SK, Bamba PS, Perkins BN, Luscinskas FW (2001) Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol 167:2323–2330
Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, Montresor A, Bolomini-Vittori M, Feigelson SW, Kirchhausen T, Laudanna C, Shakhar G, Alon R (2009) Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity 30:384–396
Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, Bruhl ML von, Gartner F, Khandoga AG, Legate KR, Pless R, Hepper I, Lauber K, Walzog B, Massberg S (2013) Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern-recognition and motility programs. Nat Immunol 14:41–51
Stefanidakis M, Newton G, Lee WY, Parkos CA, Luscinskas FW (2008) Endothelial CD47 interaction with SIRPgamma is required for human T-cell transendothelial migration under shear flow conditions in vitro. Blood 112:1280–1289
Sullivan DP, Seidman MA, Muller WA (2013) Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am J Pathol 182:1031–1042
Svensson L, Howarth K, McDowall A, Patzak I, Evans R, Ussar S, Moser M, Metin A, Fried M, Tomlinson I, Hogg N (2009) Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med 15:306–312
Vestweber D (2012) Novel insights into leukocyte extravasation. Curr Opin Hematol 19:212–217
Vijver E van de, Maddalena A, Sanal O, Holland SM, Uzel G, Madkaikar M, Boer M de, Leeuwen K van, Koker MY, Parvaneh N, Fischer A, Law SK, Klein N, Tezcan FI, Unal E, Patiroglu T, Belohradsky BH, Schwartz K, Somech R, Kuijpers TW, Roos D (2012) Hematologically important mutations: leukocyte adhesion deficiency (first update). Blood Cells Mol Dis 48:53–61
Vockel M, Vestweber D (2013) How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood 122:2512–2522
Voisin MB, Woodfin A, Nourshargh S (2009) Monocytes and neutrophils exhibit both distinct and common mechanisms in penetrating the vascular basement membrane in vivo. Arterioscler Thromb Vasc Biol 29:1193–1199
Wang Q, Doerschuk CM, Mizgerd JP (2004) Neutrophils in innate immunity. Semin Respir Crit Care Med 25:33–41
Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, Maxwell PH, Sorokin L, Nourshargh S (2006) Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med 203:1519–1532
Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, Kubes P (1997) A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest 99:2782–2790
Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA (2008) Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell 32:849–861
Author information
Authors and Affiliations
Corresponding author
Additional information
Our work was supported by funding from the NHLBI, NIH (HL-36028) and American Heart Association Postdoctoral Fellowship (11POST7730055). The project was also supported by the National Research Fund, Luxembourg and co-funded under the Marie Curie Actions of the European Commission (FP7-COFUND).
Rights and permissions
About this article
Cite this article
Leick, M., Azcutia, V., Newton, G. et al. Leukocyte recruitment in inflammation: basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res 355, 647–656 (2014). https://doi.org/10.1007/s00441-014-1809-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-014-1809-9