
Abstract
Aflatoxins are toxic secondary metabolites produced by members of the genus Aspergillus, most notably A. flavus. Non-aflatoxigenic strains of A. flavus are commonly used for biocontrol of the aflatoxigenic strains to reduce aflatoxins in corn, cotton, peanuts and tree nuts. However, genomic differences between aflatoxigenic strains and non-aflatoxigenic strains have not been reported in detail, though such differences may further elucidate the evolutionary histories of certain biocontrol strains and help guide development of other useful strains. We recently reported the genome and transcriptome sequencing of A. flavus WRRL 1519, a strain isolated from almond that does not produce aflatoxins or cyclopiazonic acid due to deletions in the biosynthetic gene clusters. Continued bioinformatics analyses focused on comparing strain WRRL 1519 to the aflatoxigenic strain NRRL 3357. The genome assembly of strain WRRL 1519 was improved by anchoring 84 of the 127 scaffolds to the putative nuclear chromosomes of strain NRRL 3357. The five largest areas of extrachromosomal mismatches observed between WRRL 1519 and NRRL 3357 were not similar to any of the mismatches that were observed with pairwise comparisons of NRRL 3357 to other non-aflatoxigenic strains NRRL 21882, NRRL 30797 or NRRL 18543. Comparisons of predicted secondary metabolite gene clusters uncovered two other biosynthetic gene clusters in which strain WRRL 1519 had large deletions compared to the homologous clusters in NRRL 3357. Additionally, there was a marked overrepresentation of repetitive sequences in WRRL 1519 compared to other inspected A. flavus strains. This is the first report of detection of a large number of putative retrotransposons in any A. flavus strain, initially suggesting that retrotransposons may contribute to the natural occurrence of genetic variation and biocontrol strains. However, the transposons may not be significantly associated with the chromosomal differences. Future experimentation and continued bioinformatics analyses will potentially illuminate causes of the differences and may reveal whether transposon activity in A. flavus can lead to random natural occurrences of non-aflatoxigenic strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungi of the genus Aspergillus are greatly important in health, agriculture and food production. Common species such as Aspergillus flavus and A. fumigatus may be opportunistic animal and plant pathogens, particularly of immunocompromised humans and of crops such as corn, cotton and nuts (de Lucca 2007; Goldman and Osmani 2008; Amaike and Keller 2011; Mousavi et al. 2016). A. nidulans is an important model organism for eukaryotic genetics and cell biology (Nierman et al. 2005; Goldman and Osmani 2008). A. oryzae is used to ferment many popular Japanese fermented foods and beverages and in the production of heterologous proteins, while A. niger and A. terreus are used for industrial production of citric acid and lovastatin (Goldman and Osmani 2008; Machida et al. 2008; Bennett 2010). A. flavus is a cosmopolitan saprophyte and opportunistic pathogen that is a common producer of aflatoxins, potent hepatotoxins and carcinogens (Richard and Payne 2003). Due to food safety implications, there is significant interest in measuring the diversity of this species in different populations and identifying stable non-aflatoxigenic strains (Cotty 1990, 1994; Cotty and Mellon 2006; Moore et al. 2009), which have been reported to comprise 15.8–78.9% of natural fungal isolates (Hua et al. 2012; Jamali et al. 2012; Solorzano et al. 2014; Divakara et al. 2015) across the world. Most biocontrol A. flavus strains have aberrant aflatoxins biosynthetic gene clusters. For example, strain NRRL 21882 (“Afla-Guard”) does not produce aflatoxins, because of a large deletion of the aflatoxins biosynthesis gene cluster and part of the adjacent sugar utilization cluster (Chang et al. 2005). Strain NRRL 30797 (“K49”) has a premature TGA stop codon in the polyketide synthase gene aflC (Chang et al. 2012) and strain NRRL 18543 (“Af36”) has deletions of 17–61 bp in genes aflU, aflC, aflR and aflaV (Adhikari et al. 2016). The evolutionary processes responsible for the occurrence of non-aflatoxigenic A. flavus strains are still unclear. However, recombination events may have a major influence on genetic variability (Geiser et al. 1998; Olarte et al. 2012; Moore 2014). Moore et al. (2009) propose that lineage-specific recombination events among the late aflatoxins synthesis pathway gene (aflE to aflP, but especially aflW and aflX) led to gene losses and phylogenetic clades by which non-aflatoxigenic strains can be grouped; later generations can regain toxicity by recombination with chromosome III of an aflatoxigenic parent. Along the same lines, Chang et al. (2012) also claim that an ancestral recombination event between two morphotypes resulted in the progenitors of NRRL 18543 and NRRL 30797. The boom in genomic sequencing of filamentous fungi in the past decade not only permits comparisons of different species, it also allows us to delve into the genomic differences among strains within a species (Nierman et al. 2005). Thereby, we can link genotypes to desirable phenotypes, such as improved industrial metabolite production, lignocellulose degradation (Emtiazi et al. 2001; Raulo et al. 2016) and biological control of mycotoxigenic strains (Richard and Payne 2003). We aim to identify genetic variations in addition to aberrant aflatoxin biosynthesis clusters that may distinguish effective biocontrol A. flavus strains.
Genetic variability and diversity within a population positively correlate with population fitness and a better ability of the population to adapt to changes in the environment (Reed and Frankham 2003). Sources of such genotypic differences include mutations, polyploidy, homologous recombination, transposable elements (TEs) and individual migration in and out of populations. TEs are of special interest because of their contributions to gene regulation and genome structural variation (Argueso et al. 2008; Feschotte 2008; Bourque 2009; Klein and O’Neil 2018). Transposons are often over-represented in heterochromatic areas such as in and near the centromere (Pimpinelli et al. 1995; Round et al. 1997; Peterson-Burch et al. 2004; Tsukahara et al. 2012; Klein and O’Neil 2018). Their high copy numbers can cause them to comprise 0.02–29.8% of fungal genomes, 3–45% of metazoan genomes and up to 80% of plant genomes (Daboussi and Capy 2003; Bennetzen 2005; Hua-Van et al. 2005; Castanera et al. 2016).
There are two categories of TEs: class I retrotransposons that have a ‘copy and paste’ mechanism in which the TE is replicated and integrated in different locations of the genome via an RNA intermediate, and class II DNA transposons that ‘cut and paste’ themselves throughout a genome. Retrotransposons can be further classed as having long terminal repeats (LTRs), long interspersed nuclear elements (LINE) or short interspersed nuclear elements. Codon usage of retrotransposons in some fungal hosts is noted to be atypical of the organism, indicating horizontal gene transfer origins (Hansen et al. 1988; Oliver 1992). The Gypsy superfamily of LTR retrotransposons is commonly found in all eukaryotic phyla (Wicker et al. 2007), but the subfamily Tf1/sushi is only reported in fungi and a few vertebrates, suggesting limited horizontal transfer from fungi to vertebrates (Miller et al. 1999; Butler et al. 2001). In fungi, the effects of TEs on strain differentiation and speciation are not well-defined. However, they are suspected to have effects on the chromosomal rearrangements and duplications via transposon activity or recombination in Fusarium oxysporum (Hua-Van et al. 2000; Davière et al. 2001), Magnaporthe grisea (Thon et al. 2006), Phanerochaete chrysosporium (Larrondo et al. 2007) and Aspergillus spp. (Clutterbuck et al. 2008; Lind et al. 2017). TEs also can serve as a “fossil” record of genome differentiation. The strain-specific distributions of transposons Vader and ANiTa1 in industrial strains of A. niger indicate they were active during domestication, yielding different genome organizations (Braumann et al. 2007, 2008). However, TEs are not always a clear cause of genomic rearrangements (Davière et al. 2001).
Several TE families are detectable in Aspergillus, totaling 1.2–2.5% of the genomes of A. nidulans, A. fumigatus and A. oryzae (Clutterbuck et al. 2008). Three Aspergillus Tf1/sushi subfamily TEs are known: Afut in A. fumigatus (Neuveglise et al. 1996), AFLAV in non-aflatoxigenic A. flavus NRRL 6541 (Okubara et al. 2003; Hua et al. 2007) and AoLTR in A. oryzae RIB40 (Jin et al. 2014). All of these TEs are 6000–7799 bp sequences flanked by two 282–669 bp repeat sequences; have two open reading frames (ORF1 and ORF2) staggered by a − 1 frameshift; and encode a putative Gag-Pol polyprotein with domains for Gag capsid protein, aspartic proteinase, reverse transcriptase, RNase H, and integrase, which are universal structural features of active Gypsy retrotransposons (Wicker et al. 2007). A screen of over 50 A. flavus field isolates reveals that about half had incomplete gag and pol regions; none had a full AFLAV-like sequence (Hua et al. 2007). A. oryzae additionally has Crawler, a Mariner/Tc1-type transposon (Ogasawara et al. 2009). Uncharacterized degenerate retrotransposons dane1 and dane2 are also found in A. nidulans (NCBI GenBank accessions AF295689.1 and AF295688.1, respectively).
The newly reported non-aflatoxigenic strain WRRL 1519 is missing nearly 42 kb of the beginning of the 75 kb aflatoxins synthesis gene cluster and does not have a complete cyclopiazonic synthesis gene cluster (Yin et al. 2018). In this study, we continued bioinformatics analyses of WRRL 1519, comparing it to the aflatoxigenic strain NRRL 3357, and here we report that the genome organization of strain WRRL 1519 differed from than that observed in other inspected non-aflatoxigenic A. flavus strains. In an attempt to identify the cause of these differences, we further modeled the association of these differences with the increased number of repetitive elements in the WRRL 1519 strain.
Methods
Comparisons of predicted proteomes
Programs were used with default settings unless otherwise stated. Genomic sequences of the aflatoxigenic A. flavus strain NRRL 3357, and the non-aflatoxigenic A. flavus strains NRRL 21882, NRRL 30797, NRRL 18543 and WRRL 1519 were retrieved from the NCBI Genome database in January 2018 (Nierman et al. 2015; Weaver et al. 2017; Yin et al. 2018; Table 1). The annotated proteome of the aflatoxigenic A. flavus strain NRRL 3357 was available from the same database. We predicted the proteomes of the non-aflatoxigenic NRRL 21882, NRRL 30797, NRRL 18543 and WRRL 1519 using Augustus version 3.2.3 (Stanke et al. 2006) and GeneMark-ES (Suite version 4.33) trained on A. oryzae (Ter-Hovhannisyan et al. 2008). The resulting GTF files were combined into one annotation file using an in-house Python 2.7 script.
Predicted protein sequences of A. flavus strains NRRL 3357, NRRL 21882, NRRL 30797, NRRL 18543 and WRRL 1519 were compared for homology (as indicated by high sequence similarity) by searching against the proteomes using HMMER 3.1b2 (Johnson et al. 2010) with an E value cutoff of 1−50. Using the annotated NRRL 3357 proteome, predicted protein sequences of the non-aflatoxigenic strains were assigned homology. Then, these assignments were used to map the relative locations and directions of the scaffolds along the eight putative chromosomes of NRRL 3357. Scaffolds with at least 20 homologous genes in agreement were visualized using Circos v 0.69 (Krzywinski et al. 2009). The method was validated by homology matching strain NRRL 3357 to itself (Supplementary Fig. 1A). MUMmer v 4.0.0 (Marçais et al. 2018) was additionally used to identify nucleotide macrosynteny between aflatoxigenic NRRL3357 and the other non-aflatoxigenic A. flavus strains.
Prediction of secondary metabolite gene clusters, repetitive sequences, candidate retrotransposons, regulatory sequences and secretory protein-coding genes
Since the aflatoxigenic cluster is partially missing in strain WRRL 1519, we searched for other differences in putative secondary metabolite gene clusters predicted by antiSMASH 3.0 (Weber et al. 2015) using Easyfig 2.2.2 (Sullivan et al. 2011). Clusters were compared between strains NRRL 3357 and WRRL 1519 to identify if other secondary metabolite clusters had large deletions. Regulatory sequences (upstream a maximum of 3 kbp and not overlapping an open reading frame) and secreted proteins were predicted using RSAT server (Nguyen et al. 2018) with the JASPAR 2018 core non-redundant transcription factor DNA-binding preferences matrix (Khan et al. 2018) and TargetP 1.1 with the non-plant option (Emanuelsson et al. 2007), respectively.
RepeatMasker version 4.0.7 (Smit et al. 2018) was used to identified all repetitive elements using the fungal-specific repetitive sequences database from RepBase volume 23, issue 5 (https://www.girinst.org/). Lower-scoring sequences in the same loci as another higher-scoring one were considered redundant and removed. We modeled random placement of all repetitive sequences in the WRRL 1519 genome using a custom Python script: weighted by scaffold size, the script randomly assigned the 883 repetitive sequences to genomic locations on a scaffold at least one nucleotide larger in size. We then counted the number of randomized loci within a specified range of nucleotides bordering predicted protein-coding genes, regulatory sequences, other repetitive sequences and secretory protein-coding genes. Ten thousand permutations were performed for each instance. The randomized model was compared to the actual counts of repetitive sequences within the borders of the sequences of interest. Genomic densities and locations of sequences of interest were visualized using Circos.
Reverse transcriptases and retrotransposons identified from A. flavus sequences and deposited in NCBI GenBank were queried against the genomic sequences of inspected strains using HMMER with an E value cutoff of 1−200. Additional LTR sequences were search with a 1−50 E value threshold. Sequence similarities of retrieved homologous sequences were visualized using MUSCLE 3.8.31 (Edgar 2004) and BOXSHADE 3.21 (https://embnet.vital-it.ch/software/BOX_form.html). Candidates were queried against the NCBI non-redundant protein database (O’Leary et al. 2016; accessed March 2018) using NCBI BLAST (Altschul et al. 1990; https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Nucleotides&PROGRAM=blastn&PAGE_TYPE=BlastSearch&BLAST_SPEC=). The gene environment of the eleven retrotransposon-like sequences in WRRL 1519 was characterized based on alignment to annotated NRRL 3357 genes.
Results
Strain WRRL 1519 was different in genomic organization from other A. flavus strains
Most of the predicted protein in all non-aflatoxigenic strains were assigned homology to at least one NRRL 3357 protein (Table 1). However, 512 NRRL 3357 genes were commonly not assigned homology to any of the non-aflatoxigenic strains (Fig. 1). The latter all corresponded to short sequences, 50–91 amino acids in length, that were mostly annotated as “conserved” and/or “hypothetical”. All could be identified as homologous to something if the E value threshold was lowered to 1−10. There were 1, 902 NRRL 3357 proteins that did not match to themselves when queried against the NRRL 3357 proteome or to anything in the predicted proteomes of the biocontrol strains, mostly consisting of conserved and hypothetical proteins, as well as some transcription factors, transmembrane transporters, kinases, enzymes and domain consensus motifs.

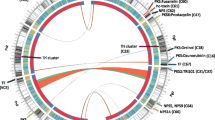
The similarities of predicted protein-coding genes between NRRL 3357 and non-aflatoxigenic strains were examined to map the respective scaffolds to the chromosomal arms of NRRL 3357. The 16 largest scaffolds of the A. flavus NRRL 3357 genome assembly comprised the 16 arms of the eight chromosomes of A. flavus. The largest 16 or 17 scaffolds of strains NRRL 21882, NRRL 30797and NRRL 18543 were homologous to these chromosomal arms (Supplementary Fig. 1B–D). Only strain NRRL 21882 did not have a clear match to the p arm of chromosome VII (NRRL 3357 chromosome EQ963487.1). The genome sequence of WRRL 1519 had 127 scaffolds of which we were able to align 84 to the nuclear genome of NRRL 3357 (Fig. 2a). Scaffold 98 was the mitochondrial genome. Scaffolds 59, 72, 73, 82, 84, 88, 90–92 and 94–127 could not be placed confidently as they had too few homologous genes or unclear alignments. All non-aflatoxigenic strains had a few genes that matched to those the 17th largest scaffold of NRRL 3357 EQ963488.1. Predicted protein-coding genes from nucleotide 37,001 to 51,632 of scaffold 65 from WRRL 1519 mapped to EQ963488.1. However, the gene sequences from 57,085 to 134,489 mapped to EQ963475.1, so this scaffold was assigned to chromosome III. Similarly, one 2636 nucleotide gene in scaffold 0 was homologous to a gene in extrachromosomal scaffold EQ965297.1, but most of the WRRL 1519 sequences aligned with NRRL chromosomal scaffolds EQ963473.1 and EQ963481.1. For the other non-aflatoxigenic strains, the 17th or 18th largest scaffold of the genomes matched to EQ963488.1. The orders and orientations of the placed scaffolds relative to the NRRL 3357 chromosomal scaffolds strain are shown in Fig. 2, Supplementary Table 1 and Supplementary Fig. 2. Results from the protein similarity searches between NRRL 3357 and WRRL 1519 are shown in Supplementary Table 2. MUMmer results with nucleotide-based similarity searches were in accordance in that the strains NRRL 21882, NRRL 30797 and NRRL 18543 were noticeably more similar in genome organization to NRRL 3357 than WRRL 1519 (Supplementary Fig. 3).

Scaffold ordering of strain WRRL 1519 to the chromosomes of NRRL 3357. Scaffolds comprising the chromosomal arms of NRRL 3357 are drawn in 16 different colors on the right and are labeled as in the genome assembly (see Supplementary Table 1). The outer track enumerates chromosome nucleotide length in kilobase pairs, and protein-coding gene density is shown by a gray histogram on the inner track. On the left, scaffolds of WRRL 1519 are connected to the chromosomes by lines representing high sequence similarity between two predicted protein-coding genes. Used scaffolds have at least 20 genes homologous to one NRRL 3357 scaffold. Beige scaffolds have deposited nucleotide sequences that run in the same direction as the deposited sequence of the aligned NRRL 3357 chromosomal arm; black ones run in the opposite direction
Most of the scaffolds of the non-aflatoxigenic strains aligned well with the putative chromosomal arms of aflatoxigenic NRRL 3357. Extrachromosomal differences were defined as a gene in a particular non-aflatoxigenic scaffold having best homology to a gene in a NRRL 3357 chromosomal arm to which the scaffold is not aligned (Table 1). For example, scaffold 30 of strain WRRL 1519 is homologous to a large region of chromosomal arm EQ963477.1. However, the best overall match for this scaffold was to EQ963472.1. These differences could be the results of lower E value assignment to non-homologous genes by coincidence, to true extrachromosomal paralogs or to true homologs with different relative chromosomal placements. For most of these extrachromosomal differences, the assigned NRRL 3357 homolog was the only hit retrieved from the PHMMER searches.
Strains NRRL 21882, NRRL 30797 and NRRL 18543 had 134 extrachromosomal mismatches common among them. Strain WRRL 1519 shared 122 of these mismatches. The mismatched genes were most frequently annotated as transcription factors, transporters/permeases, non-ribosomal peptide synthetases and polyketide synthases. There were five large sections of extrachromosomal differences between WRRL 1519 and NRRL 3357 clearly visible in both Fig. 2 and Supplementary Fig. 3: between WRRL 1519 scaffold 3 and NRRL 3357 scaffold EQ963484.1 (chromosome VI, p arm), scaffold 30 and scaffold EQ963477.1 (chromosome V, q arm), scaffold 15 and scaffold EQ963482.1 (chromosome IV, p arm), scaffold 40 and scaffold EQ963479.1 (chromosome VI, q arm), and scaffold 13 and scaffold EQ963477.1 (chromosome V, q arm). The mismatches were not associated with significant differences in the structures of predicted secondary metabolite gene clusters (Supplementary Table 3). However, two WRRL 1519 gene clusters aligned to NRRL 3357 chromosomes EQ963476.1 (chromosome VII, q arm) and EQ963486.1 (chromosome VIII, p arm) had large deletions (Fig. 3). The former is predicted to yield a terpenoid non-ribosomal peptide product, the latter a non-ribosomal peptide product.

Alignments of predicted secondary metabolite gene clusters of strains NRRL 3357 (top) and WRRL 1519 (bottom) between a chromosome EQ963476.1 and scaffold 11 and b chromosome EQ963486.1 and scaffold 7. Percent identity by tblastx between subsequences is shown as indicated by the key. Genes with predicted functions in the NRRL 3357 proteome are green; others are purple. Automatic gene annotation of WRRL 1519 may have resulted in incorrect start and stop positions
Strain WRRL 1519 had an increased number of repetitive elements
Repetitive elements may affect genome structure, so we searched for such sequences using RepeatMasker and HMMER. RepeatMasker revealed that strain WRRL 1519 (886 elements comprising 1.54% of the genome) had an increased number of fungal-specific repetitive elements relative to the other strains (NRRL 3357—603 elements, 0.43% of the genome; NRRL 21882—477 elements, 0.28% of the genome; NRRL 30797—533 elements, 0.36% of the genome; NRRL 18543—533 elements, 0.34% of the genome) (Fig. 4). Gypsy and Mariner/Tc1-like sequences were the most populous in all strains. Among the repetitive sequences in the Gypsy superfamily, AFLAV-like repeats were masked nearly 6 times more frequently in the WRRL 1519 (59 occurrences as opposed to 6–10 in other A. flavus strains).

Frequency of repetitive sequence elements in the A. flavus genomes. A total of 603, 477, 533, 533 and 886 non-redundant repetitive sequences were detected in strains NRRL 3357, NRRL 21882, NRRL 30797, NRRL 18543 and WRRL 1519, respectively
We attempted to identify complete putative reverse transcriptase and retrotransposon nucleotide sequences by querying specific sequences against the genomes of the inspected strains. Eleven candidate retrotransposons and six reverse transcriptases were found in WRRL 1519. In contrast, strains NRRL 3357, NRRL 21882, NRRL 30797 and NRRL 18543 had a maximum of one detected retrotransposon (NCBI GenBank accession AFLA_063980) and/or five reverse transcriptases (accessions AFLA_001110, AFLA_018200, AFLA_053840 and AFLA_114250). Genomic sequences from non-aflatoxigenic strains NRRL 21882, NRRL 30797, NRRL 18543 and WRRL 1519 did not share homology with the one queried LINE-1 retrotransposon (accession AFLA_063980 found in NRRL 3357). Only WRRL 1519 had sequences homologous to AFLAV (accession AY485786.2). The length of this query sequence was 7779 bp; most of the candidate retrotransposons were at least 6000 bp (Supplementary Table 4). The candidate retrotransposons in strain WRRL 1519 most similar to AFLAV were scaffold_113, scaffold_8b and scaffold_18a (Supplementary Fig. 4), suggesting that most of the candidates were derived from other non-AFLAV-like TEs, or the insertions were considerably older and have accumulated more mutations. Additionally, five of the eleven candidates were located in putatively pericentromeric scaffolds; most of the AFLAV sequences detected by RepeatMasker or HMMER were located on scaffolds predicted to be near the centromeres or the telomeres (Supplementary Fig. 2). Three of the candidates had canonical open reading frames at least 300 bp long: scaffold_8b (303 bp), scaffold_1 (390 bp) and scaffold_113 (6000 bp). According to NCBI BLAST results, the latter two open reading frames contained sequences likely to be related to Gag-Pol polyproteins. Moreover, BLAST results against the NCBI database indicated that most of the candidates were highly similar to subsequences on scaffolds SC005 and SC009 of A. oryzae RIB40 (E value < 1−247), corresponding to the 6000 bp retrotransposon AoLTR. Separate from the LTRs reported in Table 5, several sequences similar to both the 5′ and 3′ LTRs of AFLAV were identified in WRRL 1519 scaffolds 1 (two instances), 8 (four instances), 18, and 38. None were found in the other A. flavus strains.
Repetitive sequences were associated each other, but not with the differences in genome organization
The genetic neighborhood of the candidate retrotransposons included genes that were largely unannotated, except for candidate scaffold_22 which was surrounded by genes annotated as citrate synthase, transcription factor, sugar transporter and ABC multidrug transporter. Candidate retrotransposons on scaffolds 0, 1, 8 and 89 were in areas with chromosomal mismatches and genes that did not have homology to NRRL 3357 genes. Candidate scaffold_0 was located at the interface of where scaffold 0 stops matching to mostly proteins in chromosome EQ963481.1 (chromosome II, p arm) and begins matching those in EQ963473.1 (chromosome II, q arm). Candidate scaffold_1 neighbors a single extrachromosomal mismatch to EQ963480.1 (chromosome V, p arm), by a scaffold that mostly matches EQ963478.1 (chromosome II, q arm). This mismatch adds a copy of a kinesin family protein-like coding gene. AFLAV-like sequences on scaffold 8 were located in a region (nucleotides 155,663 to 341,186) where only 10 out of 44 predicted genes had homology to those in NRRL 3357. The AFLAV candidate on scaffold 89 was located at the end of a mismatch to a region of EQ963481.1 (chromosome II, p arm) NRRL 3357 that is not represented in other WRRL 1519 scaffolds.
Noting that five of the AFLAV candidates were within 100,000 bp of apparent extrachromosomal mismatches, we suspected that TE activity may be responsible for chromosomal shifts. We inspected the genomic loci of all extrachromosomal predicted protein-coding genes, regulatory sequences, repetitive elements and secretory genes and performed permutation tests to see if the repetitive elements were generally associated with any of these sequences of interest. Over half of the WRRL 1519 repeat sequences were clearly clustered with one another within 10,000 bp (Figs. 5, 6a). However, permutations in which the loci of repetitive sequences were randomized did not reveal significant associations with extrachromosomal mismatches for any of the strains (Fig. 6b).


Genomic locations of genes, regulatory and repetitive sequences and secondary metabolite gene clusters on NRRL 3357 (left) and WRRL 1519 (right) scaffolds constituting chromosomes a I–III and b IV–VIII. Densities of predicted protein-coding genes, regulatory sequences, repetitive sequences and secretory protein-coding genes are represented by the gray (max height = 19), green (max height = 231), red (max height = 14) and purple (max height = 9) histograms, respectively. Bin size (25,000 bp) is the same for all histograms. The innermost layer depicts genomic locations of secondary metabolite gene cluster colored by product type (pink, polyketide /non-ribosomal peptide; orange, siderophore; yellow, terpene; blue, indole; black, other). Scaffold length is tracked in kilobase pairs. (Color figure online)

Association of repetitive sequences with a other repetitive sequences and b extrachromosomal mismatches. The percentage of total repeats for a strain within a specified range of the borders of any repetitive sequences or extrachromosomal mismatch in the strain’s genome detected from the proteomic alignments is shown. Repeats from the strains and the randomized model are indicated by colored shapes. a The noticeable deviation of the A. flavus strains from the randomized pattern indicates that the repetitive elements were non-randomly associated with one another. b Repeat sequences were not significantly associated with extrachromosomal mismatches. Repeats similarly appeared randomly associated with protein-coding genes, regulatory sequences and secondary metabolite gene clusters (data not shown)
Discussion
The first published genome of A. oryzae was sequenced to 9X coverage depth, initially yielding six scaffolds and ten contigs (24 contigs total) organized into eight chromosomes by Southern hybridization to chromosomal probes and fingerprinting; the assembly was validated by cloning and optical mapping (Machida et al. 2005). A. oryzae and A. flavus are closely related to A. flavus. Therefore, when aflatoxigenic A. flavus NRRL 3357 was sequenced, the 16 largest scaffolds (out of 79 scaffolds) were organized into eight chromosomes by sequence comparison with the A. oryzae chromosome map. The overall genomic organizations between the species were similar except for a large translocation between chromosomes II and VI (Payne et al. 2006, 2008). In turn, the genome of Aspergillus parasiticus SU-1 was also mapped to the 16 scaffolds of A. flavus by sequence similarity (Linz et al. 2014).
Thus, using the alignment of aflatoxigenic A. flavus NRRL 3357, we were able to organize the genomic scaffolds of the four non-aflatoxigenic strains into hypothetical chromosomes. Most of the genome assemblies of the non-aflatoxigenic strains had at least one scaffold matched to a putative NRRL 3357 chromosomal arm. However, the p arm of the chromosome VII was not found in the genome assembly of NRRL 21882. None of the scaffolds had a similar size to the arm (~ 350 kbp) and the only four matched protein-coding genes were on four different scaffolds. However, this chromosomal arm was found be represented by scaffold NKQQ01000002.1 in an older genome assembly of NRRL 21882 (NCBI genome assembly ID GCA_002217635.1), so the newer assembly simply may be incomplete. We were able to align most of the WRRL 1519 genome assembly to that of NRRL 3357, providing a guide to future improvement of the current genome assembly. Our current results are purely computational and the scaffold ordering should be verified by deep sequencing.
In addition to building a better hypothetical genome assembly, we were able to compare the organization of protein-coding gene loci in the genomes of NRRL 3357 and WRRL 1519. There was a notably higher number of extrachromosomal mismatches between NRRL 3357 and WRRL 1519 than between NRRL 3357 and any other tested non-aflatoxigenic strains. The biological significance, if any, of these apparent chromosomal rearrangements is not clear. None of the secondary metabolite biosynthesis gene clusters at those locations were noticeably different between strains. However, single genes or regulatory sequences may be missing or disrupted due to the difference in genome organization that we have not detected. The apparent deletions in two predicted biosynthetic gene clusters of WRRL 1519 were not associated with extrachromosomal mismatches, and likely indicate a difference in the secondary metabolite repertoires of NRRL 3357 and WRRL 1519.
The cause of the extrachromosomal mismatches is also not clear. There was a marked increase in transposon-like sequences in WRRL 1519. TEs can cause insertional mutations in fungi and induce differences in genome structure among strains, driving evolution (Nishimura et al. 2000; Braumann et al. 2008; Lind et al. 2017). TEs may land within an open reading frame or regulatory sequence. TE activity may be actively repressed by host-directed repeat-induced point mutations and such mutations may “leak” over to neighboring genes (Irelan et al. 1994; Fudal et al. 2009). Generally, TEs may cluster preferentially near genes encoding effectors (secreted cysteine-rich proteins that modulate host-pathogen interactions), increasing the evolutionary speed of genes involved in pathogenicity; likewise TEs cluster near the immune genes of plants (Raffaele and Kamoun 2012; Dong et al. 2015; Seidl and Thomma 2017). In Aspergillus, some TEs are associated with gene clusters (Lind et al. 2017).
Transposon insertion and activity can potentially affect virulence of phytopathogenic fungi. The location of a class I Mariner/Tc1 transposon in the promotor or coding sequence of an avirulence gene allows M. oryzae to overcome gene-for-gene resistance (Kang et al. 2001; Zhou et al. 2007). Xu et al. propose that frequent transposon activity is responsible for the evolution of a pathogenic M. oryzae ancestor to the endophytic Harpophora oryzae (Xu et al. 2014). Active and inactive transposons from several plant pathogenic fungi including F. oxysporum (Anaya and Roncero 1996; Daboussi 1997; Mes et al. 2000; Rep et al. 2005), Verticillium dahlia (Amyotte et al. 2012), M. oryzae (Ikeda et al. 2001; Chadha and Sharma 2014) and Pseudocercospora fijiensis (Chang et al. 2016) are associated with increased genome size, genome instability and disease aggressiveness. Still active transposons can often be stimulated by stress conditions (Anaya and Roncero 1996; Ikeda et al. 2001; Ogasawara et al. 2009; Amyotte et al. 2012; Chadha and Sharma 2014).
Scientific publications make special note of their appearance often found near effectors, virulence genes, and gene clusters (Nishimura et al. 2000; Braumann et al. 2008; Raffaele and Kamoun 2012; Dong et al. 2015; Lind et al. 2017; Seidl and Thomma 2017). We have shown that repetitive sequences are significantly likely to be within 10,000 bp of one another, may be associated with chromosomal differences and that genome-wide associations with extrachromosomal mismatches and other sequences of interest are not obviously different from what could be randomly expected, even in biocontrol strain WRRL 1519 which exhibited a noticeably different genome organization. Payne et al. (2006, 2008) note that the frequencies of three types of TEs are consistently greater in the atoxigenic A. oryzae than in A. flavus, implying that TEs may present a method by which some non-aflatoxigenic strains may arise and differentiate from their aflatoxigenic ancestors. Chang and Ehrlich (2010), reporting that NRRL 3357 has low copy numbers of elements Tao1, Crawler and AFLAV, further state that mobile elements may contribute to the differentiation of A. flavus and A. oryzae. In light of our results, we also hypothesize that transposons may play some role in shaping the genomes of some non-aflatoxigenic A. flavus strains that can be used for biocontrol of aflatoxins. Whether transposons may lead to the nonrandom natural evolution of fungal strains in nature is an intriguing question and is one we are pursuing in future work.
References
Adhikari BN, Bandyopadhyay R, Cotty PJ (2016) Degeneration of aflatoxin gene clusters in Aspergillus flavus from Africa and North America. AMB Express 6(1):62. https://doi.org/10.1186/s13568-016-0228-6
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Amaike S, Keller NP (2011) Aspergillus flavus. Annu Rev Phytopathol 49:107–133. https://doi.org/10.1146/annurev-phyto-072910-095221
Amyotte SG, Tan X, Pennerman K, Jimenez-Gasco Mdel M, Klosterman SJ, Ma LJ, Dobinson KF, Veronese P (2012) Transposable elements in phytopathogenic Verticillium spp.: insights into genome evolution and inter- and intra-specific diversification. BMC Genom 13:314. https://doi.org/10.1186/1471-2164-13-314
Anaya N, Roncero M (1996) Stress-induced rearrangement of Fusarium retrotransposon sequences. Mol Genet Genom 253(1–2):89–94. https://doi.org/10.1007/s004380050300
Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA (2008) Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci USA 105(33):11845–11850
Bennett JW (2010) An overview of the genus Aspergillus. In: Machida M, Gomi K (eds) Aspergillus: molecular biology and genomics.Caister Academic Press, Poole, pp 1–18.
Bennetzen JL (2005) Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev 15(6):621–627
Bourque G (2009) Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr Opin Genet Dev 19(6):607–612. https://doi.org/10.1016/j.gde.2009.10.013
Braumann I, van den Berg M, Kempken F (2007) Transposons in biotechnologically relevant strains of Aspergillus niger and Penicillium chrysogenum. Fungal Genet Biol 44(12):1399–1414
Braumann I, van den Berg MA, Kempken F (2008) Strain-specific retrotransposon-mediated recombination commercially used Aspergillus niger strain. Mol Genet Genom 280:319. https://doi.org/10.1007/s00438-008-0367-9
Butler M, Goodwin T, Simpson M, Singh M, Poulter R (2001) Vertebrate LTR retrotransposons of the Tf1/sushi group. J Mol Evol 52(3):260–274
Castanera R, López-Varas L, Borgognone A, LaButti K, Lapidus A, Schmutz J, Grimwood J, Pérez G, Pisabarro AG, Grigoriev IV, Stajich JE, Ramírez L (2016) Transposable elements versus the fungal genome: impact on whole-genome architecture and transcriptional profiles. PLOS Genet 12(6):e1006108. https://doi.org/10.1371/journal.pgen.1006108
Chadha S, Sharma M (2014) Transposable elements as stress adaptive capacitors induce genomic instability in fungal pathogen Magnaporthe oryzae. PLOS One 9(4):e94415. https://doi.org/10.1371/journal.pone.0094415
Chang PK, Ehrlich KC (2010) What does genetic diversity of Aspergillus flavus tell us about Aspergillus oryzae? Int J Food Microbiol 138(3):189–199. https://doi.org/10.1016/j.ijfoodmicro.2010.01.033
Chang P-K, Horn BW, Dorner JW (2005) Sequence breakpoints in the aflatoxin biosynthesis gene cluster and flanking regions in nonaflatoxigenic Aspergillus flavus isolates. Fungal Genet Biol 42(11):914–923
Chang P-K, Abbas HK, Weaver MA, Ehrlich KC, Scharfenstein LL, Cotty PJ (2012) Identification of genetic defects in the atoxigenic biocontrol strain Aspergillus flavus K49 reveals the presence of a competitive recombinant group in field populations. Int J Food Microbiol 154(3):192–196. https://doi.org/10.1016/j.ijfoodmicro.2012.01.005
Chang TC, Salvucci A, Crous PW, Stergiopoulos I (2016) Comparative genomics of the Sigatoka disease complex on banana suggests a link between parallel evolutionary changes in Pseudocercospora fijiensis and Pseudocercospora eumusae and increased virulence on the banana host. PLOS Genet 12(8):e1005904. https://doi.org/10.1371/journal.pgen.1005904
Clutterbuck AJ, Kapitonov VV, Jurka J (2008) Transposable elements and repeat-induced point mutation in Aspergillus nidulans, Aspergillus fumigatus, and Aspergillus oryzae. In: Goldman GH, Osmani SA (eds) The Aspergilli: genomics, medical aspects, biotechnology, and research methods. CRC Press, Boca Raton, pp 343–355
Cotty PJ (1990) Effects of atoxigenic strains of Aspergillus flavus on aflatoxin contamination of developing cotton seeds. Plant Dis 74(3):233–235. https://doi.org/10.1094/PD-74-0233
Cotty PJ (1994) Influence of field application of an atoxigenic strain of Aspergillus flavus on the populations of A. flavus infecting cotton bolls and on the aflatoxin content of cottonseed. Phytopathol 84:1270–1277. https://doi.org/10.1094/Phyto-84-1270
Cotty PJ, Mellon JE (2006) Ecology of aflatoxin producing fungi and biocontrol of aflatoxin contamination. Mycotoxin Res 22(2):110–117. https://doi.org/10.1007/BF02956774
Daboussi MJ (1997) Fungal transposable elements and genome evolution. Genetica 100:253–260
Daboussi MJ, Capy P (2003) Transposable elements in filamentous fungi. Annu Rev Microbiol 57:275–299
Davière JM, Langin T, Daboussi MJ (2001) Potential role of transposable elements in the rapid reorganization of the Fusarium oxysporum genome. Fungal Genet Biol 34(3):177–192
de Lucca AJ (2007) Harmful fungi in both agriculture and medicine. Rev Iberoam Micol 24(1):3–13
Divakara ST, Aiyaz M, Moore GG, Venkataramana M, Hariprasad P, Nayaka SC, Niranjana SR (2015) Analysis of genetic and aflatoxin diversity among Aspergillus flavus isolates collected from sorghum seeds. J Basic Microbiol 55(11):1255–1264. https://doi.org/10.1002/jobm.201400951
Dong S, Raffaele S, Kamoun S (2015) The two-speed genomes of filamentous pathogens: Waltz with plants. Curr Opin Genet Dev 35:57–65. https://doi.org/10.1016/j.gde.2015.09.001
Dusa A (2016) venn: draw venn diagrams. R package version 1.2. https://www.cran.r-project.org/package=venn. Accessed 25 May 2018
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2(4):953–971
Emtiazi G, Naghavi N, Bordbar A (2001) Biodegradation of lignocellulosic waste by Aspergillus terreus. Biodegradation 12(4):259–263
Feschotte C (2008) The contribution of transposable elements to the evolution of regulatory networks. Nat Rev Genet 9(5):397–405. https://doi.org/10.1038/nrg2337
Fudal I, Ross S, Brun H, Besnard AL, Ermel M, Kuhn ML, Balesdent MH, Rouxel T (2009) Repeat-induced point mutation (RIP) as an alternative mechanism of evolution toward virulence in Leptosphaeria maculans. Mol Plant Microbe Interact 22(8):932–941. https://doi.org/10.1094/MPMI-22-8-0932
Geiser DM, Pitt JI, Taylor JW (1998) Cryptic speciation and recombination in the aflatoxin-producing fungus Aspergillus flavus. Proc Natl Acad Sci USA 95(1):388–393
Goldman GH, Osmani SA (2008) The Aspergilli: genomics, medical aspects, biotechnology, and research methods. CRC Press, Boca Raton
Hansen LJ, Chalker DL, Sandmeyer SB (1988) Ty3, a yeast retrotransposon associated with tRNA genes, has homology to animal retroviruses. Mol Cell Biol 8(12):5245–5256
Hua SS, Tarun AS, Pandey SN, Chang L, Chang PK (2007) Characterization of AFLAV, a Tf1/Sushi retrotransposon from Aspergillus flavus. Mycopathologia 163(2):97–104
Hua SS, McAlpin CE, Chang PK, Sarreal SB (2012) Characterization of aflatoxigenic and non-aflatoxigenic Aspergillus flavus isolates from pistachio. Mycotoxin Res 28(1):67–75. https://doi.org/10.1007/s12550-011-0117-4
Hua-Van A, Davière JM, Kaper F, Langin T, Daboussi MJ (2000) Genome organization in Fusarium oxysporum: clusters of class II transposons. Curr Genet 37(5):339–347
Hua-Van A, Le Rouzic A, Maisonhaute C, Capy P (2005) Abundance, distribution and dynamics of retrotransposable elements and transposons: similarities and differences. Cytogenet Genome Res 110(1–4):426–440
Ikeda K, Nakayashiki H, Takagi M, Tosa Y, Mayama S (2001) Heat shock, copper sulfate and oxidative stress activate the retrotransposon MAGGY resident in the plant pathogenic fungus Magnaporthe girsea. Mol Genet Genom 266(2):318–325. https://doi.org/10.1007/s004380100560
Irelan JT, Hagemann AT, Selker EU (1994) High frequency repeat-induced point mutation (RIP) is not associated with efficient recombination in Neurospora. Genet 138(4):1093–1103
Jamali M, Ebrahimi MA, Karimipour M, Shams-Ghahfarokhi M, Dinparast-Djadid N, Kalantari S, Pilehvar-Soltanahmadi Y, Amani A, Razzaghi-Abyaneh M (2012) An insight into the distribution, genetic diversity, and mycotoxin production of Aspergillus section Flavi in soils of pistachio orchards. Folia Microbiol (Praha) 57(1):27–36. https://doi.org/10.1007/s12223-011-0090-5
Jin FJ, Hara S, Sato A, Koyama Y (2014) Discovery and analysis of an active long terminal repeat-retrotransposable element in Aspergillus oryzae. J Gen Appl Microbiol 60(1):1–6
Johnson LS, Eddy SR, Portugaly E (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform 11(1):431
Kang S, Lebrun MH, Farrall L, Valent B (2001) Gain of virulence caused by insertion of a Pot3 transposon in a Magnaporthe grisea avirulence gene. Mol Plant Microbe Interact 14(5):671–674
Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G, Baranasic D, Arenillas DJ, Sandelin A, Vandepoele K, Lenhard B, Ballester B, Wasserman WW, Parcy F, Mathelier A (2018) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46(D1):D1284. https://doi.org/10.1093/nar/gkx1188
Klein SJ, O’Neill RJ (2018) Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosome Res 26(1–2):5–23. https://doi.org/10.1007/s10577-017-9569-5
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19(9):1639–1645. https://doi.org/10.1101/gr.092759.109
Larrondo LF, Canessa P, Vicuña R, Stewart P, Vanden Wymelenberg A, Cullen D (2007) Structure and transcriptional impact of divergent repetitive elements inserted within Phanerochaete chrysosporium strain RP-78 genes. Mol Genet Genom 277(1):43–55
Lind AL, Wisecaver JH, Lameiras C, Wiemann P, Palmer JM, Keller NP, Rodrigues F, Goldman GH, Rokas A (2017) Drivers of genetic diversity in secondary metabolic gene clusters within a fungal species. PLOS Biol 15(11):e2003583. https://doi.org/10.1371/journal.pbio.2003583
Linz JE, Wee J, Roze LV (2014) Aspergillus parasiticus SU-1 genome sequence, predicted chromosome structure, and comparative gene expression under aflatoxin-inducing conditions: evidence that differential expression contributes to species phenotype. Eukaryot Cell 13(8):1113–1123. https://doi.org/10.1128/EC.00108-14
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, Yamagata Y, Anazawa H, Hata Y, Koide Y, Komori T, Koyama Y, Minetoki T, Suharnan S, Tanaka A, Isono K, Kuhara S, Ogasawara N, Kikuchi H (2005) Genome sequencing and analysis of Aspergillus oryzae. Nature 438(7071):1157–1161
Machida M, Yamada O, Gomi K (2008) Genomics of Aspergillus oryzae: Learning from the history of Koji mold and exploration of its future. DNA Res 15(4):173–183. https://doi.org/10.1093/dnares/dsn020
Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A (2018) MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol 14(1):e1005944. https://doi.org/10.1371/journal.pcbi
Mes JJ, Haring MA, Cornelissen BJC (2000) Foxy: an active family of short interspersed nuclear elements from Fusarium oxysporum. Mol Genet Genom 263(2):271–280. https://doi.org/10.1007/PL00008681
Miller K, Lynch C, Martin J, Herniou E, Tristem M (1999) Identification of multiple Gypsy LTR-retrotransposon lineages in vertebrate genomes. J Mol Evol 49:358–366
Moore GG (2014) Sex and recombination in aflatoxigenic Aspergilli: global implications. Front Microbiol 5:32. https://doi.org/10.3389/fmicb.2014.00032
Moore GG, Singh R, Horn BW, Carbone I (2009) Recombination and lineage-specific gene loss in the aflatoxin gene cluster of Aspergillus flavus. Mol Ecol 18(23):4870–4887. https://doi.org/10.1111/j.1365-294X.2009.04414.x
Mousavi B, Hedayati MT, Hedayati N, Ilkit M, Syedmousavi S (2016) Aspergillus species in indoor environments and their possible occupational and public health hazards. Curr Med Mycol 2(1):36–42. https://doi.org/10.18869/acadpub.cmm.2.1.36
Neuveglise C, Sarfati J, Latge JP, Paris S (1996) Afut1, a retrotransposon-like element from Aspergillus fumigatus. Nucleic Acids Res 24(8):1428–1434
Nguyen NTT, Contreras-Moreira B, Castro-Mondragon JA, Santana-Garcia W, Ossio R, Robles-Espinoza CD, Bahin M, Collombet S, Vincens P, Thieffry D, van Helden J, Medina-Rivera A, Thomas-Chollier M (2018) RSAT 2018: Regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res 46(w1):w209–w214. https://doi.org/10.1093/nar/gky317
Nierman WC, May G, Kim HS, Anderson MJ, Chen D, Denning DW (2005) What the Aspergillus genomes have told us. Med Mycol 43(Suppl 1):S3–S5
Nierman WC, Yu J, Fedorova-Abrams ND, Losada L, Cleveland TE, Bhatnagar D, Bennett JW, Dean R, Payne GA (2015) Genome sequence of Aspergillus flavus NRRL 3357, a strain that causes aflatoxin contamination of food and feed. Genome Announc 3(2):pii:e00168–15. https://doi.org/10.1128/genomeA.00168-15
Nishimura M, Hayashi N, Jwa NS, Lau GW, Hamer JE, Hasebe A (2000) Insertion of the LINE retrotransposon MGL causes a conidiophore pattern mutation in Magnaporthe grisea. Mol Plant Microbe Interact 13(8):892–894
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O’Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44(D1):D733–D745. https://doi.org/10.1093/nar/gkv1189
Ogasawara H, Obata H, Hata Y, Takahashi S, Gomi K (2009) Crawler, a novel Tc1/mariner-type transposable element in Aspergillus oryzae transposes under stress conditions. Fungal Genet Biol 46(6–7):441–449. https://doi.org/10.1016/j.fgb.2009.02.007
Okubara PA, Tibbot BK, Tarun AS, McAlpin CE, Hua SS (2003) Partial retrotransposon-like DNA sequence in the genomic clone of Aspergillus flavus, pAF28. Mycol Res 107(Pt 7):841–846
Olarte RA, Horn BW, Dorner JW, Monacell JT, Singh R, Stone EA, Carbone I (2012) Effect of sexual recombination on population diversity in aflatoxin production by Aspergillus flavus and evidence for cryptic heterokaryosis. Mol Ecol 21(6):1453–1476. https://doi.org/10.1111/j.1365-294X.2011.05398.x
Oliver R (1992) Transposons in filamentous fungi. In: Stahl U, Tudzynski P (eds) Molecular Biology of Filamentous Fungi. In: Proceedings of the EMBO-Workshop, Berlin, August 24–29, 1991, Volume 4 of EMBO Workshop. Weinheim, Germany: VCH Verlagsgesellschaft, pp 3–11
Payne GA, Pritchard BL, Brown D, Yu J, Nierman WC, Dean RA, Bhatnagar D, Cleveland TE, Machida M (2006) Whole genome comparison of A. flavus and A. oryzae. Med Mycol 44(S1):S9–S11. https://doi.org/10.1080/13693780600835716
Payne GA, Yu J, Nierman WC, Machida M, Bhatnagar D, Cleveland TE, Dean RA (2008) A first glance into the genome sequence of Aspergillus flavus. In: Goldman GH, Osmani SA (eds) The Aspergilli: genomics, medical aspects, biotechnology, and research methods. CRC Press, Boca Raton, pp 15–23
Peterson-Burch BD, Nettleton D, Voytas DF (2004) Genomic neighborhoods for Arabidopsis retrotransposons: a role for targeted integration in the distribution of the Metaviridae. Genome Biol 5(10):R78
Pimpinelli S, Berloco M, Fanti L, Dimitri P, Bonaccorsi S, Marchetti E, Caizzi R, Caggese C, Gatti M (1995) Transposable elements are stable structural components of Drosophila melanogaster heterochromatin. Proc Natl Acad Sci USA 92(9):3804–3808
R Core Team (2015) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. Accessed 25 May 2018
Raffaele S, Kamoun S (2012) Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat Rev Microbiol 10(6):417–430. https://doi.org/10.1038/nrmicro2790
Raulo R, Kokolski M, Archer DB (2016) The roles of the zinc finger transcription factors XlnR, ClrA and ClrB in the breakdown of lignocellulose by Aspergillus niger. AMB Express 6:5. https://doi.org/10.1186/s13568-016-0177-0
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Rep M, van der Does HC, Cornelissen BJ (2005) Drifter, a novel, low copy hAT-like transposon in Fusarium oxysporum is activated during starvation. Fungal Genet Biol 42(6):546–553
Richard JL, Payne GA (2003) Mycotoxins: risks in plant, animal and human systems. Council for agricultural science and technology (CAST), Ames, IA. https://www.international-food-safety.com/pdf/Mycotoxins%20-%20Risks%20in%20Plant,%20Animals%20and%20Human%20Systems.pdf. Accessed 12 Apr 2018
Round EK, Flowers SK, Richards EJ (1997) Arabidopsis thaliana centromere regions: Genetic map positions and repetitive DNA structure. Genome Res 7(11):1045–1053
Seidl MF, Thomma BPHJ (2017) Transposable elements direct the coevolution between plants and microbes. Trend Genet 33(11):842–851. https://doi.org/10.1016/j.tig.2017.07.003
Smit AFA, Hubley R, Green P (2015) RepeatMasker Open-4.0. (2013–2015). https://www.repeatmasker.org. Accessed 20 May 2018
Solorzano CD, Abbas HK, Zablotowicz RM, Chang PK, Jones WA (2014) Genetic variability of Aspergillus flavus isolates from a Mississippi corn field. Sci World J 2014:356059. https://doi.org/10.1155/2014/356059
Stanke M, Schöffmann O, Morgenstern B, Waack S (2006) Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform 7:62
Sullivan MJ, Petty NK, Beatson SA (2011) Easyfig: a genome comparison visualizer. Bioinformatics 27(7):1009–1010. https://doi.org/10.1093/bioinformatics/btr039
Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M (2008) Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 18(12):1979–1990. https://doi.org/10.1101/gr.081612.108
The Inkscape Team (2015) Inkscape 0.91. https://inkscape.org/en/release/0.91/. Accessed 25 May 2018
Thon MR, Pan H, Diener S, Papalas J, Taro A, Mitchell TK, Dean RA (2006) The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus Magnaporthe oryzae. Genome Biol 7(2):R16
Tsukahara S, Kawabe A, Kobayashi A, Ito T, Aizu T, Shin-i T, Toyoda A, Fujiyama A, Tarutani Y, Kakutani T (2012) Centromere-targeted de novo integrations of an LTR retrotransposon of Arabidopsis lyrata. Genes Dev 26(7):705–713. https://doi.org/10.1101/gad.183871.111
Weaver MA, Scheffler BE, Duke M, Ballard L, Abbas HK, Grodowitz MJ (2017) Genome sequence of three strains of Aspergillus flavus for the biological control of aflatoxin. Genome Announc 5(44):pii:e01204–17. https://doi.org/10.1128/genomeA.01204-17
Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, Breitling R, Takano E, Medema MH (2015) AntiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acid Res 43(W1):W237–W243. https://doi.org/10.1093/nar/gkv437
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8(12):973–982
Xu XH, Su ZZ, Wang C, Kubicek CP, Feng XX, Mao LJ, Wang JY, Chen C, Lin FC, Zhang CL (2014) The rice endophyte Harpophora oryzae genome reveals evolution from a pathogen to a mutualistic endophyte. Sci Rep 4:5783. https://doi.org/10.1038/srep05783
Yin G, Hua SST, Pennerman KK, Yu J, Bu L, Sayre RT, Bennett JW (2018) Genome sequence and comparative analyses of atoxigenic Aspergillus flavus WRRL 1519. Mycologia 110(3):482–493. https://doi.org/10.1080/00275514.2018.1468201
Zhou E, Jia Y, Singh P, Correll JC, Lee FN (2007) Instability of the Magnaporthe oryzae avirulence gene AVR-Pita alters virulence. Fungal Genet Biol 44(10):1024–1034
Funding
This work was funded by the USDA-ARS Non-Assistance Cooperative Agreement (no. 58-2030-6-053). Use of a company or product name by the U.S. Department of Agriculture does not imply approval or recommendation of the product to the exclusion of others that may also be suitable. We also greatly appreciate additional funding received from the Rutgers University Educational Opportunity Fund (J Gonzalez) and the Mycological Society of America John W. Rippon Award (KK Pennerman).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
KK Pennerman declares that she has no conflict of interest. J Gonzalez declares that she has no conflict of interest. LF Chenoweth declares that she has no conflict of interest. JW Bennett declares that she has no conflict of interest. G Yin declares that she has no conflict of interest. SST Hua declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2018_1474_MOESM1_ESM.eps
Supplementary Figure 1. Scaffold matching of NRRL 3357 to (A) itself, (B) NRRL 21882, (C) NRRL 30797 and (D) NRRL 18543. Chromosomes of NRRL 3357 are drawn in 16 different colors on the left side of each diagram. The outer track enumerates chromosome nucleotide length in kilobase pairs, and protein-coding gene density is shown by a histogram on the inner track. Scaffolds and chromosomes are connected to the NRRL 3357 chromosomes by lines representing high sequence similarity between two protein-coding genes. There is one extrachromosomal mismatch between NRRL 3357 chromosomes EQ963477.1 to EQ963476.12 due to a match to an apparent paralog. Many of the mismatches between NRRL 3357 and a non-aflatoxigenic strain are the same for different strains. (EPS 9861 KB)
438_2018_1474_MOESM5_ESM.pdf
Supplementary Figure 2. Scaffold organization of strain WRRL 1519. The scaffolds are ordered according to their relative placement to the centromeres. Blue and orange colors indicate the nucleotide sequence direction of the scaffold relative to the deposited NRRL 3357 genomic sequence. An asterisk denotes scaffolds on which one or more AFLAV-like sequences were found by either HMMER or RepeatMasker. Scaffolds 59, 72, 73 and 113 in which AFLAV-like sequences were detected are not represented because they were not confidently aligned to the NRRL 3357 chromosomes. The diagram is not to scale. (PDF 193 KB)
438_2018_1474_MOESM6_ESM.pdf
Supplementary Figure 3. Macrosynteny of A. flavus (A) NRRL 21882, (B) NRRL 30797, (C) NRRL 18543 and (D) WRRL 1519 scaffolds to NRRL 3357 chromosomes. Scaffolds of the strains are listed in the same relative orders as in Figure 2 and Supplementary Figure 1. (PDF 163 KB)
438_2018_1474_MOESM7_ESM.pdf
Supplementary Figure 4. Full alignment of WRRL 1519 candidate retrotransposons to retrotransposon AFLAV AY485786.2 from A. flavus NRRL 3357. Candidate retrotransposons are labeled according to Supplementary Table 4. Black and gray shading indicates strength of nucleotide consensus. The LTR regions, ORF1, ribosomal-1 frameshift site and ORF2 of AY485786.2 are highlighted in yellow, blue, orange and green, respectively. (PDF 1161 KB)
438_2018_1474_MOESM8_ESM.xlsx
Supplementary Table 1. Summary of non-aflatoxigenic A. flavus genome scaffolds matching to putative NRRL 3357 chromosomes. (XLSX 24 KB)
438_2018_1474_MOESM9_ESM.xlsx
Supplementary Table 2. Summary of results of sequence similarity searches of WRRL 1519 putative proteins against the annotated NRRL 3357 proteome. (XLSX 1323 KB)
Rights and permissions
About this article

Cite this article
Pennerman, K.K., Gonzalez, J., Chenoweth, L.R. et al. Biocontrol strain Aspergillus flavus WRRL 1519 has differences in chromosomal organization and an increased number of transposon-like elements compared to other strains. Mol Genet Genomics 293, 1507–1522 (2018). https://doi.org/10.1007/s00438-018-1474-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-018-1474-x