
Abstract
Phytophthora sojae is an oomycete soil-borne plant pathogen that causes the serious disease Phytophthora root rot in soybean, leading to great loss of soybean production every year. Understanding the genetic basis of this plant–pathogen interaction is important to improve soybean disease resistance. To discover genes or QTLs underlying naturally occurring variations in soybean P.sojae resistance, we performed a genome-wide association study using 59,845 single-nucleotide polymorphisms identified from re-sequencing of 279 accessions from Yangtze-Huai soybean breeding germplasm. We used two models for association analysis. The same strong peak was detected by both two models on chromosome 13. Within the 500-kb flanking regions, three candidate genes (Glyma13g32980, Glyma13g33900, Glyma13g33512) had SNPs in their exon regions. Four other genes were located in this region, two of which contained a leucine-rich repeat domain, which is an important characteristic of R genes in plants. These candidate genes could be potentially useful for improving the resistance of cultivated soybean to P.sojae in future soybean breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soybean (Glycine max) is one of the most important commercial crops across the world. About 30 kinds of major disease have been reported in soybean. Root and stem rot (Phytophthora root rot, PRR) was ranked among the five most serious diseases of soybean in the United States during 2003–2005 (Wrather and Koenning 2006). Soybean root and stem rot disease is caused by the soil-borne oomycete pathogen Phytophthora sojae (Kaufmann and Gerdemann) (Schmitthenner 1985; Zhu et al. 2003) and can occur throughout the entire period of soybean growth, resulting in huge losses in agricultural production (Wrather and Koenning 2006).
There are many methods to control this disease; seed coating agents, reducing soil moisture and screening for resistance resources are among the most effective (Dorrance et al. 2003; Dorrance et al. 2007). To date, special soybean genotypes conferring complete immunity to every strain of P.sojae have not been found. However, partial resistance is more durable for the changing diversities of P.sojae strains. Most research has focused on partial resistance to PRR (Dorrance et al. 2003; Lee et al. 2014; Sun et al. 2014a). So far, 21 Rps (resistance to P.sojae) genes/alleles have been detected on four chromosomes. Ten Rps genes/alleles were located on the short arm of chromosome 3, including Rps1 (containing five alleles; Rps1a, Rps1b, Rps1c, Rps1d and Rps1k), Rps7, Rps9, RpsYu25, RpsYD29, RpsUN1, and an unnamed Rps gene in the Japanese cultivar ‘Waseshiroge’ (Demirbas et al. 2001; Weng et al. 2001; Gao et al. 2005; Sugimoto et al. 2011; Sun et al. 2011; Wu et al. 2011a; Lin et al. 2013; Zhang et al. 2013). Rps3 (containing five alleles; Rps3a, Rps3b and Rps3c) has been mapped on chromosome 13, and is linked with Rps8 (Demirbas et al. 2001; Sandhu et al. 2005; Gordon et al. 2006; Lee et al. 2013). Rps2 and RpsUN2 have been mapped on chromosome 16 (Demirbas et al. 2001; Lin et al. 2013). RpsJS, Rps4, Rps5 and Rps6 are linked and located on chromosome 18 (Demirbas et al. 2001; Sandhu et al. 2004; Lee et al. 2013, 2014; Sun et al. 2014b). Among these Rps genes/alleles, only RpsJS and RpsYD29 have been finely mapped (Zhang et al. 2013; Sun et al. 2014b) and only one gene with a coiled coil-nucleotide binding site–leucine rich repeat has been cloned (Gao et al. 2005).
All previous studies on PRR resistance in soybean have been performed using traditional linkage mapping. Association mapping is another effective approach to connect structural genomics and phenomics in plants, if information on population structure and linkage disequilibrium (LD) is available (Jin et al. 2010). With high-density simple nucleotide polymorphisms (SNPs), genome-wide association mapping has been successfully used to analyze complex trait variation in Arabidopsis (Atwell et al. 2010; Horton et al. 2012), rice (Huang et al. 2010; Yang et al. 2014b), maize (Kump et al. 2011; Tian et al. 2011; Yang et al. 2014a), and soybean (Wang et al. 2008; Hao et al. 2012a; Wen et al. 2014; Zhang et al. 2014; Zhao et al. 2015) to identify alleles and genes for disease resistance and agronomic traits (Wen et al. 2014; Zhao et al. 2015). Here, we performed a genome-wide association study (GWAS) of soybean using 59,854 high-quality SNPs to identify significant markers and genes associated with PRR resistance. Our results could help to further enhance durable resistance based on the genes we identified and improve our understanding of the genetic basis of partial resistance to P.sojae.
Materials and methods
Sampling and genotyping
A total of 472 soybean accessions from Yangtze-Huai soybean breeding germplasm population were obtained from National Center for Soybean Improvement, Nanjing city, Jiangsu province, China.
The sequenced RAD (restriction association site DNA) markers were used in the present study. All the genotyping work was done at BGI Tech, Shenzhen, China. Soybean genomic DNA was extracted from young leaves using a modified cetyltrimethylammonium bromide (CTAB) method (Allen et al. 2006), with average concentration 90 ug/ul. The sequences of the 472 accessions were obtained using an Illumina HiSeq 2000 instrument and through utilizing the MSG (multiplexed shotgun genotyping) method (Andolfatto et al. 2011). DNA fragments between 400 and 600 bp were generated by TaqI digestion, giving a total of 1322.73 million paired-end reads of 90 bp (including 6 bp index) read length (110.87 Gb of sequence) with most at an approximately × 3.86 depth and 4.57 % coverage. All the sequence reads were aligned against the reference genome of Williams 82 (genome assembly 1 annotation version 1.1) (Schmutz et al. 2010) using the SOAP2 short read alignment software (Li et al. 2009) with parameters including sequence similarity, pair-end relationships, and sequence quality. RealSFS (Yi et al. 2010) was used for the population SNP calling, based on the Bayesian estimation of the site frequency at every site. The fastPHASE software (Scheet and Stephens 2006) was used for genotyping the SNP imputation after the heterozygous alleles were turned into missing alleles. In this study, we used 279 accessions with 59,845 SNPs for GWAS study (see Additional file 1).
Partial resistance evaluation
An isolate of P. sojae, P7076 (with virulence to 1a,1b,1c,1d,1k,3a), was obtained from the Key laboratory of Monitoring and Management of Plant Diseases and Insects, Ministry of Agriculture, Nanjing Agricultural University, China. The 472 soybean accessions were determined using the modified hypocotyl inoculation technique (Sun et al. 2011). There are 279 susceptible materials, which were used as the slant board assay. The method was previously published (Wu et al. 2011b). In brief, to avoid the difference of materials, ten 7-day-old seedlings for each accession were placed on trays, respectively. And wounded 20 mm below the crown using a scalpel. Lesion lengths were measured from the inoculation site to the edge of the lesion margin. Details of the procedures have been described previously (Sun et al. 2014a).
The soybean accessions were evaluated, and compared with two controls (var. ‘Conrad’ and var. ‘Sloan’) with known partial resistance characteristics. Conrad has high levels of partial resistance to P. sojae, whereas Sloan is moderately susceptible (Burnham et al. 2003). The experiment was repeated three times.
Genetic diversity and polymorphism information content (PIC)
Nei’s gene diversity index (H) (Nei 1973), and the polymorphism information content (PIC) per locus were calculated with an in-house R script. Nei’s gene diversity (H) was calculated based on the formula
where n i is the allele frequency at the ith locus, n is the number of alleles this locus and N is the total number of accessions.
The PIC for each marker was calculated based on the formula
where P is the relative frequency of the jth pattern for SNP marker i (Botstein et al. 1980).
Population genetic study
Structure and maximum likelihood (ML) tree was applied to infer the population stratification of our materials. The population structure was inferred using the software program STRUCTURE 2.2 (Pritchard et al. 2000) based on 3113 SNPs with minor allele frequency (MAF) >20 % and physical distance >60 kb (Wang 2014). The burn in period was 100,000-iteration followed by 200,000 using a model that allowed for admixture and correlated allele frequencies (He and Park 2013). At least four independent runs of structure were performed, setting K from 1 to 10, and an average likelihood value, L(K), across all runs was calculated for each K. The most likely number of subpopulations was then determined using the Delta K method proposed by Evanno et al. (2005). Principal component analysis (PCA) was done by using GCTA software (Yang et al. 2011). We applied a maximum likelihood method to construct the phylogenetic tree based on the SNP-based genetic distance using RAxML software (Stamatakis 2006) with 100 replicates for a bootstrap confidence analysis based on 59,845 SNPs (MAF > 0.05) by using GTR+G model which was based on the result of jModelTest (Posada 2008). Linkage disequilibrium parameter (r 2) for estimating the degree of LD between pair-wise SNPs (59,845 with MAF > 0.05) was calculated by using the software TASSEL 4.0 (Bradbury et al. 2007) with 1000 permutations. The LD decay rate was measured as the chromosomal distance at the r 2 dropped to the half of from its maximum to minimum value.
Genome-wide association study
In this study we used two different models to test associations between the SNPs (MAF > 0.05) and disease assessment criteria. The first model was general linear model (GLM), which just containing the SNP tested as a fixed effect, was used to test the association between the SNP and the disease assessment criteria. The second model is mixed linear model (MLM) where, in addition to the SNP being tested, Q matrix and kinship matrix were included as fixed and random effects, respectively. Kinship was calculated by using TASSEL 4.0. Both of those two models were performed by the software TASSEL 4.0 (Bradbury et al. 2007). To get reliable results, false discovery rate (FDR) ≤ 0.05 was used to identify the significant associations.
Results
Genetic diversity, population structure and linkage disequilibrium analysis
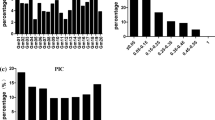
A total of 60,656 SNPs were used to analyze the genetic diversity in the 279 soybean accessions. The allele frequencies of most alleles were less than 10 % and the average MAF was 0.217 (range 0.014–0.498) (Fig. 1a). The expected heterozygosity, nucleotide diversity and PIC of the 60,656 SNPs averaged 0.306, 0.307 and 0.251, with ranges of 0.028–0.499, 0.028–0.500 and 0.027–0.375, respectively (Fig. 1).

Distribution of genetic diversity of 60,656 SNPs across 279 accession. a Distribution of MAF, b distribution of expected heterozygosity, c distribution of PIC values, and d distribution of nucleotide diversity (color figure online)
In this study, we used Bayesian model-based methods and principal component analysis to analyze the population structure. Unfortunately, estimated likelihood values, LnP (D), do not indicate the exact K value. Therefore, an ad hoc quantity (∆K) was used to overcome the difficulty interpreting real K values. Using this approach, an identifiable peak indicated the true value of K based on ∆K. For the 279 accessions, the highest value of ∆K was at K = 3 (Fig. 2a). This suggested that the 279 accessions could be divided into three major subpopulations (Fig. 2a, b). Principal component analysis gave the same result (Fig. 2c). Using the maximum likelihood method, a phylogenetic tree was constructed that also showed consistent results with STRUCTURE (Fig. 2d). Interestingly, we found that the backgrounds of the three subpopulations were consistent with three main parents of the populations. In subpopulation Q1 (including 73 accessions), most soybean accessions were derived from the main parent 86–4. Subpopulation Q2 included 75 accessions that were mainly descended from the parent TongDou. There were 99 accessions in subpopulation Q3, whose main parent was 88–48. The 32 remaining accessions showed admixture patterns. The corresponding Q-matrix at K = 3 was used for the subsequent genome-wide association analysis. The results of AMOVA indicated that 71.52 % of the genetic variation was within subpopulations, whereas 28.48 % of the total genetic variation was among subpopulations (Table 1). The population pair-wise F ST was 0.2848 (P < 0.001) between the three subpopulations (Table 1).

Population structure of 279 soybean accessions. a Determination of K value in Structure analysis. Green line is log-likelihood of the data, L(K), as a function of K (number of groups used to stratify the sample). Red line are values of ∆K, which is model value used to detect true K of the three groups (K = 3). b Model-based clustering for each of 279 accessions based on 3113 SNPs used to build the Q matrix. c PCA plots of the first two components of 279 accession. d Maximum likelihood tree of 279 accessions. The red correspond with Q1, green correspond with Q2, and blue correspond with Q3 (color figure online)
Different crops have different LD, which can extend from a few hundred base pairs to kilo-bases (http://www.extension.org). LD generally decays faster in cross-pollinated crops than in self-pollinated crops (Li et al. 2014). The LD in our materials was approximately 480 kb, where r 2 dropped to half its maximum value (r 2 = 0.47) (Fig. 3). It has been reported that there is high LD in the soybean genome (Lam et al. 2010; Hao et al. 2012b; Wen et al. 2014). The LD decay estimates in soybean are slightly higher than those in rice (75–150 kb), and much greater than in maize (1.5–10 kb) (Yan et al. 2009; Huang et al. 2010). This is consistent with an earlier suggestion that LD extends a much longer distance in self-pollinated species than in cross-pollinated species (Zhu et al. 2008).

Genome-wide average LD decay (measured as genotypic r 2) estimated in the population of 279 soybean accessions
Phenotypic analysis
In our study, 279 soybean lines were inoculated with isolates of P.sojae race P7076 to assess partial resistance by measuring lesion length. The cultivar ‘Conrad’ has high levels of partial resistance to P.sojae, whereas ‘Sloan’ is moderately susceptible (Burnham et al. 2003). Conrad and Sloan were used as control materials in the experiment to establish the feasibility of the experimental environment. We evaluated the phenotypic maximum value, minimum value and average values, and performed ANOVA using SAS 9.2. The maximum value was approximately 8–10 times the minimum value. Phenotypic analysis showed that there were significant correlations between materials. There was no significant difference between repeats (Table 2).
Genome-wide association analysis
In this study, the population containing 279 soybean accessions with 59,845 (MAF > 0.05) SNPs was used to carry out association analysis. Both the GLM model without population structure (P) and familial relatedness (K) and the MLM model (Q+K) were used for association mapping. The quantile–quantile (QQ) plot showed that the MLM model was better than GLM for reducing false positive results (Fig. 4b, d). However, the same strong peak (SNP 35123596) was found on chromosome 13 by both models (Fig. 4a, c). This SNP may be significantly associated with partial resistance to P.sojae.

Genome-wide association study of P.sojae in the population. a Manhattan plots of the MLM model for P.sojae association. The horizontal red line indicates the genome-wide significance threshold (−log10(P) > 6). b Quantile–quantile (QQ) plot of MLM model for P.sojae association. c Manhattan plots of GLM for P.sojae assocation. d Quantile–quantile plot of GLM for P.sojae association (color figure online)
Possible candidate gene identification and analysis
Based on the LD distance, we extended the region of interest 500 kb upstream and downstream of the significantly associated SNP position. Within this region, there were three SNPs (SNPs 34976382, 35564109, 35324537) in the exon parts of three genes (Glyma13g32980, Glyma13g33900, Glyma13g33512) (Table 3). Glyma13g32980 is a coat protein I (COPI)-related gene, while Glymal13g33900 and Glymal13g33512 encode a 2OG-FE(II) oxygenase family protein and pentatricopeptide (PPR) repeat-containing protein, respectively. In addition, there are two genes encoding leucine-rich repeat (LRR) domains (Glyma13g33536, Glyma13g33740) (Table 3), which are important in plant responses to various diseases (Avrova et al. 2004; Gao and Bhattacharyya 2008), were near the strong peak. Two other genes (Glyma13g33243, Glyma13g33260), which encoded a Gpi16 subunit and Zn-finger protein, respectively, were also located in this hot region. These genes are probably involved in natural variation in partial resistance to P.sojae. In the next stage, we will research the functions of these genes and examine their association with partial resistance to P.sojae in soybean.
Discussion
PRR is one of the most serious diseases in soybean, and has caused a great loss of soybean production in recent years. Here, we performed a whole-genome association study for soybean PRR disease resistance among 279 accessions. Based on the population structure, PCA and phylogenetic analysis, these 279 accessions were clearly divided into three subgroups (Fig. 2) that corresponded to three different parents. The GLM and MLM models were applied to our GWAS study. Our results were consistent with previous reports that the MLM model is superior to the GLM model (Huang et al. 2010; Yang et al. 2010; Hao et al. 2012a). We found that the MLM model greatly reduced false positives (Fig. 4b, d).
In recent years, genome-wide association mapping has been successfully used to analyze complex trait variation in several crops (Breseghello and Sorrells 2006; Zhao et al. 2007; Huang et al. 2010; Yang et al. 2010; Hao et al. 2012a; Wang et al. 2012). However, the GWAS approach had not been applied to study PRR resistance in soybean. Here, using GWAS to dissect the genetic architecture of PRR resistance in 279 accessions, we found the same strong peak on chromosome 13 using both models (Fig. 4a, c). This SNP may be significantly associated with partial resistance to P.sojae.
Within the 500-kb flanking regions of the significantly associated position, we found seven genes that are probably involved in natural variation in partial resistance to P.sojae. These genes encode a COPI (Glyma13g32980), a 2OG-Fe(II) protein (Glyma13g33900), a PPR protein (Glyma13g33512), LRR domain proteins (Glyma13g33536, Glyma13g33740), a Gpi16 subunit (Glyma13g33243) and a Zn-finger protein (Glyma13g33260) (Table 3). Three SNPs were located in exon regions within these genes (Glyma13g32980, Glyma13g33900, Glyma13g33512) (Table 3). According to previous research, the 2OG-Fe(II) domain of the DMR6 gene, through negative expression regulation of defense response genes, enhances resistance to downy mildew in Arabidopsis (van Damme et al. 2008). Here, we suggest that Glymal13g33900, which contains a 2OG-Fe(II) domain, may be related to soybean P.sojae resistance. In addition, two genes (Glyma13g33536, Glyma13g33740) encoding LRR domains (Table 3), which are characteristic of R genes that are important in plant responses to various diseases (Lupas et al. 1991; Cheng et al. 2010). Also found near the strong peak, the Gpi16 subunit plays a vital role in GPI anchoring in the yeast Saccharomyces cerevisiae, and Zn-finger proteins are important transcription factors in plant defense responses (Fraering et al. 2001; Lee et al. 2006) (Table 3).
Previous studies have refined the map locations of known loci related to SDS and SWM resistance in soybean by GWAS (Wen et al. 2014; Zhao et al. 2015). Our results demonstrate that genome-wide association mapping is also a powerful tool for developing soybean accessions with partial resistance to P.sojae. The new alleles and candidate genes we identified are probably involved in natural variation in partial resistance to P.sojae. In future research, the functions of these genes will be examined in relation to partial resistance to P.sojae in soybean.
References
Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF (2006) A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc 1:2320–2325
Andolfatto P, Davison D, Erezyilmaz D, Hu T, Mast J, Sunayama-Morita T, Stern D (2011) Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res 21:610–617
Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JDG, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631
Avrova AO, Taleb N, Rokka V-M, Heilbronn J, Campbell E, Hein I, Gilroy EM, Cardle L, Bradshaw JE, Stewart HE, Fakim YJ, Loake G, Birch PR (2004) Potato oxysterol binding protein and cathepsin B are rapidly up-regulated in independent defence pathways that distinguish R gene-mediated and field resistances to Phytophthora infestans. Mol Plant Pathol 5:45–56
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Burnham K, Dorrance A, VanToai T, St. Martin S (2003) Quantitative trait loci for partial resistance to in soybean. Crop Sci 43:1610–1617
Cheng X, Jiang H, Zhao Y, Qian Y, Zhu S, Cheng B (2010) A genomic analysis of disease-resistance genes encoding nucleotide binding sites in Sorghum bicolor. Genet Mol Biol 33:292–297
Demirbas A, Rector B, Lohnes DG, Fioritto RJ, Graef GL, Cregan PB, Shoemaker RC, Specht JE (2001) Simple sequence repeat markers linked to the soybean genes for Phytophthora resistance. Crop Sci 41:1220–1227
Dorrance AE, McClure SA, St. Martin SK (2003) Effect of partial resistance on phytophthora stem rot incidence and yield of soybean in Ohio. Plant Dis 87:308–312
Dorrance AE, Mills D, Robertson AE, Draper MA, Giesler L, Tenuta A (2007) Phytophthora root and stem rot of soybean. Plant Health Instr. doi:10.1094/PHI-I-2007-0830-07
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fraering P, Imhof I, Meyer U, Strub JM, van Dorsselaer A, Vionnet C, Conzelmann A (2001) The GPI transamidase complex of Saccharomyces cerevisiae contains Gaa1p, Gpi8p, and Gpi16p. Mol Biol Cell 12:3295–3306
Gao H, Bhattacharyya MK (2008) The soybean-Phytophthora resistance locus Rps1-k encompasses coiled coil-nucleotide binding-leucine rich repeat-like genes and repetitive sequences. BMC Plant Biol 8:29–36
Gao H, Narayanan NN, Ellison L, Bhattacharyya MK (2005) Two classes of highly similar coiled coil-nucleotide binding-leucine rich repeat genes isolated from the Rps1-k locus encode Phytophthora resistance in soybean. Mol Plant Microbe In 18:1035–1045
Gordon SG, Martin SKS, Dorrance AE (2006) Rps8 maps to a resistance gene rich region on soybean molecular linkage group F. Crop Sci 46:168–173
Hao DR, Cheng H, Yin ZT, Cui SY, Zhang D, Wang H, Yu DY (2012a) Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine max) landraces across multiple environments. Theor Appl Genet 124:447–458
Hao DR, Chao MN, Yin ZT, Yu DY (2012b) Genome-wide association analysis detecting significant single nucleotide polymorphisms for chlorophyll and chlorophyll fluorescence parameters in soybean (Glycine max) landraces. Euphytica 186:919–931
He Q, Park YJ (2013) Evaluation of Genetic Structure of Amaranth Accessions from the United States. Weed&Turfgrass Science 2:230–235
Horton MW, Hancock AM, Huang YS, Toomajian C, Atwell S, Auton A, Muliyati NW, Platt A, Seperone FG, Vilhjálmsson BJ, Nordburg M, Borevitz JO, Bergelson J (2012) Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat Genet 44:212–216
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Li W, Lin Z, Buckler ES, Qian Q, Zhang QF, Li J, Han B (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967
Jin L, Lu Y, Xiao P, Sun M, Corke H, Bao JS (2010) Genetic diversity and population structure of a diverse set of rice germplasm for association mapping. Theor Appl Genet 121:475–487
Kump KL, Bradbury PJ, Wisser RJ, Buckler ES, Belcher AR, Oropeza-Rosas MA, Zwonitzer JC, Kresovich S, McMullen MD, Ware D, Balint-Kurti PJ, Holland JB (2011) Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Gene 43:163–168
Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B, Li J, Jian M, Wang J, Shao G, Wang J, Sun SM, Zhang G (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Gene 42:1053–1059
Lee SC, Choi HW, Hwang IS, Choi DS, Hwang BK (2006) Functional roles of the pepper pathogen-induced bZIP transcription factor, CAbZIP1, in enhanced resistance to pathogen infection and environmental stresses. Planta 224:1209–1225
Lee S, Mian MAR, McHale L, Wang H, Wijeratne A, Sneller C, Dorrance A (2013) Novel quantitative trait loci for partial resistance to Phytophthora sojae in soybean PI 398841. Theor Appl Genet 126:1121–1132
Lee S, Rouf Mian MA, Sneller CH, Wang H, Dorrance AE, McHale LK (2014) Joint linkage QTL analyses for partial resistance to Phytophthora sojae in soybean using six nested inbred populations with heterogeneous conditions. Theor Appl Genet 127:429–444
Li RQ, Yu C, Li YR, Lam TW, Yiu SM, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li YH, Zhou GY, Ma JX, Jiang WK, Jin LG, Zhang ZH, Guo Y, Zhang JB, Sui Y, Zheng LT, Zhang SS, Zuo QY, Shi XH, Li YF, Zhang WK, Hu YY, Kong GY, Hong HL, Tan B, Song J, Liu ZX, Wang YS, Ruan H, Yeung CKL, Liu J, Wang HL, Zhang LJ, Guan RX, Wang KJ, Li WB, Chen SY, Chang RZ, Jiang Z, Jackson SA, Li RQ, Qiu LJ (2014) De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol 32:1045
Lin F, Zhao MX, Ping JQ, Johnson A, Zhang B, Abney TS, Hughes TJ, Ma JX (2013) Molecular mapping of two genes conferring resistance to Phytophthora sojae in a soybean landrace PI 567139B. Theor Appl Genet 126:2177–2185
Lupas A, Vandyke M, Stock J (1991) Predicting Coiled Coils from Protein Sequences. Science 252:1162–1164
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Pritchard JK, Stephens M, Donelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Sandhu D, Gao HY, Cianzio S, Bhattacharyya MK (2004) Deletion of a disease resistance nucleotide-binding-site leucine-rich-repeat-like sequence is associated with the loss of the Phytophthora resistance gene Rps4 in soybean. Genetics 168:2157–2167
Sandhu D, Schallock KG, Rivera-velez N, Lundeen P, Cianzio S, Bhattacharyya M (2005) Soybean Phytophthora resistance gene Rps8 maps closely to the Rps3 region. J Hered 96:536–541
Scheet P, Stephens M (2006) A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet 78:629–644
Schmitthenner AF (1985) Problems and Progress in Control of Phytophthora Root-Rot of Soybean. Plant Dis 69:362–368
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Sugimoto T, Yoshida S, Kaga A, Hajika M, Watanabe K, Aino M, Tatsuda K, Yamamoto R, Matoh T, Walker DR, Biggs AR, Ishimoto M (2011) Genetic analysis and identification of DNA markers linked to a novel Phytophthora sojae resistance gene in the Japanese soybean cultivar Waseshiroge. Euphytica 182:133–145
Sun S, Wu XL, Zhao JM, Wang YC, Tang QH, Yu DY, Gai JY, Xing H (2011) Characterization and mapping of RpsYu25, a novel resistance gene to Phytophthora sojae. Plant Breeding 130:139–143
Sun JT, Guo N, Lei J, Li LH, Hu GJ, Xing H (2014a) Association mapping for partial resistance to Phytophthora sojae in soybean (Glycine max (L.) Merr.). J Genet 93:355–363
Sun JT, Li LH, Zhao JM, Huang J, Yan Q, Xing H, Guo N (2014b) Genetic analysis and fine mapping of RpsJS, a novel resistance gene to Phytophthora sojae in soybean [Glycine max (L.) Merr.]. Theor Appl Genet 127:913–919
Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43:159–162
Van Damme M, Huibers RP, Elberse J, Van den Ackerveken G (2008) Arabidopsis DMR6 encodes a putative 2OG-Fe(II) oxygenase that is defense-associated but required for susceptibility to downy mildew. Plant Journal 54:785–793
Wang D (2014) Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean plant and animal genome XXII conference. Plant and Animal Genome
Wang JPM, Lee R, Goos RJ, Helms T (2008) Association mapping of iron deficiency chlorosis loci in soybean (Glycine max L. Merr.) advanced breeding lines. Theor Appl Genet 116:777–787
Wang M, Yan JB, Zhao JR, Song W, Zhang XB, Xiao YN, Zheng YL (2012) Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci 196:125–131
Wen ZX, Tan RJ, Yuan JZ, Bales C, Du WY, Zhang SC, Chilvers MI, Schmidt C, Song QJ, Cregan PB, Wang DC (2014) Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom 15:809
Weng C, Yu K, Anderson T, Poysa V (2001) Mapping genes conferring resistance to Phytophthora root rot of soybean, Rps1a and Rps7. J Hered 92:442–446
Wrather JA, Koenning SR (2006) Estimates of disease effects on soybean yields in the United States 2003 to 2005. J Nematol 38:173
Wu XL, Zhang BQ, Sun S, Zhao JM, Yang F, Guo N, Gai JY, Xing H (2011a) Identification, Genetic Analysis and Mapping of Resistance to Phytophthora sojae of Pm28 in Soybean. Agr Sci China 10:1506–1511
Wu XL, Zhou B, Zhao JM, Guo N, Zhang BQ, Yang F, Chen S, Gai J, Xing H (2011b) Identification of quantitative trait loci for partial resistance to Phytophthora sojae in soybean. Plant Breeding 130:144–149
Yan JB, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J (2009) Genetic Characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS One 4:e8451
Yang XH, Yan JB, Shah T, Warburton ML, Li Q, Li L, Gao YF, Chai YC, Fu ZY, Zhou Y, Xu ST, Bai GH, Meng YJ, Zheng YP, Li JS (2010) Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection. Theor Appl Genet 121:417–431
Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Human Genet 88:76–82
Yang N, Lu YL, Yang XH, Huang J, Zhou Y, Ali F, Wen WW, Liu J, Li JS, Yan J (2014a) Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 Agronomic traits in an enlarged maize association panel. PLoS Genet 10:e1004573
Yang W, Guo Z, Huang C, Duan L, Chen G, Jiang N, Fang W, Feng H, Xie W, Lian X, Wang G, Luo Q, Zhang Q, Liu Q, Xiong L (2014b) Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat Commun 5:5087
Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZXP, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng HC, Liu T, He WM, Li K, Luo RB, Nie XF, Wu HL, Zhao MR, Cao HZ, Zou J, Shan Y, Li SZ, Yang Q, Asan, Ni PX, Tian G, Xu JM, Liu X, Jiang T, Wu RH, Zhou GY, Tang MF, Qin JJ, Wang T, Feng SJ, Li GH, Huasang, Luosang J, Wang W, Chen F, Wang YD, Zheng XG, Li Z, Bianba Z, Yang G, Wang XP, Tang SH, Gao GY, Chen Y, Luo Z, Gusang L, Cao Z, Zhang QH, Ouyang W, Ren XL, Liang HQ, Zheng HS, Huang YB, Li JX, Bolund L, Kristiansen K, Li YR, Zhang Y, Zhang XQ, Li RQ, Li SG, Yang HM, Nielsen R, Wang J, Wang J (2010) Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329:75–78
Zhang JQ, Xia CJ, Wang XM, Duan CX, Sun SL, Wu XF, Zhu ZD (2013) Genetic characterization and fine mapping of the novel Phytophthora resistance gene in a Chinese soybean cultivar. Theor Appl Genet 126:1555–1561
Zhang D, Song HN, Cheng H, Hao DR, Wang H, Kan GZ, Jin HX, Yu DY (2014) The Acid Phosphatase-Encoding Gene GmACP1 Contributes to Soybean Tolerance to Low-Phosphorus Stress. PLoS Genet 10:e1004061
Zhao KY, Aranzana MJ, Kim S, Clare L, Shindo C, Tang CL, Toomajian C, Zheng HG, Dean C, Marjoram P, Nordborg M (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:71–82
Zhao X, Han YP, Li YH, Liu DY, Sun MM, Zhao Y, Lv C, Li DM, Yang ZJ, Huang L, Teng WL, Qiu LJ, Zheng HK, Li WB (2015) Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J 82:245–255
Zhu ZD, Wang HB, Wang XM, Chang RZ, Wu XF (2003) Distribution and virulence diversity of Phytophthora sojae in China. Agri Sci China 3:116–123
Zhu CS, Gore M, Buckler ES, Yu JM (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgments
This study was funded by Modern Agro-industry Technology Research System of China (Grant Number CARS-004-PS10), Genetically modified organisms breeding major projects (Grant Number 2015ZX08004), the key technologies R & D program of Jiangsu (Grant Number BE2013350), Program for Changjiang Scholars and Innovative Research Team in University (Grant Number PCSIRT13073), Jiangsu Collaborative Innovation Center for Modern Crop Production, National Hightech R&D Program of China (Grant Number 2012AA101106), the National Natural Science Foundation of China (Grant Number 31271750).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors, Lihong Li, Na Guo, Jingping Niu, Zili Wang, Xiaoxia Cui, Jutao Sun, Tuanjie Zhao and Han Xing declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by the author.
Additional information
Communicated by B. Yang.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, L., Guo, N., Niu, J. et al. Loci and candidate gene identification for resistance to Phytophthora sojae via association analysis in soybean [Glycine max (L.) Merr.]. Mol Genet Genomics 291, 1095–1103 (2016). https://doi.org/10.1007/s00438-015-1164-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1164-x