
Abstract
Phytophthora root rot (PRR) is among the most important soybean (Glycine max (L.) Merr.) diseases worldwide, and the host displays complex genetic resistance. A genome-wide association study was performed on 337 accessions from the Yangtze-Huai soybean breeding germplasm to identify resistance regions associated with PRR resistance using 60,862 high-quality single nucleotide polymorphisms markers. Twenty-six significant SNP-trait associations were detected on chromosomes 01 using a mixed linear model with the Q matrix and K matrix as covariates. In addition, twenty-six SNPs belonged to three adjacent haplotype blocks according to a linkage disequilibrium blocks analysis, and no previous studies have reported resistance loci in this 441 kb region. The real-time RT-PCR analysis of the possible candidate genes showed that two genes (Glyma01g32800 and Glyma01g32855) are likely involved in PRR resistance. Markers associated with resistance can contribute to marker-assisted selection in breeding programs. Analyses of candidate genes can lay a foundation for exploring the mechanism of P. sojae resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phytophthora root rot is the second highest yield-suppressing disease and results in significant economic losses worldwide (Tyler 2007; Wrather and Koenning 2009). Cultivating resistant soybean cultivars can reduce the influence of PRR. Race-specific resistance and partial resistance are two types of resistant cultivars (Sugimoto et al. 2012).
To the best of our knowledge, regarding race-specific resistance, the following 28 resistant genes have been identified: Rps1a, Rps1b, Rps1c, Rps1d, Rps1 k, Rps2, Rps3a, Rps3b, Rps3c, Rps4, Rps5, Rps6, Rps7, Rps8, Rps9, Rps10, Rps11, RpsUN1, RpsUN2, RpsYu25, RpsYD29, Rps?, RpsJS, RpsHN, Rps12, RpsWY, RpsHC18, and RpsQ (Cheng et al. 2017; Li et al. 2016b, 2017; Niu et al. 2017; Ping et al. 2015; Sahoo et al. 2017; Sugimoto et al. 2011, 2012; Sun et al. 2011, 2014; Wu et al. 2011; Zhang et al. 2013a, b; Zhong et al. 2017). Regarding partial resistance, the QTLs for P. sojae include the following: Phytoph1-1, 1-2, 2-1, 2-2, 3-1, 3-2, 4-1, 4-2, 5-1, 5-2, 5-3, 6-1, 6-2, 6-3, 6-4, 6-5, 6-6, 6-7, 6-8, 7-1, 8-1, 8-2, 8-3, 8-4, 9-1, 9-2, 9-3, 9-4, 9-5, 9-6, 10-1, 10-2, 10-3, 11-2, 11-19, 11-20, 11-21, 11-22, 12-1, 12-2, 13-1, 13-2, 13-3, 13-4, 13-5, 14-1, 14-2, 14-3, 14-5, 14-6, 14-7, 14-8, 14-9, 14-10, 15-1, 15-2, 15-3, 15-4, 15-5 etc. (Data from: https://www.soybase.org/search/qtllist_by_symbol.php). Complete resistance or partial resistance, under high disease pressure and evolution of races, may be ineffective (Dorrance et al. 2003; Schmitthenner 1985). Therefore, new resistance loci for Phytophthora sojae are necessary in the long run.
Association mapping is a method used for studying the genetics of simple and complex traits in plants (Gupta et al. 2005). Recently, this method has been used to analyze PRR. The partial resistance to P. sojae in 279 accessions from the Yangtze-Huai soybean breeding germplasm was studied, and a single locus on chromosome 13 was identified (Li et al. 2016a). The partial resistance to P. sojae in 800 lines from the Republic of Korea were studied, and two QTLs on chromosome 03 and three QTLs on chromosome 19 were identified (Rhiannon et al. 2016). Association mapping of the quality resistance of 224 germplasm accessions from the Chinese soybean mini core collection to 7 P. sojae revealed 14 marker-trait associations responsible for Phytophthora resistance (Huang et al. 2016).
Here, we used 337 accessions from the Yangtze-Huai soybean breeding germplasm to study resistance to P. sojae, and approximately 160 of the 337 accessions have been studied for partial resistance (Li et al. 2016a). A genome-wide association study (GWAS) was performed to identify significant SNP loci associated with PRR resistance using 60,862 SNP, and the possible candidate genes were validated by qRT-PCR.
Materials and method
Plant materials
In total, 337 soybean accessions from the Yangtze-Huai soybean breeding germplasm population were obtained from the National Center for Soybean Improvement, Nanjing city, Jiangsu Province, China.
Resistance evaluation
Phytophthora sojae HeN08-35 (virulence formula is 3a, 3c, 4, 5, 6 and 7) was provided by Professor Yuanchao Wang of Nanjing Agricultural University, maintained on V8 juice agar medium (10% V8 vegetable juice, 0.02% CaCO3 and 1.0% Bacto-agar) (Niu et al. 2017), and used to evaluate 337 accessions.
A modified hypocotyl inoculation technique was utilized for the disease evaluation in this experiment (Niu et al. 2017). All materials were planted in plastic pots containing vermiculite. The mycelia from 7-day-old seedlings were maintained on V8 juice agar and subsequently inoculated onto wounded hypocotyls. After the inoculation, the seedlings were placed in a high humidity mist chamber for 48 h and subsequently transferred to a greenhouse at 25 °C with a 14-h light/10-h dark photoperiod for 5 days. Then, 337 accession reactions were evaluated 5 days post inoculation (DPI) and recorded as the percentage of dead seedlings. Each accession had 30 scored plants. The standard criterion for each accession was as follows: if the percentage of dead seedlings was > 70%, the accession was considered susceptible (S); if the percentage of dead seedlings was < 30%, the accession was considered resistant (R); and if the percentage of dead seedlings was between 30 and 70%, the accession was considered intermediate (I). The intermediate reactions were considered resistant reactions (Huang et al. 2016).
SNP calling and SNP data analysis
Restriction-site-association DNA sequencing (RAD-seq) was used in this study and performed at the Beijing Genomics Institution, Shenzhen, China. The genomic DNA of the 337 accessions was extracted from young leaves using the CTAB method (Murray and Thompson 1980). All DNA fragments between 400 bp and 600 bp were obtained by TaqI digestion. The fragments were sequenced using an Illumina HiSeq 2000 instrument with a paired-end reads length of 90 bp and 6 bp of the barcode (Li et al. 2016a). All sequence reads were aligned against the reference Glyma.Wm82.a1.v1.1 (Schmutz et al. 2010) using SOAP2 software (Li et al. 2009), and SNP calling was performed by RealSFS software (Yi et al. 2010). The SNPs of the 337 accessions were filtered at a rate of missing and heterozygous allele calls ≤ 30%. Missing alleles were imputed with fastPHASE software (Scheet and Stephens 2006).
In total, 87,308 SNPs, including 60,862 SNPs with minor allele frequencies (MAF) > 5%, were studied in the association analyses. The MAF and polymorphism information content (PIC) were analyzed with an in-house R script (Li et al. 2016a).
Population analysis
For the population structure analysis, 3929 SNPs were screened by PlinkV1.07 (Purcell et al. 2007) (threshold r2 value was 0.3). Subsequently, the population structure and Q matrix were estimated using the software program Structure 2.2 (Pritchard et al. 2000), and the parameter settings are the same as those in the article (Li et al. 2016a). Kinship was calculated by using TASSEL 5.0. The degree of linkage disequilibrium (LD) between each pair of SNPs was estimated with the correlation coefficient r2 by PopLDdecay (https://github.com/BGI-shenzhen/PopLDdecay).
Association mapping
The association analysis was performed using TASSEL5.0 software (Bradbury et al. 2007). Two statistical models were considered. The first model was a general linear model (GLM) with the Q matrix as a covariate. The second model was a mixed linear model (MLM) with the Q matrix and K matrix as covariates. The appropriate model for this study was chosen by comparing the two models. The Bonferroni threshold P < 1/60,862 (− log10P > 4.78) was used as the threshold to identify significant SNP associations. Haploview 4.2 software (Barrett 2009) and the four-gamete method were used to calculate the haplotype blocks on the resistance loci.
Expression analysis of candidate genes
Two resistant and unseparated lines P240 (R) and P548 (R) seedlings were cultivated for 7 days and subsequently inoculated with isolate HeN08-35. Approximately 1-cm samples of the treated hypocotyl tissues were collected at five time points. The total RNA was extracted from the plants using an RNA Simple Total RNA Kit (TIANGEN, China). cDNA was synthesized using a Prime ScriptTM RT Reagent Kit (TaKaRa, Japan) following a standard protocol. The CDS sequences of the candidate genes were obtained from Phytozome v12.1 (Niu et al. 2017). The primers for qRT-PCR were designed using Primer Premier 5.0. In addition, the housekeeping gene GmCons4 was used as a control. For each sample, three technological replications were conducted. The primers are shown in Table 1, and qRT-PCR was conducted using a Light Cycler 480 instrument.
Results
Genetic diversity
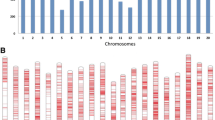
In total, 87,308 SNPs were used to analyze the genetic diversity in the 337 soybean accessions. The SNPs were unevenly distributed on 20 chromosomes, and the distribution ratio ranged from 2.47 to 7.83% (Fig. 1a). The MAF of 69.71% of the SNPs was greater than 0.05, and these SNPs were used in the association mapping (Fig. 1b). The PIC of 87,308 SNPs was averaged to 0.19, and the SNP ratio within the maximum PIC range of 0.35–0.37 was 14.49% (Fig. 1c).

Information analysis of 87,308 SNPs. a Distribution of SNPs on Chromosome; b minor allele frequency analysis; c polymorphic information content analysis
Population structure
The population structure was analyzed by Structure 2.2 software using 3929 SNPs. The deltaK value was high when k was equal to 3, while the mean LnP (K) value increased slowly as the K value increased (Fig. 2a). Therefore, deltaK acted as a grouping parameter, and this population was divided into three main subgroups. The 337 accessions were obtained from the Yangtze-Huai soybean breeding germplasm population, of these accessions, 100 accessions belonged to subgroup 1, 120 accessions belonged to subgroup 2, 95 accessions belonged to subgroup 3 and 22 accessions belonged to the mixed subgroup (Fig. 2b). The corresponding Q matrix was used in the subsequent genome-wide association analysis.

Population structure analysis in soybean. a True K was analyzed in the population. The red line indicates the change in ΔK, and the blue line indicates the change in LnP(K); b population structure analysis. Subgroup 1 is shown in red, subgroup 2 is shown in green, and subgroup 3 is shown in blue. (Color figure online)
Phenotypic data and association mapping
In this study, the resistance of 337 accessions was evaluated by hypocotyl inoculation with P. sojae HeN08-35. The results showed that 294 accessions were resistant, and 43 accessions were susceptible.
The combination of phenotype and 60,862 SNPs (MAF > 0.05) of 337 accessions was used to perform the association analysis. The quantile–quantile (QQ) plot of MLM and GLM showed that the MLM method was more appropriate for this study and could reduce the number of false positive results (Fig. 3a). Therefore, the MLM model was used for the association analysis.

Genome-wide association study of P. sojae in the population. a Quantile–Quantile plots were estimated by − log10P using two models; b Manhattan plots of MLM for resistance to P. sojae
In total, 26 association peaks were identified on Chr01 (Fig. 3b, Table 2). The phenotypic variation explained by each peak ranged from 6.14% to 11.18%, and the highest peak was Gm01_44414453.
Candidate gene prediction
The LD in our population was approximately 330 kb when r2 decreased to 0.506, which is halfway between the maximum and minimum (Fig. 4). Based on the LD distance, the position of peak Gm01_44414453 extended 330 kb upstream and downstream. Based on the haplotype block analysis of all SNPs in the 660 kb region using Haploview 4.2 software, 26 SNPs were associated with resistance loci located in three adjacent haplotype blocks in a region of approximately 441 kb (Fig. 5). This region acted as the resistance region to P. sojae HeN08-35 in our study.

Linkage disequilibrium decay of the whole genome

Haplotype blocks were divided by linkage disequilibrium. White represents an r2 value of zero, black represents an r2 value of 1; the red line indicates the location of the associated SNPs, and blue represents the locations of the genes; the gene annotations are shown on the right. (Color figure online)
In total, thirteen genes in the 441 kb were annotated using Glyma 1.0. Among these genes, Glyma01g32800 encodes serine/threonine protein kinase (STK), and Glyma01g32855 encodes the leucine-rich repeat protein kinase family proteins, which may be involved in plant disease resistance. These genes were predicted as possible candidate genes.
To confirm whether Glyma01g32800 and Glyma01g32855 were induced under the treatment of P. sojae HeN08-35, the expression patterns of these two genes were examined using a qRT-PCR analysis in P240 (R) and P548 (R). As shown in Fig. 6, compared with the control (0 h), the expression of Glyma01g32800 was up-regulated 48 h after treatment in the resistant line P240 and 24, 48, and 72 h after treatment in the resistant line P548, and the expression of Glyma01g32855 was down-regulated at four time points in P240 and P548. Thus, two genes were induced by P. sojae HeN08-35, and the expression trend of the two genes in the two accessions was similar. Thus, Glyma01g32800 and Glyma01g32855 were considered potential candidate genes.

Relative expression of Glyma01g32800 and Glyma01g32855 was induced by P. sojae HeN08-35. Seven-day-old soybean seedlings were inoculated with the isolate HeN08-35. The sampling times were 0, 24, 36, 48, and 72 h post inoculation (hpi)
Discussion
In this study, association mapping was performed using the MLM method. In total, 26 SNPs associated with resistance to P. sojae HeN08-35 were identified and divided into three approximately 441 kb adjacent haplotype blocks. This region acted as a resistance-associated region.
The method used for the resistance evaluation in the 337 accessions was based on previous research. The accessions with a percentage of dead seedlings > 70% were considered susceptible and those with a percentage ≤ 70% were considered resistant (Huang et al. 2016). We also directly analyzes the resistance SNPs using the percentage of dead seedlings as a resistance evaluation index by GWAS. The SNP with the highest phenotypic variation was Gm01_44414453 (data not shown). This finding was consistent with the findings of this study. The difference of other mapping results may be caused by the method used for the resistance evaluation.
Previous studies have shown that the markers Satt221 (nucleotide region 25819853-25819924) and BARC-044479-08708 (nucleotide region 50867367-50867390) on Chr01 are associated with partial resistance to P. sojae (Lee et al. 2013; Sun et al. 2014). The distances of the nucleotide positions between Gm01_44414438 and Satt221 and between Gm01_44550999 and BARC-044479-08708 were 18.5 Mb and 6.3 Mb, respectively. Therefore, we speculate that the 26 SNPs associated with resistance may represent new loci and require further verification.
The resistance of other soybean resources to P. sojae HeN08-35 has also been studied and reported. The resistance loci BARC-014467-01559 on Chr16 and BARC-013645-01207 on Chr20 have been identified in the Chinese soybean mini core collection (Huang et al. 2016). The complete gene RpsHN was identified in the resistance line Meng8206 (Niu et al. 2017). Although we used the same P. sojae HeN08-35, the resistance loci were different. This discrepancy may be caused by the different genetic backgrounds of the soybean resources. In addition, approximately 160 of the 337 accessions this study have been studied for partial resistance to P.sojae 7076 (Li et al. 2016a), but the resistance loci were different. These results may be caused by the different genetic backgrounds of the remaining soybean resources and different pathogenicity of P. sojae.
In wheat, the receptor-kinase-like protein Stb6 was found to control resistance to the fungal pathogen Zymoseptoria tritici and showed gene responsible for gene resistance (Saintenac et al. 2018). The Stpk-V gene is a serine/threonine kinase gene located on the Pm21 locus that confers resistance to powdery mildew (Cao et al. 2011). Thus, this protein kinase could participate in resistance. In soybean, two STK-type genes were cloned in the Rps10 fine mapping region (Zhang et al. 2013a). The expression of the serine/threonine kinase gene Glyma.03g04340 was induced by P. sojae HeN08-35 (Niu et al. 2017). Therefore, in the present study, two protein kinase genes, i.e., Glyma01g32800 and Glyma01g32855, were considered potential candidate genes, and their expression was induced by P. sojae HeN08-35.
References
Barrett JC (2009) Haploview: visualization and analysis of SNP genotype data. Cold Spring Harb Protoc 2009:pdb.ip71
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cao A, Xing L, Wang X, Yang X, Wang W, Sun Y, Qian C, Ni J, Chen Y, Liu D (2011) Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci 108:7727
Cheng Y, Ma Q, Ren H, Xia Q, Song E, Tan Z, Li S, Zhang G, Nian H (2017) Fine mapping of a Phytophthora-resistance gene RpsWY in soybean (Glycine max L.) by high-throughput genome-wide sequencing. Theor Appl Genet 130:1041–1051
Dorrance AE, Mcclure SA, Martin SK (2003) Effect of partial resistance on phytophthora stem rot incidence and yield of soybean in Ohio. Plant Dis 87:308–312
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Huang J, Guo N, Li Y, Sun J, Hu G, Zhang H, Li Y, Zhang X, Zhao J, Xing H (2016) Phenotypic evaluation and genetic dissection of resistance to Phytophthora sojae in the Chinese soybean mini core collection. BMC Genet 17:85
Lee S, Mian MAR, Mchale LK, Wang H, Wijeratne AJ, Sneller CH, Dorrance AE (2013) Novel quantitative trait loci for partial resistance to Phytophthora sojae in soybean PI 398841. Theor Appl Genet 126:1121–1132
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li L, Guo N, Niu J, Wang Z, Cui X, Sun J, Zhao T, Xing H (2016a) Loci and candidate gene identification for resistance to Phytophthora sojae via association analysis in soybean [Glycine max (L.) Merr]. Mol Genet Genomics 291:1095–1103
Li L, Lin F, Wang W, Ping J, Fitzgerald JC, Zhao M, Li S, Sun L, Cai C, Ma J (2016b) Fine mapping and candidate gene analysis of two loci conferring resistance to Phytophthora sojae in soybean. Theor Appl Genet 129:2379–2386
Li Y, Sun S, Zhong C, Wang X, Wu X, Zhu Z (2017) Genetic mapping and development of co-segregating markers of RpsQ, which provides resistance to Phytophthora sojae in soybean. Theor Appl Genet 130:1–11
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Niu J, Guo N, Sun J, Li L, Cao Y, Li S, Huang J, Zhao J, Zhao T, Xing H (2017) Fine mapping of a resistance gene RpsHN that controls Phytophthora sojae using recombinant inbred lines and secondary populations. Front Plant Sci 8:e3376
Ping J, Fitzgerald JC, Zhang C, Lin F, Bai Y, Wang D, Aggarwal R, Rehman M, Crasta O, Ma J (2015) Identification and molecular mapping of Rps11, a novel gene conferring resistance to Phytophthora sojae in soybean. Theor Appl Genet 129:445–451
Pritchard JK, Stephens M, Donelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purcell S, Neale B, Toddbrown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, Bakker PI, Daly MJ (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Rhiannon S, William R, Song Q, Perry C, Dorrance AE, Mchale LK (2016) Genome-wide association mapping of partial resistance to Phytophthora sojae in soybean plant introductions from the Republic of Korea. BMC Genom 17:607
Sahoo DK, Abeysekara NS, Cianzio SR, Robertson AE, Bhattacharyya MK (2017) A novel Phytophthora sojae resistance Rps12 gene mapped to a genomic region that contains several Rps genes. PLoS ONE 12:e0169950
Saintenac C, Lee WS, Cambon F, Rudd JJ, King RC, Marande W, Powers SJ, Bergès H, Phillips AL, Uauy C (2018) Wheat receptor-kinase-like protein Stb6 controls gene-for-gene resistance to fungal pathogen Zymoseptoria tritici. Nature 50:368–374
Scheet P, Stephens M (2006) A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet 78:629–644
Schmitthenner AF (1985) Problems and progress in control of Phytophthora root rot of soybean. Plant Dis 69:362–368
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Sugimoto T, Yoshida S, Kaga A, Hajika M, Watanabe K, Aino M, Tatsuda K, Yamamoto R, Matoh T, Walker DR (2011) Genetic analysis and identification of DNA markers linked to a novel Phytophthora sojae resistance gene in the Japanese soybean cultivar Waseshiroge. Euphytica 182:133–145
Sugimoto T, Masayasu K, Shinya Y, Isao M, Tamotsu K, Akito K, Makita H, Ryo Y, Kazuhiko W, Masataka A (2012) Pathogenic diversity of Phytophthora sojae and breeding strategies to develop Phytophthora-resistant soybeans. Breed Sci 61:511–522
Sun S, Wu XL, Zhao JM, Wang YC, Tang QH, Yu DY, Gai JY, Xing H (2011) Characterization and mapping of RpsYu25, a novel resistance gene to Phytophthora sojae. Plant Breeding 130:139–143
Sun J, Guo NA, Lei J, Lihong LI, Hu GJ, Xing H (2014) Association mapping for partial resistance to Phytophthora sojae in soybean (Glycine max (L.) Merr.). J Genet 93:355–363
Tyler BM (2007) Phytophthora sojae: root rot pathogen of soybean and model oomycete. Mol Plant Pathol 8:1–8
Wrather A, Koenning S (2009) Effects of diseases on soybean yields in the United States 1996 to 2007. Plant Health Prog. https://doi.org/10.1094/PHP-2009-0401-01-RS
Wu X, Zhou B, Zhao J, Guo N, Zhang B, Yang F, Chen S, Gai J, Xing H (2011) Identification of quantitative trait loci for partial resistance to Phytophthora sojae in soybean. Plant Breeding 130:144–149
Yi X, Liang Y, Huertasanchez E, Jin X, Zha XP, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS (2010) Sequencing of fifty human exomes reveals adaptation to high altitude. Science 329:75–78
Zhang J, Xia C, Duan C, Sun S, Wang X, Wu X, Zhu Z (2013a) Identification and candidate gene analysis of a novel phytophthora resistance gene Rps10 in a Chinese soybean cultivar. PLoS ONE 8:e69799
Zhang J, Xia C, Wang X, Duan C, Sun S, Wu X, Zhu Z (2013b) Genetic characterization and fine mapping of the novel Phytophthora resistance gene in a Chinese soybean cultivar. Theor Appl Genet 126:1555–1561
Zhong C, Sun S, Li Y, Duan C, Zhu Z (2017) Next-generation sequencing to identify candidate genes and develop diagnostic markers for a novel Phytophthora resistance gene, RpsHC18, in soybean. Theor Appl Genet 131:525–538
Acknowledgements
We thank Prof. Yuanchao Wang (Nanjing Agricultural University) for providing the isolates of P. sojae. This work was supported by National Key R&D Program of China (2017YFD0101500), Modern Agro-industry Technology Research System of China (CARS-04-PS10), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT_17R55), Jiangsu Collaborative Innovation Center for Modern Crop Production, The Natural Science Foundation of China (31571691) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX17_0560).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Niu, J., Guo, N., Zhang, Z. et al. Genome-wide SNP-based association mapping of resistance to Phytophthora sojae in soybean (Glycine max (L.) Merr.). Euphytica 214, 187 (2018). https://doi.org/10.1007/s10681-018-2262-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-018-2262-8