
Abstract
Detection of Blastocystis is routinely performed by microscopy, culture, and formyl-ether (ethyl acetate) concentration technique (FECT). Yet, these methods require special skilled personnel, are time consuming, and often involve processing that may cause misdiagnosis. The aim of this work is to demonstrate the usefulness of a newly introduced ELISA test for the detection of Blastocystis antigens in stool samples (CoproELISATM Blastocystis, Savyon Diagnostics) as a proper alternative to currently used methods, especially microscopy. A cohort of 179 fresh/frozen clinical stool samples was tested by the ELISA test, and results were compared to consensus methods comprised of microscopic examination of Lugol’s iodine staining, culture, and immunofluorescence assay (IFA). The new ELISA test was able to detect fewer than 103 cells, recognized subtypes 1, 2, 3, and 5 (comprising >95 % of human Blastocystis infections), and exhibited similar reactivity when comparing formalin-preserved samples to fresh/frozen samples. The test demonstrated 92 % sensitivity, 87 % specificity, and 89 % accuracy when culture, and IFA or microscopy consensus results were taken as reference. When the consensus was comprised of culture and IFA, the test demonstrated sensitivity, specificity, and accuracy of 82, 86, and 84 %, respectively. In contrast, the sensitivity of Lugol staining microscopy was only 18 %. This work presents a unique ELISA test that provides an alternative to the use of microscopy, currently most widely used method. The test enables high-throughput screening and diagnosis of Blastocystis, adaptation to automatic procedures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Blastocystis is an enteric protozoan parasite highly prevalent in humans and animals (Abe 2004; Alfellani et al. 2013; Ash and Orihel 1987). Infection is associated with non-specific symptoms (i.e., diarrhea, abdominal pain, weight loss, constipation, anal itching, excess gas) and was found to be associated with irritable bowel disease (Qadri et al. 1989; Krüger et al. 1994; Pasqui et al. 2004; Biedermann et al. 2002; Dogruman-Al et al. 2009). This wide array of non-specific symptoms has confounded the understanding of the potential pathogenicity of Blastocystis species. As a result, many of these infections are undiagnosed. Most common approaches to the detection of Blastocystis include direct smear, microscopy; formyl-ether (ethyl acetate) concentration technique (FECT); and xenic in vitro culture (XIVC). Yet, these methods are time and labor intensive and require skilled personnel. Blastocystis has several morphological forms (vacuolar, cyst, amoeboid, granular, multivacuolar, and avacuolar) as well as numerous morphological features associated with the parasitic growth cycle. Consequently, microscopy is difficult, resulting in lower sensitivity, particularly when parasites are found in low numbers (Stenzel and Boreham 1996). FECT destroys some of the forms during stool processing, thus reduces recovery of parasites and therefore may not be efficient (O’Gorman et al. 1993). Culture requires 2–3 days for diagnosis and may allow preferential growth of specific strains while eliminating others (Parkar et al. 2007). This is most important in view of the possibility of having mixed subtype infections (Stensvold et al. 2008), in which the pathogenic strains may be eliminated. Therefore, culture has a limited efficiency as a screening tool. Nevertheless, to date, microscopy and culture are considered as the “gold standard” methods for the detection of Blastocystis. PCR for diagnosis of Blastocystis was introduced in 2006 (Stensvold et al. 2006). Some publications refer to DNA isolation and PCR detection of Blastocystis directly from stool samples (Stensvold et al. 2006, 2007a; Eida and Eida 2008; Roberts et al. 2011; Forsell et al. 2012). The analysis of DNA extracted directly from stool samples is considered to be highly sensitive, providing the means for genotyping and subtyping. Nevertheless, it is aggravated by the presence of stool-associated PCR inhibitors and protozoan nucleases. In addition to apoptosis-related DNA fragmentation and the substantial genetic heterogeneity, the task of finding suitable loci that can be used for detection and differentiation is challenging (Chou and Tai 1996; Nasirudeen and Tan 2004). Also, Stensvold et al. (2007b) showed that conventional PCR was not significantly more sensitive than short-term XIVC and permanent staining. Diagnostic RT-PCR assays have been recently introduced, however, so far have not been used for screening (Stensvold 2013). While molecular DNA genotyping in stool has been described for many intestinal protozoa (Limor et al. 2002; Verweij et al. 2004; Peek et al. 2004), in the case of Blastocystis, PCR was used principally for subtyping of cultured isolates (Scicluna et al. 2006; Yoshikawa et al. 2003, 2004), phylogenetic studies, and ribotyping (Scicluna et al. 2006; Noël et al. 2003; Abe 2004; Rivera and Tan 2005; Rivera 2008). The genus Blastocystis includes highly diversified isolates, and it was recently proposed that any of the isolates from mammals and birds may be assigned to one of 17 subtypes (Alfellani et al. 2013). It was shown that variations of commonly found Blastocystis subtypes of human infections further confound and limit detection efficacy (Stensvold et al. 2007b; Vennila et al. 1999). Genotyping and subtyping of isolates may be used to differentiate between symptomatic and asymptomatic infections. However, isolates that have been associated with symptomatic infections in humans have also been found in asymptomatic carriers, making subtyping unsuitable for the determination of pathogenicity.
The aim of this work was to evaluate the analytical and clinical utility of a newly introduced ELISA-based test (CoproELISATM Blastocystis, Savyon Diagnostics, Israel) for the detection of Blastocystis antigens in fresh/frozen and preserved stool samples. This ELISA is intended to be used for the detection of Blastocystis antigens in specimens collected from patients with gastrointestinal (GI) symptoms and is suggested to be utilized as a proper alternative for the diagnosis and screening of Blastocystis infections.
Materials and methods
Clinical samples
Fecal specimens were obtained from routine fecal examinations in the microbiology laboratory of Numune Education and Research Hospital and Gazi University Hospital (both from Ankara, Turkey) from September 1 to October 30, 2012 and from Clalit Health Services Microbiology Laboratory, Nesher Regional Laboratory (Nesher, Israel). Samples were stored at −20 °C and were preserved with or without Formalin/SAF. The clinical samples were used under the authorization of the Gazi University Clinical Research Ethics Committee in Turkey and the Clalit Health Services Ethics Committee in Israel, according to the origin of the samples.
Culture
In Turkey, pea-sized fecal samples were cultured in 8-ml culture medium (Ringer’s solution containing 10 % horse serum and 0.05 % asparagine) (Dogruman-Al et al. 2009, 2010; Roberts et al. 2013) at 37 °C for 3 days followed by standard microscopic analysis. Negative samples were reanalyzed by microscopy at 4-, 5-, and 7-day culture. A sample was designated as negative if Blastocystis spp. growth was not obtained within 7 days. In Israel, all fresh fecal samples were inoculated into two culture systems upon receipt. A pea-sized stool sample was put into two different diphasic systems. One was an in-house growth medium consisting of a modified Boeck and Drbohlav’s diphasic growth media consisted of egg base with an overlay of 3-ml Ringer-Lockes solution (Ash and Orihel 1987). The other medium was HY ENTAMOEBA KIT, purchased from Hy Laboratories Ltd. (Rehovot, Israel). Tubes were incubated at 37 °C in anaerobic conditions, and a drop of sediment was examined every 2 days by microscopy.
Microscopy
All stool samples were examined by direct wet smear, prepared by mixing a small amount of stool (about 2 mg) with a drop of 0.85 % NaCl and a drop of Lugol’s iodine on the same slide. These mixtures were covered by a 22 × 22-mm coverslip and were screened by microscopy (Forsell et al. 2012).
Molecular genotyping
Extraction of genomic DNA from clinical isolates was carried out by QIAamp DNA Mini Kit (Qiagen, Germany). (1) Amplification: A set of primers was used for PCR amplification and sequencing. These primers consisted of the forward primer BhRDr (GAGCTTTTTAACTGCAACAACG) and the reverse primer RD5 (ATCTGGTTGATCCTGCCAGT) (Scicluna et al. 2006), synthesized by Integrated DNA Technologies Ltd. (Israel). The primers were used in a standard PCR reaction using a FastTaq DNA polymerase (Roche Ltd, Germany) comprising denaturation at 95 °C for 3 min, 30 cycles of 1 min each at 94, 55, and 72 °C for 60 s, followed by a final extension step at 72 °C for 2 min. Amplicons of 619 bp long were observed after electrophoresis on a 1 % agarose gels. (2) Sequencing: DNA sequence analysis was performed on all PCR-positive samples. The PCR products were purified using QIAquick™ PCR Purification Kit (Qiagen). The PCR products were then sequenced in both directions by Hy Laboratories Ltd. (Rehovot, Israel). The SSU rDNA sequences were then compared with those available in GenBank using the BLASTN program run on the National Center for Biotechnology Information server (http://www.ncbi.nlm.nih.gov/BLAST). Following PCR and sequencing, clinical Blastocystis isolates were identified and classified according to Stensvold et al. (2007c).
Immunofluorescence assay (IFA)
Fecal specimens were evaluated using Blasto-Fluor (Antibodies Inc., Davis, CA, USA), a commercially available FITC-tagged antibody stain specific for Blastocystis prepared from whole cell Blastocystis antigen (subtype 3). Staining was performed by combining 200 μl of fecal sample, 200 μl of phosphate buffered saline (PBS), and 4 μl of stain, followed by incubation for 60 min at 37 °C. The sample was viewed under a fluorescence microscope using 495- and 515-nm excitation and emission filters, respectively.
CoproELISATM Blastocystis ELISA
All procedures were according to the manufacturer instructions (http://savyondx.com/_Uploads/dbsAttachedFiles/CoproELISA_Blastocystis_V4.pdf). The CoproELISATM Blastocystis ELISA test is based on a mixture of rabbit polyclonal antibodies raised against a pool of Blastocystis subtypes (1, 2, 3, and 5).
Analytical sensitivity assay
A titration (102–5 × 104 cells) was performed on subtype-defined clinical isolates using the kit’s stool diluent. The negative control included all the test components excluding cells. The specific limit of detection (LoD) for each isolate was quantified separately.
Preservative compatibility assay
The suitability of the assay to be used with fresh/frozen or preserved specimens was determined using a cohort of 23 positive fecal samples (fresh/frozen or preserved in 10 % formalin) for 6 days.
Clinical performance
A cohort of 179 stool samples from symptomatic patients were collected and tested by the ELISA. Microscopic examination of Lugol’s iodine staining, culture, and IFA were used as a consensus reference. In a cohort of 89 stool samples, the performances of the ELISA and standard microscopic examination of Lugol’s iodine staining were assessed separately, each against a consensus reference composed of culture and IFA.
Cross-reactivity
Cross-reactivity with other GI pathogens was determined using stool specimens which have been confirmed as positive by routine ova and parasite (O&P) analysis, culture, or enzyme immunoassays (EIA). The organisms tested included Entamoeba strains, Endolimax nana, Dientamoeba fragilis, Cryptosporidium spp., Giardia lamblia, Clostridium difficile, Helicobacter pylori, Salmonella spp., Shigella spp., and Campylobacter jejuni.
Results
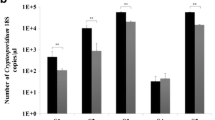
As previously reported, infections in humans are caused by certain Blastocystis subtypes which are more prevalent than others. Figure 1a demonstrates the analytical sensitivity of subtypes commonly found in human blastocystosis. The calculated LoDs were 230, 380, 250, and 1100 cells for subtypes 1, 2, 3, and 5, respectively.

Analytical performance of CoproELISA™ Blastocystis. a Analytical sensitivity of the test for Blastocystis subtypes 1 (white up-pointing triangle), 2 (black circle), 3 (black square), and 5 (black up-pointing triangle) representing limits of detection (LoD) of 230, 380, 250, and 1100 cells, respectively. b Linear correlation between absorbance values (OD, A 450/620) achieved with fresh vs. formalin-fixed samples (R 2 > 0.96). Similar results were obtained with SAF-fixed samples (data not shown)
The compatibility of the ELISA test with common preservation media and procedures was assessed (Fig. 1b). High correlation (R 2 = 0.96) was found in the readouts of fresh/frozen vs. 10 % formalin-fixed samples, thus indicating that the test is compatible with both fresh/frozen and formalin-fixed samples. In a different set of experiments (data not shown), similar results were obtained with SAF preservation. Furthermore, neither formalin nor SAF had influence on the signal of negative samples, i.e., no increase in background signals was observed.
The clinical performance of the ELISA test in a cohort of 179 stool samples from symptomatic patients is presented in Table 1. The ELISA exhibited 92 % sensitivity, 87 % specificity, and 89 % accuracy as compared to the consensus techniques. Tables 2 and 3 present the performances of the ELISA and the standard microscopic examination of Lugol’s iodine staining in a cohort of 89 stool samples, respectively, both assessed against a consensus reference composed of culture and IFA. While the ELISA demonstrated sensitivity, specificity, and accuracy levels of 82, 86, and 84 %, respectively, the commonly used microscopic analysis demonstrated sensitivity, specificity, and accuracy levels of 18, 100, and 64 %, respectively.
Finally, the ELISA was examined for cross-reactivity with relevant GI parasites and bacteria as listed above and had shown no reactivity with any of the tested pathogens.
Discussion
Blastocystosis has been associated with a myriad of non-specific and confounding symptoms, also exhibited by infection with other GI pathogens. The shared symptoms together with the current limitations of existing diagnostic techniques make the management of Blastocystis challenging and provide ambiguous interpretation in regard to its pathogenicity.
The purpose of this work was to evaluate the suitability of a newly marketed ELISA (and currently the only commercial test in this field) to be adopted in clinical laboratories for routine diagnosis and screening of Blastocystis in stool specimens. In this regard, an efficient screening assay is expected to (i) exhibit high performance parameters, (ii) be easy to perform, (iii) be compatible with fresh/frozen and fixed specimens, (iv) be relatively rapid (offering same day results), and (v) enable the use of automatic processing.
Assessment of the analytical sensitivity of the test for common human-associated Blastocystis subtypes revealed LoDs in the range of 200–1100 cells/well. The theoretical analytical sensitivity of microscopic examination is 1 cell/HPF (high-power field), which is equivalent to 1000 cells/well in ELISA. This lower LoD of the ELISA as compared to microscopy elucidates the favorable clinical performance of the ELISA, when the performances of both methods were examined against culture and IFA. Subtype variations of commonly found Blastocystis subtypes in human infections is considered to further confound and limit detection efficacy. Subtypes 1, 2, 3, and 5 were reported to account for over 95 % of human blastocystosis cases. The observation that these Blastocystis subtypes are well detected increases the ELISA utility for diagnosis and screening.
Storage conditions may influence the detection of Blastocystis spp. These conditions affect the ability of Blastocystis to maintain its morphology during laboratory processing, are important for the detection of co-infections, and permit the characterization of infection intensity (Stensvold et al. 2007b; Stensvold 2013). Formalin/SAF fixation is a routine preservation method which is used both for shipment of samples as well as for preservation of samples by the laboratory itself for batching or confirmation. The compatibility of the ELISA test with routine procedures was shown by its capability to provide reliable detection of Blastocystis spp. in fresh/frozen specimens as well as formalin/SAF-preserved stool samples. This is important since maintaining detection capability in the presence of preservatives as well as freeze/thaw cycles is not obvious. Preservative agents and freeze/thaw cycles affect parasitic recovery and abrogate antigenicity. As reflected in Fig. 1b, detection of Blastocystis spp. by the ELISA test is independent of storage conditions or preservation medium and emphasizes the suitability of this ELISA test in this aspect.
The clinical performance of the test was examined against a consensus of three available methods, namely microscopic examination of Lugol’s iodine staining (Stenzel and Boreham 1996), culture (Tan 2004), and IFA (Tan 2008). Overall performance of the ELISA indicated a high degree of sensitivity (92 %), specificity (87 %), and accuracy (89 %) providing the clinician with a comparative and efficient tool by which to assess the clinical probability of Blastocystis infection. We have previously reported on the performance and clinical usefulness of the FITC-conjugated anti-Blastocystis antibody for usage in IFA (Dogruman-Al et al. 2010). As reported, the IFA stain exhibited high sensitivity and specificity levels (86.7 and 97.3 %, respectively) when using culture as the gold standard. We have therefore used culture and IFA as a consensus in a second set of experiments where we assessed the clinical performance of ELISA vs. regular microscopic analysis (the most prevalent method for detection of Blastocystis spp. in routine laboratory setup). This assessment revealed that while ELISA exhibited 82 % sensitivity, Lugol staining microscopy was only able to detect 18 % of the positive samples. This is in agreement with previous reports exemplifying the insensitivity of microscopy.
The relatively high prevalence of Blastocystis that can be found in the colonization state calls for cross-reaction study with other human intestinal pathogens in order to ensure specific detection of Blastocystis as being the cause of the disease especially in cases of mixed infections. This is important in particular in view of the similar clinical symptoms between many of the pathogens. The lack of cross-reactivity with other GI pathogens emphasizes the ability of the ELISA to specifically detect the Blastocystis in these cases or alternatively to eliminate it as the cause of illness.
All in all, our results suggest that the CoproELISATM Blastocystis test may provide a reliable screening tool for blastocystosis, thus overcoming few of the drawbacks of current techniques for the diagnosis of Blastocystis. The test exhibits superior analytical and clinical performances over routinely used microscopic techniques and allows for the detection of the most prevalent human Blastocystis subtypes from a myriad of sample processing types. It enables medium-high-throughput testing capabilities as well as adaptation to automation, offering sample-to-answer results within the same day and without the need for highly skillful personnel. We surmise that the ELISA assay may possess a niche in the clinical setting for rapid screening and detection of Blastocystis in the clinical microbiology laboratory.
References
Abe N (2004) Molecular and phylogenetic analysis of Blastocystis isolates from various hosts. Vet Parasitol 25:235–242
Alfellani MA, Taner-Mulla D, Jacob AS, Imeede CA, Yoshikawa H, Stensvold CR, Clark CG (2013) Genetic diversity of Blastocystis in livestock and zoo animals. Protist 164:497–509
Ash LR, Orihel TC (1987) Parasites: a guide to laboratory procedures and identification. ASCP Press, Chicago, pp 121–122
Biedermann T, Hartmann K, Sing A, Przybilla B (2002) Hypersensitivity to non-steroidal anti-inflammatory drugs and chronic urticaria cured by treatment of Blastocystis hominis infection. Br J Dermatol 146:1113–1114
Chou CF, Tai JH (1996) Simultaneous extraction of DNA and RNA from nuclease-rich pathogenic protozoan Trichomonas vaginalis. Biotechniques 20:790–791
Dogruman-Al F, Kustimur S, Yoshikawa H, Tuncer C, Simsek Z, Tanyuksel M, Araz E, Boorom K (2009) Blastocystis subtypes in irritable bowel syndrome and inflammatory bowel disease in Ankara, Turkey. Mem Inst Oswaldo Cruz 104:724–727
Dogruman-Al F, Simsek Z, Boorom K, Ekici E, Sahin M, Tuncer C, Kustimur S, Altinbas A (2010) Comparison of methods for detection of Blastocystis infection in routinely submitted stool samples, and also in IBS/IBD patients in Ankara, Turkey. PLoS One 5:1–7
Eida AM, Eida MM (2008) Identification of Blastocystis hominis in patients with irritable bowel syndrome using microscopy and culture compare to PCR. Parasitol United J 2:87–92
Forsell J, Granlund M, Stensvold CR, Clark CG, Evengård B (2012) Subtype analysis of Blastocystis isolates in Swedish patients. Eur J Clin Microbiol Infect Dis 31:1689–1696
Krüger K, Kamilli I, Schattenkirchner M (1994) Blastocystis hominis as a rare arthritogenic pathogen. A case report. Z Rheumatol 53:83–85
Limor JR, Lal AA, Xiao L (2002) Detection and differentiation of Cryptosporidium parasites that are pathogenic for humans by real time PCR. J Clin Microbiol 40:2335–2338
Nasirudeen AM, Tan KS (2004) Caspase-3-like protease influence but is not essential for DNA fragmentation in Blastocystis undergoing apoptosis. Eur J Cell Biol 83:477–482
Noël C, Peyronnet C, Gerbod D, Edgcomb VP, Delgado-Viscogliosi P, Sogin ML, Capron M, Viscogliosi E, Zenner L (2003) Phylogenetic analysis of Blastocystis isolates from different hosts based on the comparison of small-subunit rRNA gene sequences. Mol Biochem Parasitol 126:119–123
O’Gorman MA, Orenstein SR, Proujansky R, Wadowsky RM, Putnam PE, Kocoshis SA (1993) Prevalence and characteristics of Blastocystis hominis infection in children. Clin Pediatr 32:91–96
Parkar U, Traub RJ, Kumar S, Mungthin M, Vitali S, Leelayoova S, Morris K, Thompson RC (2007) Direct characterization of Blastocystis from faeces by PCR and evidence of zoonotic potential. Parasitology 134:359–367
Pasqui AL, Savini E, Saletti M, Guzzo C, Puccetti L, Auteri A (2004) Chronic urticaria and Blastocystis hominis infection: a case report. Eur Rev Med Pharmacol Sci 8:117–120
Peek R, Reedeker FR, van Gool T (2004) Direct amplification and genotyping of Dientamoeba fragilis from human stool specimens. J Clin Microbiol 42:631–635
Qadri SM, al-Okaili GA, al-Dayel F (1989) Clinical significance of Blastocystis hominis. J Clin Microbiol 27:2407–2409
Rivera WL (2008) Phylogenetic analysis of Blastocystis isolates from animal and human hosts in the Philippines. Vet Parasitol 1:178–182
Rivera WL, Tan MA (2005) Molecular characterization of Blastocystis isolates in the Philippines by riboprinting. Parasitol Res 96:253–257
Roberts T, Barratt J, Harkness J, Ellis J, Stark D (2011) Comparison of microscopy, culture, and conventional polymerase chain reaction for detection of Blastocystis sp. in clinical stool samples. Am J Trop Med Hyg 84:308–312
Roberts T, Stark D, Harkness J, Ellis J (2013) Subtype distribution of Blastocystis isolates identified in a Sydney population and pathogenic potential of Blastocystis. Eur J Clin Microbiol Infect Dis 32:335–343
Scicluna SM, Tawari B, Clark CG (2006) DNA barcoding of Blastocystis. Protist 157:77–85
Stensvold CR (2013) Blastocystis: genetic diversity and molecular methods for diagnosis and epidemiology. Trop Parasitol 3:26–34
Stensvold R, Brillowska-Dabrowska A, Nielsen HV, Arendrup MC (2006) Detection of Blastocystis hominis in unpreserved stool specimens by using polymerase chain reaction. J Parasitol 92:1081–1087
Stensvold CR, Traub RJ, von Samson-Himmelstjerna G, Jespersgaard C, Nielsen HV, Thompson RC (2007a) Blastocystis: subtyping isolates using pyrosequencing technology. Exp Parasitol 116:111–119
Stensvold CR, Arendrup MC, Jespersgaard C, Mølbak K, Nielsen HV (2007b) Detecting Blastocystis using parasitologic and DNA-based methods: a comparative study. Diagn Microbiol Dis 59:303–307
Stensvold CR, Suresh GK, Tan KS, Thompson RC, Traub RJ, Viscogliosi E, Yoshikawa H, Clark CG (2007c) Terminology for Blastocystis subtypes—a consensus. Trends Parasitol 23:93–96
Stensvold CR, Nielsen HV, Mølbak K, Smith HV (2008) Pursuing the clinical significance of Blastocystis—diagnostic limitations. Trends Parasitol 25:23–29
Stenzel DJ, Boreham PF (1996) Blastocystis hominis revisited. Clin Microbiol Rev 9:563–584
Tan KS (2004) Blastocystis in humans and animals: new insights using modern methodologies. Vet Parasitol 126:121–144
Tan KS (2008) New insights on classification, identification, and clinical relevance of Blastocystis spp. Clin Microbiol Rev 21:639–665
Vennila GD, Suresh Kumar G, Khairul Anuar A, Rajah S, Saminathan R, Sivanandan S, Ramakrishnan K (1999) Irregular shedding of Blastocystis hominis. Parasitol Res 85:162–164
Verweij JJ, Blange RA, Tempeton K, Schinkel J, Brienen EA, van Rooyen MA, van Lieshout L, Polderman AM (2004) Simultaneous detection of Entamoeba histolytica, Giardia lamblia and Cryptosporidium parvum in fecal samples using multiplex real-time PCR. J Clin Microbiol 42:1220–1223
Yoshikawa H, Abe N, Wu Z (2003) Genomic polymorphism among Blastocystis isolates and development of PCR based identification of zoonotic isolates. J Eukaryot Microbiol 50:710–711
Yoshikawa H, Wu Z, Kimata I, Iseki M, Ali IK, Hossain MB, Zaman V, Haque R, Takahashi Y (2004) Polymerase chain reaction-based genotype classification among human Blastocystis hominis populations isolated from different countries. Parasitol Res 92:22–29
Acknowledgments
The authors wish to acknowledge Mira Barak, Shifra Ken-Dror, and Elsa Pavlotzky from Central Laboratories Haifa and Western Galilee, Clalit Health Services (Israel), for their contribution in this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dogruman-Al, F., Turk, S., Adiyaman-Korkmaz, G. et al. A novel ELISA test for laboratory diagnosis of Blastocystis spp. in human stool specimens. Parasitol Res 114, 495–500 (2015). https://doi.org/10.1007/s00436-014-4208-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-014-4208-y