
Abstract
The molecular alterations of pancreatic acinar cell carcinomas (ACCs) and mixed acinar-neuroendocrine carcinomas (MANECs) are not completely understood, and the possible role of c-MYC amplification in tumor development, progression, and prognosis is not known. We have investigated c-MYC gene amplification in a series of 35 ACCs and 4 MANECs to evaluate its frequency and a possible prognostic role. Gene amplification was investigated using interphasic fluorescence in situ hybridization analysis simultaneously hybridizing c-MYC and the centromere of chromosome 8 probes. Protein expression was immunohistochemically investigated using a specific monoclonal anti-c-myc antibody. Twenty cases had clones with different polysomies of chromosome 8 in absence of c-MYC amplification, and 5 cases had one amplified clone and other clones with chromosome 8 polysomy, while the remaining 14 cases were diploid for chromosome 8 and lacked c-MYC amplification. All MANECs showed c-MYC amplification and/or polysomy which were observed in 54% pure ACCs. Six cases (15.3%) showed nuclear immunoreactivity for c-myc, but only 4/39 cases showed simultaneous c-MYC amplification/polysomy and nuclear protein expression. c-myc immunoreactivity as well as c-MYC amplification and/or chromosome 8 polysomy was not statistically associated with prognosis. Our study demonstrates that a subset of ACCs shows c-MYC alterations including gene amplification and chromosome 8 polysomy. Although they are not associated with a different prognostic signature, the fact that these alterations are present in all MANECs suggests a role in the acinar-neuroendocrine differentiation possibly involved in the pathogenesis of MANECs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acinar cell carcinomas (ACCs) of the pancreas are a heterogeneous group of cancers showing different morphological features and clinical presentations [1]. In addition, about 10–20% of cases showing a significant neuroendocrine component (> 30% of the tumor burden) are defined mixed acinar-neuroendocrine carcinomas (MANECs). Together with mixed ductal-neuroendocrine carcinomas, they belong to the group of mixed neuroendocrine/non-neuroendocrine neoplasms (MiNENs) [2, 3]. Approximately 50% of ACCs/MANECs are metastatic at the time of diagnosis, and about 40% of cases recur as local and/or metastatic disease after surgical resection [4, 5]. The prognosis is poor with 5-year survival rates ranging between 25 and 50% without difference between ACCs and MANECs [5]. Although several attempts have been made to search for morphological, immunohistochemical, and molecular prognostic factors, tumor stage still seems to be the best prognosticator in resectable cases [5].
Our knowledge on molecular alterations of ACCs has been greatly expanded in the last years, and several molecular alterations involved in tumor development and progression have been recently identified [1]. Some of them are typical of ACCs, like alterations in the APC/β-catenin pathway and fusion in RAF genes [6, 7], while others involve genes (i.e., TP53 and BRCA2) which play a crucial role in the development and progression of a wide spectrum of different cancers [1]. Interestingly, recently published data have suggested that some molecular alterations, like those involving TP53, may identify ACC subtypes showing a more aggressive behavior [8].
It has been recently reported that about 17% of ACCs show c-MYC amplification, but the prognostic role of this alteration remains to be clarified [9]. The oncogene c-MYC is a transcription factor implicated in about one third of human malignancies by promoting tumor growth increasing DNA replication and transcription, protein synthesis, cellular metabolism, and proliferation [10]. c-MYC overexpression is frequently associated with poor clinical outcome [11], and it has been demonstrated that c-MYC plays a pivotal role in the molecular mechanisms underlying the aggressiveness of pancreatic ductal adenocarcinoma [10, 11].
In the present study, we have investigated c-MYC gene amplification in a series of 39 pancreatic ACCs/MANECs in order to evaluate its frequency and a possible prognostic role.
Materials and methods
Cases
Thirty-nine cases were selected from our previously reported series of 62 well-characterized pancreatic ACCs [5]. Tumor selection mainly depended on the availability of sufficient material to perform immunohistochemical and fluorescence in situ hybridization (FISH) analyses together with complete clinical information. The main clinicopathologic characteristics are summarized in Table 1. All tissues were fixed in buffered formalin (formaldehyde 4% w/v and acetate buffer 0.05 M) and routinely processed to paraffin wax.
Immunohistochemistry
For immunohistochemistry, 3-μm-thick sections were mounted on poly-L-lysine-coated slides, deparaffinized, and hydrated through graded alcohols to water. After endogenous peroxidase activity inhibition, performed by dipping sections in 3% hydrogen peroxide for 10 min, incubation with primary rabbit monoclonal anti-c-myc antibody (Y69, Abcam, Cambridge, UK) was carried out at 4 °C for 18–20 h, followed by the avidin–biotin complex (ABC) procedure. Immunoreactions were developed using 0.03% 3,3’diaminobenzidine tetrahydrochloride and then sections were counterstained with Harris’ hematoxylin.
Fluorescence in situ hybridization
Interphasic FISH analysis was performed on 3–4-μm-thick sections used for conventional histologic examination as reported in the guidelines of the European Cytogeneticists Association [12], and the cytogenetic interpretation of data agrees with the International System for human Cytogenetic Nomenclature (ISCN) [13]. FISH analysis was performed using direct viewing on a standard fluorescence microscope at × 100 magnification. FISH results were evaluated on representative areas of each tumor identified on hematoxylin- and eosin-stained slides. To ensure a representative sample and to permit an assessment of the extent of tumor heterogeneity, c-MYC amplification and chromosome 8 polysomies were scored in more than 200 interphasic nuclei from at least five to eight separate areas of each tumor by two independent operators (BB and MGT). Only experiments with 90% hybridization efficiency were considered. c-MYC amplification was investigated simultaneously hybridizing c-MYC (red signal) and the centromere of chromosome 8 (green signal) probes (Zytolight SPEC MYC/CEN8 Dual Color Probe, Zytovision GmbH, Bremerhaven, Germany). Cases were defined as amplified when the ratio (R) between red (c-MYC) and green (CEN8) signals was > 2.0. Cases were defined as polysomic for chromosome 8 when at least 20% of neoplastic cells showed three or more copies of CEN8 signals (green signals).
Statistical analysis
Comparisons of continuous data were performed using Student’s t tests, and discrete variables were compared with χ2 test or Fisher’s exact test. Univariate survival analysis was performed using Kaplan–Meier curves and log-rank test. Data were statistically analyzed using MedCalc® Version 12.5.0.0 and GraphPad Prism Version 5.00 software, and p value < 0.05 was considered significant.
Results
ACCs and MANECs were more frequently observed in males (29 cases) than in females (10 cases), and the average age at diagnosis was 59.7 years (range 33–84 years). Tumors were more frequently located in the pancreatic head (15 cases) followed by the tail (12 cases) and the body region (12 cases). The mean size was 8 cm with a range between 1.6 and 29 cm. Thirty-five out of 39 (89%) cases were pure ACCs, while four cases showing a neuroendocrine cell population > 30% were defined as MANECs [3]. The mean follow-up time was 33 months (range 6–135 months). Twenty-seven patients died of disease after a mean follow-up time of 18.6 months, while 11 patients were alive at the last follow-up control (mean follow-up time of 67.5 months). One patient was lost to follow-up.
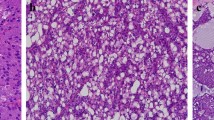
Six cases (15.3%) showed nuclear immunoreactivity for c-myc, in a cell population ranging from 10 to 80% neoplastic cells (Fig. 1).

c-myc nuclear immunoreactivity in the majority of neoplastic cells of a pancreatic acinar cell carcinoma
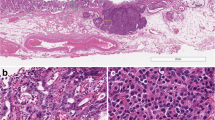
All 39 cases were scored for both c-MYC amplification (Fig. 2a) and chromosome 8 polysomy (Fig. 2b): in detail, 20 ACCs had clones with different polysomies of chromosome 8 in absence of c-MYC amplification, and 5 cases had one amplified clone and other clones with chromosome 8 polysomy. The remaining 14 cases were diploid for chromosome 8 and lacked c-MYC amplification. FISH data of five c-MYC amplified cases are reported in Table 2. The clones with c-MYC amplification ranged from 33.1 to 77.4% of neoplastic cells. The ratio of c-MYC and chromosome 8 centromere ranged from 2.22 to 2.87 indicating presence of low level of c-MYC amplification in all cases. Polysomic cases showed different levels of polysomies ranging from three to ten chromosome 8 resulting in gain of MYC copies. Figure 2b shows a polysomic ACC showing nuclei with six to ten chromosome 8. Interestingly, all MANECs showed c-MYC amplification and/or polysomy, which were observed in 19 out 35 (54%) pure ACCs. Four out of the 39 cases investigated showed simultaneous c-MYC amplification/polysomy and a nuclear protein expression. As in most cases of mixed acinar-neuroendocrine carcinomas [3, 5], the two components were not clearly separated or identifiable on morphological analysis and their identification was performed using immunohistochemistry.

FISH analysis using MYC probe (red signals) and chromosome 8 centromere (green signals). A subgroup of acinar cell carcinomas and all mixed acinar-neuroendocrine carcinomas showed c-MYC amplification (a) and/or chromosome 8 polysomy (b)
c-myc immunoreactivity as well as c-MYC amplification and/or chromosome 8 polysomy was not statistically associated with prognosis (p = 0.04) (Fig. 3).

C-myc protein expression (a) was not statistically associated with prognosis as well as c-MYC amplification (b), chromosome 8 polysomy (c), or their combination (d)
Discussion
The molecular signature of ACCs is different from that of pancreatic ductal adenocarcinomas and neuroendocrine neoplasms and more frequently includes alterations in the APC/β-catenin pathway, while gene alterations frequently involved in ductal adenocarcinomas like mutations in KRAS, DPC4, and p16 are absent or very rarely present [1, 14]. Alterations of TP53 gene (mutation of one allele and loss of the other alleles) were recently found to be associated with worse survival [8] suggesting the possibility that specific subtypes of ACC may show specific molecular features with prognostic relevance. In this context, the recent identification that a subset of ACC shows c-MYC amplification [9] has suggested us to explore the prognostic role of c-MYC alterations in pancreatic ACCs.
c-MYC, whose function is tightly controlled by growth factor-dependent signals in normal adult cells [15, 16], plays a pivotal role in organogenesis and, in particular, in pancreatic acinar cell development and maturation [17]. However, its expression can be deregulated and enhanced via multiple mechanisms in tumor cells and is implicated in the pathogenesis, progression, and aggressiveness of several human tumors. In particular, c-MYC protein expression and c-MYC gene activation by amplification have been found to be associated with tumor aggressiveness and poor prognosis of several cancers [11, 15, 18,19,20] including pancreatic ductal adenocarcinoma (PDAC) [10, 11]. Most of genetic and epigenetic alterations playing a role in the pathogenesis and progression of PDACs involve c-MYC activations. c-MYC overexpression occurs in about 40% of advance PDACs, although comprehensive genetic analysis demonstrated that c-MYC is generally amplified at low levels [21]. Mechanisms involved in c-MYC deregulation in PDACs include genetic events or transcriptional, post-transcriptional signaling, or post-translational mechanisms. Genetic aberrations include mutation of the TGFβ-inhibitory elements controlling c-MYC promoter or c-MYC amplification. Transcriptional mechanisms include the activation of transcription factors inducing c-MYC transcription or enhancement of c-MYC transcriptional elongation by CDK9-mediated phosphorylation of RNA-polymerase. Alterations in post-transcriptional signaling include the attenuation of c-MYC-inhibiting miRNAs in absence of TP53 functions; post-translational mechanisms include CK2-mediated phosphorylation of c-MYC, which prevents proteasome degradation resulting in the reduction of c-MYC ubiquitination and degradation [10].
In 20 cases of our series, we found chromosome 8 polysomy in absence of c-MYC amplification and in five cases both c-MYC amplification and chromosome 8 polysomy. This result suggests that c-MYC activation by gene amplification and/or polysomy is involved in the pathogenesis of at least a subset of ACCs. Interestingly, we found that all MANECs showed c-MYC amplification and/or polysomy which, on the contrary, were observed in only 54% of pure ACCs. In this context, it is interesting to recall that c-MYC has been found to regulate neuroendocrine trans-differentiation of prostate adenocarcinoma resulting in the formation of the aggressive neuroendocrine-differentiated subtype [22,23,24,25]. In addition, c-MYC has been demonstrated to play a pivotal role in regulating ductal-neuroendocrine plasticity of pancreatic ductal adenocarcinoma leading to a neuroendocrine differentiation, which contributes to poor outcome and therapeutic resistance [26]. Taking together, these findings may suggest that activation of c-MYC may lead to an acinar-neuroendocrine differentiation responsible of MANEC development. Starting from this observation, further studies are needed to confirm this hypothesis.
In general, we have found low level of c-MYC amplification in our series, and this may explain the discordance observed between FISH and immunohistochemistry considering that the latter is a less sensitive method. Immunohistochemical expression of MYC seems to predict well c-MYC alterations when more than 50% of nuclei are MYC positive [27]. In our series, among the five cases immunoreactive for MYC, only two showed more than 50% of positive nuclei and both cases showed c-MYC polysomy.
Regarding a possible prognostic role of c-MYC alterations, it is worth noting that pancreatic ACCs and MANECs are a group of aggressive carcinomas showing poor prognosis with 5-year survival rates ranging between 25 and 50%. To date, tumor stage seems to be the only prognosticator in resectable cases [5]. However, among resected cases, the search for prognostic factors useful to stratify patients in different prognostic categories is a hot topic in pancreatic pathology. To the best of our knowledge, there are no prognostic factors for surgical resected pancreatic ACCs and MANECs, although in recent years, several attempts have been made to search for them. One of our aims was to check whether c-MYC amplification could be used as prognostic marker. Although we have observed a trend of worse survival in patients with c-MYC amplification than in patients without it, we did not find a statistical meaning.
In conclusion, our study demonstrates that a subset of ACCs show c-MYC alterations including gene amplification and chromosome 8 polysomy. Although they are not associated with a different prognostic signature, the fact that these alterations are present in all MANECs suggests a role in the acinar-neuroendocrine differentiations possibly involved in the pathogenesis of MANECs.
References
La Rosa S, Sessa F, Capella C (2015) Acinar cell carcinoma of the pancreas: overview of clinicopathologic features and insights into the molecular pathology. Front Med 2:41. https://doi.org/10.3389/fmed.2015.00041
La Rosa S, Sessa F, Uccella S (2016) Mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs): unifying the concept of a heterogeneous group of neoplasms. Endocr Pathol 27:284–311. https://doi.org/10.1007/s12022-016-9432-9
Klöppel G, Couvelard A, Hruban RH, Klimstra DS, Komminoth P, Osamura RY, Perren A, Rindi G (2017) Neoplasms of the neuroendocrine pancreas. Introduction. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J (eds) WHO classification of tumours of endocrine organs. IARC Press, Lyon, pp 211–214
Wisnoski NC, Townsend CM Jr, Nealon WH, Freeman JL, Riall TS (2008) 672 patients with acinar cell carcinoma of the pancreas: a population-based comparison to pancreatic adenocarcinoma. Surgery 144:141–148. https://doi.org/10.1016/j.surg.2008.03.006
La Rosa S, Adsay V, Albarello L, Asioli S, Casnedi S, Franzi F, Marando A, Notohara K, Sessa F, Vanoli A, Zhang L, Capella C (2012) Clinicopathologic study of 62 acinar cell carcinomas of the pancreas: insights into the morphology and immunophenotype and search for prognostic markers. Am J Surg Pathol 36:1782–1795. https://doi.org/10.1097/PAS.0b013e318263209d
Furlan D, Sahnane N, Bernasconi B, Frattini M, Tibiletti MG, Molinari F, Marando A, Zhang L, Vanoli A, Casnedi S, Adsay V, Notohara K, Albarello L, Asioli S, Sessa F, Capella C, La Rosa S (2014) APC alterations are frequently involved in the pathogenesis of acinar cell carcinoma of the pancreas, mainly through gene loss and promoter hypermethylation. Virchows Arch 464:553–564. https://doi.org/10.1007/s00428-014-1562-1
Chmielecki J, Hutchinson KE, Frampton GM, Chalmers ZR, Johnson A, Shi C, Elvin J, Ali SM, Ross JS, Basturk O, Balasubramanian S, Lipson D, Yelensky R, Pao W, Miller VA, Klimstra DS, Stephens PJ (2014) Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 4:1398–1405. https://doi.org/10.1158/2159-8290.CD-14-0617
La Rosa S, Bernasconi B, Frattini M, Tibiletti MG, Molinari F, Furlan D, Sahnane N, Vanoli A, Albarello L, Zhang L, Notohara K, Casnedi S, Chenard MP, Adsay V, Asioli S, Capella C, Sessa F (2016) TP53 alterations in pancreatic acinar cell carcinoma: new insights into the molecular pathology of this rare cancer. Virchows Arch 468:289–296. https://doi.org/10.1007/s00428-015-1882-9
Bergmann F, Aulmann S, Sipos B, Kloor M, von Heydebreck A, Schweipert J, Harjung A, Mayer P, Hartwig W, Moldenhauer G, Capper D, Dyckhoff G, Freier K, Herpel E, Schleider A, Schirmacher P, Mechtersheimer G, Klöppel G, Bläker H (2014) Acinar cell carcinomas of the pancreas: a molecular analysis in a series of 57 cases. Virchows Arch 465:661–672. https://doi.org/10.1007/s00428-014-1657-8
Hessmann E, Schneider G, Ellenrieder V, Siveke JT (2016) MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene 35:1609–1618. https://doi.org/10.1038/onc.2015.216
Nesbit CE, Tersak JM, Prochownik EV (1999) MYC oncogenes and human neoplastic disease. Oncogene 18:3004–3016. https://doi.org/10.1038/sj.onc.1202746
Hastings RJ, Bown N, Tibiletti MG, Debiec-Rychter M, Vanni R, Espinet B, van Roy N, Roberts P, van den Berg-de-Ruiter E, Bernheim A, Schoumans J, Chatters S, Zemanova Z, Stevens-Kroef M, Simons A, Heim S, Salido M, Ylstra B, Betts DR, Tumour Best Practice meeting, Eurogentest (2016) Guidelines for cytogenetic investigations in tumours. Eur J Hum Genet 24:6–13. https://doi.org/10.1038/ejhg.2015.35
Shaffer LG, McGowan-Jordan J, Schmid M (2013) ISCN an international system for human cytogenetic nomenclature. Published in collaboration with ‘Cytogenetic and Genome Research’. Karger, Basel ISBN:978-3-318-02253-7
Jäkel C, Bergmann F, Toth R, Assenov Y, van der Duin D, Strobel O, Hank T, Klöppel G, Dorrell C, Grompe M, Moss J, Dor Y, Schirmacher P, Plass C, Popanda O, Schmezer P (2017) Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability. Nat Commun 8:1323. https://doi.org/10.1038/s41467-017-01118-x
Eilers M, Eisenman RN (2008) Myc’s broad reach. Genes Dev 22:2755–2766. https://doi.org/10.1101/gad.1712408
Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8:976–990. https://doi.org/10.1038/nrc2231
Sánchez-Arévalo Lobo VJ, Fernández LC, Carrillo-de-Santa-Pau E, Richart L, Cobo I, Cendrowski J, Moreno U, del Pozo N, Megías D, Bréant B, Wright CV, Magnuson M, Real FX (2018) c-Myc downregulation is required for preacinar to acinar maturation and pancreatic homeostasis. Gut 67:707–718. https://doi.org/10.1136/gutjnl-2016-312306
Skoudy A, Hernández-Muñoz I, Navarro P (2011) Pancreatic ductal adenocarcinoma and transcription factors: role of c-Myc. J Gastrointest Cancer 42:76–84. https://doi.org/10.1007/s12029-011-9258-0
Dang CV (2012) MYC on the path to cancer. Cell 149:22–35. https://doi.org/10.1016/j.cell.2012.03.003
Kress TR, Sabò A, Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15:593–607. https://doi.org/10.1038/nrc3984
Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U (2002) c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol 15:462–469
Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, Dhir R, Nelson JB, de la Taille A, Allory Y, Gerstein MB, Perner S, Pienta KJ, Chinnaiyan AM, Wang Y, Collins CC, Gleave ME, Demichelis F, Nanus DM, Rubin MA (2011) Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 1:487–495. https://doi.org/10.1158/2159-8290
Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, Cheung C, Yuen KS, Gao D, Chen Y, Eilers M, Mosquera JM, Robinson BD, Elemento O, Rubin MA, Demichelis F, Rickman DS (2016) N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30:563–577. https://doi.org/10.1016/j.ccell.2016.09.005
Wang J, Kim J, Roh M, Franco OE, Hayward SW, Wills ML, Abdulkadir SA (2010) Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 29:2477–2487. https://doi.org/10.1038/onc.2010.10
Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, Baertsch R, Sokolov A, Meyerowitz JG, Mathis C, Cheng D, Stuart JM, Shokat KM, Gustafson WC, Huang J, Witte ON (2016) N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 29:536–547. https://doi.org/10.1016/j.ccell.2016.03.001
Farrell AS, Joly MM, Allen-Petersen BL, Worth PJ, Lanciault C, Sauer D, Link J, Pelz C, Heiser LM, Morton JP, Muthalagu N, Hoffman MT, Manning SL, Pratt ED, Kendsersky ND, Egbukichi N, Amery TS, Thoma MC, Jenny ZP, Rhim AD, Murphy DJ, Sansom OJ, Crawford HC, Sheppard BC, Sears RC (2017) MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat Commun 8:1728. https://doi.org/10.1038/s41467-017-01967-
Nwanze J, Siddiqui MT, Stevens KA, Saxe D, Cohen C (2017) MYC immunohistochemistry predicts myc rearrangements by FISH. Front Oncol 7:209. https://doi.org/10.3389/fonc.2017.00209
Author information
Authors and Affiliations
Contributions
Design and conception: SLR, BB, MGT; data gathering and analysis: SLR, BB, MGT, AV, LZ, KN, LA, PB, AS, FS; manuscript preparation: SLR, MGT, FS. All authors have read and approved the final version of this manuscript.
Corresponding author
Ethics declarations
This study was performed according to the clinical standards of the 1975 and 1983 Declaration of Helsinki and was approved by the Ethical Committee of the Ospedale di Circolo (ASST Sette Laghi), Varese, Italy (n.1277/10).
Conflict of interest
The authors declare that they have not conflict of interest.
Rights and permissions
About this article

Cite this article
La Rosa, S., Bernasconi, B., Vanoli, A. et al. c-MYC amplification and c-myc protein expression in pancreatic acinar cell carcinomas. New insights into the molecular signature of these rare cancers. Virchows Arch 473, 435–441 (2018). https://doi.org/10.1007/s00428-018-2366-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-018-2366-5