
Abstract
Pancreatic acinar cell carcinomas (PACs) are rare but are distinct aggressive neoplasms that phenotypically differ from pancreatic ductal adenocarcinomas (PDACs) and pancreatic neuroendocrine neoplasms (PNENs). Despite recent work on the genetic changes of PACs, their molecular pathogenesis is still poorly understood. In this study, we focus on a comparative genomic hybridization analysis. Based on frequent chromosomal imbalances, the involvement of DCC and c-MYC in the pathogenesis of PACs is further investigated. Moreover, we examine markers harboring potential therapeutic relevance (K-RAS, BRAF, EGFR, MGMT, HSP90, L1CAM, Her2). PACs revealed a microsatellite stable, chromosomal unstable genotype, defined by recurrent chromosomal losses of 1p, 3p, 4q, 5q, 6q, 8p, 9p, 11q, 13q, 16q, and 18, as well as gains of 1q, 7, 8q, 12, 17q, and 20q. Subsets of PAC displayed reduction/loss of DCC (79 %) and c-MYC-amplification (17 %). Significant EGFR expression occurred in 42 %, HSP90 expression in 98 %, L1CAM expression in 72 %, and loss of MGMT in 26 %. Two cases carried a K-RAS mutation. Mutations of EGFR or BRAF were not detected. All cases were Her2/neu-negative. PACs display characteristic chromosomal imbalances which are distinctly different from those in pancreatic ductal adenocarcinomas and pancreatic neuroendocrine neoplasms. Our findings suggest that DCC and c-MYC alterations may play an important role in the pathogenesis of PACs. Furthermore, EGFR, MGMT, HSP90, and L1CAM may be useful as therapeutic markers and predictors of response to therapy in a subset of PACs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pancreatic acinar cell carcinomas (PACs) are rare pancreatic exocrine tumors which most commonly occur in adults in the sixth and seventh decades of life but show a wide age range and may affect children as well [1–5]. At the time of diagnosis, approximately 50 % of PACs have metastasized, most frequently involving regional lymph nodes and/ or the liver [4, 6]. The prognosis of these neoplasms, even if they are resectable, is therefore poor. Local, and especially metastatic, relapse occurs in up to 42 % of patients [1–4, 6]. Radiotherapy and chemotherapy are only effective in a limited number of patients [6]. Five-year survival rates range between 25 and 50 %, depending on stage [7], and survival times between 3 and 123 months have been reported [1–3].
PACs are usually solid tumors, composed of cells expressing trypsin and other pancreatic enzymes [4, 8]. Rare cystic variants are acinar cell cystadenocarcinomas [9] and acinar cell carcinomas containing substantial (>30 %) proportions of cells showing neuroendocrine and/or ductal differentiation [8, 10, 11].
The molecular mechanisms leading to the initiation and progression of PACs are not known. Few published, mostly small, series have shown that mutations that are common in pancreatic ductal adenocarcinoms (PDACs), i.e., K-RAS, p16, and SMAD4/DPC4, are rare in PACs [12–19]. Alterations of the β-catenin/adenomatous polyposis coli (APC) pathway have been reported in a subset of PACs [8, 12, 19, 20], paralleling findings in pancreatoblastoma [21]. An involvement of the β-catenin/APC pathway in a significant subset of PACs was supported by a more recent study, in which APC loss and hypermethylation were found in up to 56 % of PACs, by far outnumbering APC mutations [20]. Moreover, using whole exome sequencing analyses, mutations of single genes were shown to generally occur in less than 30 % of PACs, repeatedly affecting, among others, GNAS, JAK1, BRAF, and RB1 [19]. In a few PACs, microsatellite instability was detected [12, 18], including five cases in which an epigenetic analysis revealed CpG island promoter methylation of MSH6 [18]. Chromosomal imbalances were investigated in only eight cases [22, 23].
In the present study, we aimed to characterize a comprehensive series of PACs, using comparative genomic hybridization (CGH). Detected chromosomal imbalances were compared with CGH findings in salivary gland acinar cell carcinomas (shown in Supplement), as well as with earlier reported findings in PDACs and pancreatic neuroendocrine neoplasms (PNENs) to see whether they have a common cytogenetic background. Furthermore, loci of recurrent chromosomal imbalances were used to further evaluate candidate genes possibly involved in the pathogenesis of PACs. This included DCC on 18q and c-MYC on 8q. Finally, molecular and immunohistochemical analyses were performed to investigate putative therapeutic markers and targets in PACs. For these analyses, K-RAS, EGFR, Her2/neu, BRAF, MGMT, L1CAM, and HSP90 were included as they were reported to harbor therapeutic significance in clinical and preclinical studies in other, more frequent tumor entities.
Materials and methods
Pancreatic acinar cell carcinomas
Formalin-fixed, paraffin-embedded tissues samples from 57 PACs were retrieved from the archives of the Institute of Pathology, University of Heidelberg, Germany, and the files of the Consultation Center for Pancreatic and Endocrine Tumors of the Department of Pathology, Technical University Munich, Germany. The 57 tumor samples were obtained from 17 women and 40 men, with a median age of 61 years (range 7 to 79 years). Primary tumors were located in the pancreatic head (n = 14), body (n = 7), tail (n = 8), body and tail (n = 2), or the entire gland (n = 2). One tumor was found in heterotopic pancreatic tissue in the wall of the stomach. In the remaining cases, the location within the pancreas could not be clearly defined. The tumor size ranged from 2 to 21.5 cm (mean 8.5 cm). Mitotic count revealed between 0 and 47 mitoses per 10 high-power fields (mean 12.3). Metastatic tissues were obtained from the liver (n = 5), peritoneum (n = 2), and lymph nodes (n = 3). The corresponding primary tumors were available in six metastatic cases. Forty-six patients had a solid PAC, following the diagnostic criteria recommended by the World Health Organization [7]. Two patients had an acinar cell cystadenocarcinoma, and nine patients had mixed acinar neuroendocrine carcinoma (MACNEC). The study was approved by the ethics committee of the University of Heidelberg (no. 207/2005).
Comparative genomic hybridization
CGH analyses were performed as previously described [24]. Furthermore, in a Medline search, CGH findings in PDACs and PNENs (including neuroendocrine tumors and neuroendocrine carcinomas) were identified. Only those cases were included in the analysis whose CGH could be assigned to specific tumor cases. Series in which the chromosomal imbalances were collectively presented were not considered.
Fluorescence in situ hybridization
For fluorescence in situ hybridization, dewaxed slides were incubated in 1 M NaSCN, 80 °C for 30 min, digested using proteinase K (100 μg/ml, 15 min) and dehydrated. After co-denaturation with a spectrum-orange-labeled c-MYC DNA probe (RCPI-753J1555) at 85 °C for 5 min, slides were hybridized in a humid chamber at 37 °C for 16–24 h. Post-hybridization washes were done in 50 % formamid/2× SSC, pH 7, 42 °C for 3× 5 min and 2× SSC, pH 7, 42 °C for 3× 5 min after which slides were coverslipped using Vectashield mounting medium containing 4,6-diamidino-2-phenylindole and visualized on an epifluorescence microscope. For analysis, at least 50 cells in two representative areas of the tumors were counted. Low-level amplifications of c-MYC were assumed in cases exceeding four signals per nucleus, and amplifications were assumed in cases exceeding seven signals per nucleus.
Immunohistochemistry
Immunohistochemical analyses, using the avidin-biotin complex method, were performed with primary antibodies directed against EGFR (clone 31G7; 1:25; Zymed Laboratories, San Francisco, CA, USA), HSP90 (anti-Hsp90; 1:70; Millipore, Billerica, MA, USA), MGMT (1:30; clone MT3.1, Thermo Scientific, Fremont, USA), the mismatch repair gene products MLH1 (1:100; clone G168-15; BD Transduction Laboratories), MSH2 (1:100; AB2; Oncogene Research, Cambridge, MA, USA), and MSH6 (1:200; clone 44; BD Transduction Laboratories), PMS2 (1.50; clone A16-4, BD Pharmingen, San Diego, CA, USA), L1CAM (1:100; L1-14.10; [25]), Her2/neu (polyclonal rabbit, 1:500; clone A0485; DAKO), DCC (1:100; G97-449; BD Pharmingen), and BRAF V600E (1:5; VE1; Spring Bioscience, Pleasanton, CA, USA). If necessary, antigen retrieval was achieved by microwave pretreatment in citrate buffer (HSP90, p53, MSH6) or by microwave pretreatment in 1 mM EDTA (MGMT, MLH1, MSH2). Immunohistochemical stains were considered as positive in few cells if less than 10 % of the tumor cells showed immunoreactivity, as focally positive if 10 to 70 % of the tumor cells showed immunoreactivity, and as positive if more than 70 % of the tumor cells showed immunoreactivity.
Mutational analyses
Mutational analyses of K-RAS (exons 1 and 2) and EGFR (exons 18–21) were performed as previously described [26, 27]. Primer sequences are listed in Supplemental Table 1.
Microsatellite instability analysis
Microsatellite instability analyses were conducted as previously described [28], using the marker panels CAT25, BAT25, and BAT26. High-level microsatellite instability (MSI) was diagnosed if at least two of three markers showed MSI.
Statistical analyses
For statistical analyses, the Wilcoxon rank sum test and the Pearson’s chi-squared test were used to test continuous and countable data, respectively. Hierarchical clustering was performed as described in a previous study [29]. Furthermore, oncogenetic tree models were used to assess dependencies between chromosomal imbalances occurring in the course of tumor development and progression, as previously reported [30, 31].
Results
CGH findings
CGH analyses of 32 primary tumors and 9 metastases revealed chromosomal imbalances in 39 samples (Table 1; Fig. 1). Chromosomal losses were slightly more prevalent than gains (average 7.5 versus 5.6 changes per case). Losses affected most frequently the chromosomes 1p (n = 23), 18q (n = 21), 4q (n = 17), 11q (n = 17), 9p (n = 16), 5q (n = 13), 6q (n = 13), 8p (n = 11), 18p (n = 11), 3p (n = 10), 13q (n = 10), and 16q (n = 10). Gains were most frequently found at chromosomes 1q (n = 23), 20q (n = 18), 12p (n = 14), 12q (n = 14), 8q (n = 10), 17q (n = 10), 7p (n = 9), and 7q (n = 9).

Chromosomal imbalances in 41 PACs (32 primary tumors and 9 metastases). Lines to the left of ideogram represent chromosomal losses, lines to the right, chromosomal gains, thin lines, primary tumors, and fat lines, metastases
In PACs with a size less than 4 cm, gains of 3q22–29 occurred more frequently (22 %; p = 0.049) than in larger tumors. Loss of 18p11 and 18q11 accumulated in tumors larger than 4 cm (44 and 38 %, respectively) but was not found in smaller tumors (p = 0.019 and 0.035, respectively). Imbalances that occurred significantly more frequent in highly proliferative tumors (cutoff ten mitoses per ten high-power fields) included gain of 8q (0 versus 38 %; p = 0.004), loss of 6q22 (6 versus 46 %, p = 0.008), 6q23–26 (6 versus 54 %; p = 0.002), and 6q27 (6 versus 46 %; p = 0.008). On the other hand, gain of 7p15 and 7p21–22 was restricted to tumors of lower proliferative activity (28 and 33 versus 0 %; p = 0.038 and p = 0.020, respectively). Several chromosomal imbalances were significantly more frequent in the 10 metastases than in the 31 primary tumors. Among others, this included gains of 12p11–13, 12q12–14, 12q23–24, and 22q11–13, as well as loss of 4q28 (p ≤ 0.001). Furthermore, gains of 8q24 and 20q11–12 were more frequent in metastases (p = 0.030 and p = 0.002, respectively). Moreover, the direct comparison of six primary tumors and their corresponding metastases as expected revealed a significantly higher average number of chromosomal imbalances in metastases (18.625; range 10–25) than in their corresponding primaries (8.5; range 4–15) (p = 0.001).
Hierarchical clustering revealed two main groups, which could be further subdivided (Fig. 2). In general, primary tumors and corresponding metastases were located within the same clusters. Clinical and pathological parameters such as sex, age, tumor size, spread at the time of diagnosis, mitotic count, or necrosis did not significantly correspond to the described clusters. Furthermore, of note, no specific pattern could be attributed to solid PACs versus cystic PACs or MACNECs.

Hierarchical cluster analyses of 41 PACs. MACNECs are depicted in red
As shown by the oncogenetic tree model and a heat map (Fig. 3), loss of 1p depicted as an early change, followed by and frequently associated with loss of 18q. Further early changes included gain of 1q, showing frequent association with gains of 20q and 12q. In a third branch of the tree model, loss of 11q frequently concurred with loss of 6q and 16q.

Oncogenetic tree models (A–C) and heat maps (a–c) show different clusters in the progression of PACs (A, a), PDACs (B, b), and PNENs (C, c)
Comparison with reported chromosomal imbalances in pancreatic ductal adenocarcinomas and pancreatic neuroendocrine neoplasms
For PDACs (none of which was reported to be associated with IPMN), the reviewed CGH analyses showed chromosomal imbalances in 94 of 111 tumors [32–36]. Most frequent findings were gains on chromosomes 20q (38 %), 8q (37 %), 1q (31 %), 5p (28 %), 7p (27 %), 12p (26 %), 3q (23 %), 7q (23 %), 20p (23 %), and 13q (21 %), Frequent losses affected chromosomes 18q (62 %), 17p (41 %), 8p (38 %), 6q (33 %), 9p (33 %), 3p (27 %), 4q (25 %), and 12q (20 %) [32–36]. The tree model analysis and the heat map analysis revealed that in PDACs, one cluster was characterized by losses of 18q and 4q, associated with gain of 1q, losses of 9p and 6q, as well as loss of 8q and gain of 20q. A second cluster displayed loss of 17p, followed by gains of 5p, 7p, and 8q (Fig. 3).
In PNENs, chromosomal imbalances were reported in 104 of 124 cases, comprising 98 functionally active and 26 inactive PNENs [37–41]. Chromosomal gains were most frequently found on 7q (40 %), 17q (36 %), 14q (34 %), 7p (32 %), 9q (32 %), 5q (31 %), 20q (29 %), 12q (28 %), 17p (28 %), and 5p (25 %). Chromosomal losses most frequently affected 6q (31 %), 11q (30 %), 11p (25 %), and 3q (21 %) [37–41]. The tree model analysis revealed one cluster characterized by gains of 20q, 17, 12q, 14q, 5, and 7 and a second cluster characterized by losses of 6q and 11q, as well as gain of 9q (Fig. 3).
Comparing the CGH findings in PACs (present and previous data [22, 23]) with those in PDACs showed that both entities share some chromosomal imbalances in similar frequency, including gains of 7, 8q, 12p, and 20 and losses of 3p, 4q, 6q, 8p, 9p, and 18q. For others, however, significantly higher frequencies in PACs were found for gains of 1q, 9q, 12q, and 22p, as well as losses of 1p, 5q, 11, 16q, and 21p (p ≤ 0.001). In contrast, loss of 17p was far more common in PDACs (p < 0.001).
Comparison of PACs with PNENs revealed comparable frequencies for gains of 5q, 7, 12q, 17q, and 20q and losses of 6q and 11, but significantly higher frequencies were seen in PACs for gains of 1q, 3, 8q, 10, and 22p, as well as losses of 1p, 4q, 5q, 9, 14q, 16q, 17q, and 18. Gain of 14q occurred more frequent in PNENs (p ≤ 0.001).
Molecular and immunohistochemical results
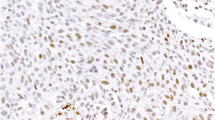
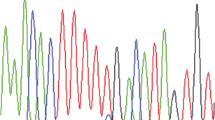
A total of 57 tumor tissues were available for additional molecular and immunohistochemical tests (Supplementary Tables 2 and 3), depending on the DNA quality and the size of the tumor samples. Fluorescence in situ hybridization (FISH) revealed amplification of c-MYC (up to ten signals per nucleus) in one case, low-level amplifications (between five and seven signals per nucleus) in 6 cases and a normal c-MYC copy number in 34 tumor samples. Immunohistochemical stains showed diffuse expression of DCC in non-neoplastic tissues. In contrast to this, 17 of 56 available tumors (30.4 %) showed complete loss of DCC expression, and 27 tumors (48.2 %) revealed significant reduction of DCC expression, displaying only very faint staining intensities. Only 12 tumors (21.4 %) showed regularly maintained DCC immunoreactivity, comparable to that in non-neoplastic tissues. Microsatellite instability was detected in 2 of 42 informative tumor tissues, revealing MSI in at least 2 of 3 markers. No tumor revealed MSI of only one marker. Aberrant expression of the mismatch repair gene products MLH1, MSH2, and MSH6 was not observed. In one of the microsatellite unstable tumors, MSH6 staining was considered not informative because of the complete absence of staining in tumor and control tissue. Additional PMS2 staining in the two microsatellite unstable tumors revealed lack of nuclear PMS2 expression in case 40, suggesting a PMS2 deficiency as cause of the MSI phenotype, and a reduction of PMS2 staining compared to surrounding non-tumorous cells in case 10. Mutational analyses of K-RAS comprising exons 1 and 2 revealed mutations in 2 of 41 tumor tissues tested (2 %), affecting codon 12 (GGT 12 GAT) and codon 13 (GGC 13 GAC) (Fig. 4). Membranous immunohistochemical expression of EGFR was observed in 19/45 tumors (42 %), with positive tumor cells ranging between 80 and 100 % in 14 cases, 40 % in 1 case, and 5 % in 4 cases. There were no EGFR mutations found in exon 18 (41 informative cases), exon 19 (n = 40), exon 20 (n = 31), and exon 21 (n = 26). Immunostaining with the BRAF mutation specific antibody V600E or with Her2/neu was negative in 42 tumors. Loss of nuclear MGMT expression was lost in 13/51 tumors (26 %) (Fig. 5). High immunohistochemical expression of L1CAM was recognized in 39/54 tumors (72 %), with diffuse staining in 29 cases and focal staining in 10 cases. Finally, cytoplasmic HSP90 expression was observed in 47/48 cases (98 %). Seven cases, including one cytoplasmic-negative tumor, showed nuclear HSP90 immunoreactivity. There were no significant differences between PACs and MACNECs. None of the above molecular findings correlated with the cytogenetic clusters.

Molecular and immunohistochemical characteristics of PACs: a FISH analysis showing c-MYC amplifications; b immunohistochemical loss of DCC expression in liver metastasis (regular DCC expression in adjacent liver tissue, depicting red); c PAC (case 40) with loss of PMS2 expression in tumor cells, while intratumoral lymphocytes show regular nuclear PMS2 positivity (depicting red); d, e k-ras mutations in two cases (indicated by arrows)

Putative therapeutic targets and predictors in PACs: a membrane-bound expression of EGFR; b loss of nuclear MGMT in one tumor (regular MGMT expression in another tumor (insert)); c L1CAM positive tumor; d expression of HSP90
Discussion
PAC is a phenotypically well-characterized neoplasm of the pancreas, but its carcinogenesis is not yet well understood. By analyzing a comprehensive series of PACs with various molecular tests, we show that these tumors, including cystic PACs and MACNECs, have a microsatellite stable, but chromosomal highly unstable (CIN) genotype, defined by a number of recurrent chromosomal imbalances, including losses of 1p, 3p, 4q, 5q, 6q, 8p, 9p, 11q, 13q, 16q, 18p, and 18q, as well as gains of 1q, 7p, 7q, 8q, 12p, 12q, 17q, and 20q. Hierarchical clustering of the CGH findings revealed subgroups which, however, showed no correlations with clinical or pathological features such as sex, age, tumor size, spread at the time of diagnosis, mitotic count, or necrosis. In particular, no differences were detected between pure solid PACs, cystic PACs, and MACNECs, indicating that these subtypes have the same cytogenetic background.
As PACs may not only display a mixed acinar and neuroendocrine differentiation but rarely may also contain ductal elements [4, 8, 11], we compared the CGH findings in PACs with those previously reported in PDACs and PNENs. This revealed some cytogenetic overlap between PACs, PDACs, and PNENs, including, among others, gains of 7 and 20q, as well as loss of 6q. However, the cytogenetic differences outnumbered the similarities. Imbalances highly characteristic for PACs included gains of 1q and 22p, as well as losses of 1p, 5q, and 16q. In contrast, loss of 17p was highly characteristic of PDACs, as was gain of 14q for PNENs. Our findings therefore confirm the notion, as mentioned above, that PACs are genetically clearly distinct from PDACs and PNENs. As shown in the Supplement, PACs furthermore differ cytogenetically from salivary gland acinar cell carcinomas, which only infrequently display few chromosomal imbalances.
In the present series of PACs, several chromosomal imbalances were associated with tumor size exceeding 4 cm and with metastatic tumor, indicating a possible role during tumor progression. Thus, chromosomal aberrations found more frequently in metastases than in primaries included gains of 12p11–13, 12q12–14, 12q23–24, 20q11–12, and 22q11–13, as well as loss of 4q28. Gain of 20q12 was also observed to occur significantly more frequent in nodal metastases than in primary tumors in gastric carcinomas [42]. A possible candidate gene in this region is TNFRSF6B, which is located at 20q12.3 and was found to be overexpressed in gastric carcinomas with nodal metastases [43]. Whether TNFRSF6B, a member of the tumor necrosis factor superfamily binding FasL and inhibiting apoptosis [44], plays a role in PACS remains to be analyzed. In breast carcinomas, gain of 12q24 correlated with distant metastases and poor clinical outcome [45], and in stomach [42, 46, 47] and lung carcinomas [48, 49], loss of 4q was associated with metastases. However, candidate genes in these areas remain to be identified.
Guided by the frequently affected chromosomal regions identified in our CGH analyses, we examined a number of putative candidate genes. Loss of 18q was one of the most frequent findings in our series (51 % of cases) and in previous studies (five of eight cases) [22, 23]. PDACs, in which monosomy 18 is the most common cytogenetic aberration, frequently harbor mutations of the SMAD4 gene on chromosome 18q21.1, resulting in loss or reduced function of the Smad4 protein [34, 50]. As previously shown, deregulation of Smad4 plays no significant role in PACs, since loss of Smad4 was found in only 1 of 41 cases [12, 13, 17, 18], and SMAD4 mutations were detected in only few tumors [19]. However, 79 % of the tumors of the present series showed complete loss or significant reduction of the deleted in colorectal carcinoma (DCC) protein. The DCC gene is located at 18q21.3 and encodes for a transmembrane protein functioning as receptor for the axonal chemoattractant netrin-1 [51–53]. As loss of DCC occurs independently from loss of Smad4 in a subset of PDACs, this genetic change was thought to be a late carcinogenic event in these tumors, being associated with distant metastases [52]. Our data suggest that, in contrast to PDAC, dysregulation of DCC may be an early step in the development of PACs and thus may play a crucial role in their pathogenesis.
Gains of 8q were another frequent finding, accumulating in metastatic PACs. Using FISH, we show that c-MYC located at 8q24 is amplified (high or low) in 17 % of the tumors. The c-MYC oncogene is involved in the control of cell proliferation and differentiation and is commonly deregulated in PDACs [54]. In PDACs, overexpression of the MYC protein could be correlated with the histomorphological grade of the tumors, whereas no correlations have been detected between c-MYC expression and tumor stage or metastatic lesions [54].
PACs have frequently metastasized at the time of diagnosis or develop metastases during follow-up [1–4, 6]. When compared with PDACs, higher resectability rates can be achieved for PACs [1–3]. Moreover, some PAC patients seem to benefit from radiotherapy and chemotherapy [2, 1, 6]. However, tumor relapse is common [6] and, in general, therapeutic response rates are disappointing, urging the need for additional therapeutic targets in PACs [55]. Due to the rarity of PACs, prospective therapeutic trials are difficult to perform. We tested molecular markers that displayed promising clinical or preclinical results in other, more frequent tumors. Expression of EGFR was detected by immunohistochemistry in 42 % of the PACs. This suggests that some PACs and MACNECs might be eligible for an anti-EGFR-directed therapy [56]. It is known that patients may not benefit from this therapy, if their tumors bear K-RAS mutations [56]. However, as K-RAS mutations were detected in only two cases of our series of PACs (both in EGFR-positive tumors), this adverse effect may not play a significant role in PACs. Mutations of EGFR were not detected; therefore, a therapy with EGFR kinase inhibitors is unlikely to show any response [57].
Loss of nuclear MGMT expression was seen in 26 % of the PACs. In PNENs, loss of expression of MGMT by immunohistochemistry was shown to be a good indicator of MGMT deficiency, the latter being associated with a better response to chemotherapy with alkylating substances [58]. Further analyses, including evaluation of MGMT methylation, should be performed in PACs and MACNECs, to investigate whether the loss of MGMT immunoreactivity, which has been observed in a relatively high percentage of these tumors, might be indicative of MGMT deficiency which would open options for therapy.
HSP90 expression was observed in 98 % of PACs. In a previous study, we demonstrated HSP90 expression in 95 % of PNENs and provided functional evidence that HSP90 targeting shows significant therapeutic effects in vitro [59]. Thus, treatment of PNEN cell lines with HSP90 inhibitors, 17-AAG and 17 DMAG, resulted in reduction of cell viability, cell cycle arrest, and increased apoptosis [59]. Furthermore, HSP90 inhibition led to degradation and inactivation of several oncogenic client proteins and an increased therapeutic efficacy of the conventional chemotherapeutic agents 5-FU and doxorubicin [59]. HSP90 expression has also been shown in various other neoplasms, including PDACs [60]. In non-neoplastic pancreatic tissue, including acinar cells, HSP90 is expressed at a lower level [59]. Data from preclinical trials with HSP90 inhibitors are promising [61–63], and phase III clinical trials have been initiated [62].
Finally, we detected L1CAM expression in 72 % of the tumors of the present series. The adhesion molecule L1CAM (CD171) is expressed in a variety of neoplasms, including among others PDAC [64], anaplastic pancreatic cancer [65], PNEN [66], ovarian cancer [67], and colon cancer [68]. L1CAM expression has been associated with poor prognosis [69, 66, 70, 71]. In PDAC, it was shown to account for chemoresistance and to contribute to a more migratory phenotype [72, 73]. In a preclinical in vivo setting, targeted therapy with an antibody directed against L1CAM achieved promising therapeutic effects in ovarian cancer, resulting in effective inhibition of intraperitoneal tumor growth and tumor dissemination [74].
Using a BRAF V600E mutation-specific antibody, no tumor showed immunoreactivity, and there was no expression of Her2/neu. Although BRAF mutations were previously reported in three PACs [19], it seems unlikely that therapies directed against activated BRAF or Her2/neu, as established for other tumors like malignant melanoma or breast carcinoma [75, 76] will play a significant therapeutic role in PAC.
In summary, we show that PACs (including MACNECs) display a microsatellite stable, chromosomal unstable genotype, characterized by recurrent chromosomal imbalances, which, despite some overlap, allows clear discrimination of PACs from PDACs and PNENs. We show that PACs are frequently affected by alterations of DCC and, in a smaller number, also harbor c-MYC amplifications. We provide molecular evidence that PACs might be amenable for well-established therapies using EGFR antagonists or alkylating substances, as well as for innovative therapies directed against L1CAM or HSP90, whereas therapeutic strategies aiming at EGFR kinases, BRAF, or Her2/neu are unlikely to play a significant role in these tumors. Finally, our CGH data on salivary gland acinar cell tumors (shown in Supplement) demonstrate that these tumors are genetically distinct from PACs, suggesting profound differences in the cytogenetic background and molecular pathogenesis of these two acinar cell neoplasms.
References
Wisnoski NC, Townsend CM Jr, Nealon WH, Freeman JL, Riall TS (2008) 672 patients with acinar cell carcinoma of the pancreas: a population-based comparison to pancreatic adenocarcinoma. Surgery 144(2):141–148. doi:10.1016/j.surg.2008.03.006
Schmidt CM, Matos JM, Bentrem DJ, Talamonti MS, Lillemoe KD, Bilimoria KY (2008) Acinar cell carcinoma of the pancreas in the United States: prognostic factors and comparison to ductal adenocarcinoma. J Gastrointest Surg 12(12):2078–2086. doi:10.1007/s11605-008-0705-6
Kitagami H, Kondo S, Hirano S, Kawakami H, Egawa S, Tanaka M (2007) Acinar cell carcinoma of the pancreas: clinical analysis of 115 patients from pancreatic cancer registry of Japan pancreas society. Pancreas 35(1):42–46. doi:10.1097/mpa.0b013e31804bfbd3
Klimstra DS, Heffess CS, Oertel JE, Rosai J (1992) Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol 16(9):815–837
Osborne BM, Culbert SJ, Cangir A, MacKay B (1977) Acinar cell carcinoma of the pancreas in a 9-year-old child: case report with electron microscopic observations. South Med J 70(3):370–372
Lowery MA, Klimstra DS, Shia J, Yu KH, Allen PJ, Brennan MF, O’Reilly EM (2011) Acinar cell carcinoma of the pancreas: new genetic and treatment insights into a rare malignancy. Oncologist 16(12):1714–1720. doi:10.1634/theoncologist.2011-0231
Klimstra DS, Hruban RH, Klöppel G, Morohoshi T, Ohike N (2010) Acinar cell neoplasms of the pancreas. In: Bosman FT, Carneiro F, Hruban RH, Theise ND (eds) WHO classification of tumours of the digestive system. International Agency for Research on Cancer, Lyon, pp 314–318
La Rosa S, Adsay V, Albarello L, Asioli S, Casnedi S, Franzi F, Marando A, Notohara K, Sessa F, Vanoli A, Zhang L, Capella C (2012) Clinicopathologic study of 62 acinar cell carcinomas of the pancreas: insights into the morphology and immunophenotype and search for prognostic markers. Am J Surg Pathol 36(12):1782–1795. doi:10.1097/PAS.0b013e318263209d
Colombo P, Arizzi C, Roncalli M (2004) Acinar cell cystadenocarcinoma of the pancreas: report of rare case and review of the literature. Hum Pathol 35(12):1568–1571
Stelow EB, Shaco-Levy R, Bao F, Garcia J, Klimstra DS (2010) Pancreatic acinar cell carcinomas with prominent ductal differentiation: Mixed acinar ductal carcinoma and mixed acinar endocrine ductal carcinoma. Am J Surg Pathol 34(4):510–518. doi:10.1097/PAS.0b013e3181cfcac7
Ohike N, Kosmahl M, Kloppel G (2004) Mixed acinar-endocrine carcinoma of the pancreas A clinicopathological study and comparison with acinar-cell carcinoma. Virchows Arch 445(3):231–235. doi:10.1007/s00428-004-1037-x
Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, Brennan M, Cameron JL, Klimstra DS (2002) Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol 160(3):953–962
Rigaud G, Moore PS, Zamboni G, Orlandini S, Taruscio D, Paradisi S, Lemoine NR, Kloppel G, Scarpa A (2000) Allelotype of pancreatic acinar cell carcinoma. Int J Cancer 88(5):772–777
Terhune PG, Heffess CS, Longnecker DS (1994) Only wild-type c-Ki-ras codons 12, 13, and 61 in human pancreatic acinar cell carcinomas. Mol Carcinog 10(2):110–114
Hoorens A, Lemoine NR, McLellan E, Morohoshi T, Kamisawa T, Heitz PU, Stamm B, Ruschoff J, Wiedenmann B, Kloppel G (1993) Pancreatic acinar cell carcinoma. An analysis of cell lineage markers, p53 expression, and Ki-ras mutation. Am J Pathol 143(3):685–698
Pellegata NS, Sessa F, Renault B, Bonato M, Leone BE, Solcia E, Ranzani GN (1994) K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res 54(6):1556–1560
Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, Bassi C, Lemoine NR, Scarpa A (2001) Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. Br J Cancer 84(2):253–262. doi:10.1054/bjoc.2000.1567
de Wilde RF, Ottenhof NA, Jansen M, Morsink FH, de Leng WW, Offerhaus GJ, Brosens LA (2011) Analysis of LKB1 mutations and other molecular alterations in pancreatic acinar cell carcinoma. Mod Pathol 24(9):1229–1236. doi:10.1038/modpathol.2011.83
Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JR, Herman JG, Poh W, Pelosof L, Wolfgang CL, Vogelstein B, Kinzler KW, Hruban RH, Papadopoulos N, Wood LD (2014) Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol 232(4):428–435. doi:10.1002/path.4310
Furlan D, Sahnane N, Bernasconi B, Frattini M, Tibiletti MG, Molinari F, Marando A, Zhang L, Vanoli A, Casnedi S, Adsay V, Notohara K, Albarello L, Asioli S, Sessa F, Capella C, La Rosa S (2014) APC alterations are frequently involved in the pathogenesis of acinar cell carcinoma of the pancreas, mainly through gene loss and promoter hypermethylation. Virchows Arch. doi:10.1007/s00428-014-1562-1
Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee JH, Yeo CJ, Cameron JL, Hruban RH (2001) Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol 159(5):1619–1627
Kitoh H, Ryozawa S, Harada T, Kondoh S, Furuya T, Kawauchi S, Oga A, Okita K, Sasaki K (2005) Comparative genomic hybridization analysis for pancreatic cancer specimens obtained by endoscopic ultrasonography-guided fine-needle aspiration. J Gastroenterol 40(5):511–517. doi:10.1007/s00535-005-1577-0
Taruscio D, Paradisi S, Zamboni G, Rigaud G, Falconi M, Scarpa A (2000) Pancreatic acinar carcinoma shows a distinct pattern of chromosomal imbalances by comparative genomic hybridization. Genes Chromosomes Cancer 28(3):294–299
Rieker RJ, Aulmann S, Penzel R, Schnabel PA, Blaeker H, Esposito I, Morresi-Hauf A, Otto HF, Hecker E, Dienemann H, Schirmacher P, Mechtersheimer G (2005) Chromosomal imbalances in sporadic neuroendocrine tumours of the thymus. Cancer Lett 223(1):169–174. doi:10.1016/j.canlet.2004.10.027
Huszar M, Moldenhauer G, Gschwend V, Ben-Arie A, Altevogt P, Fogel M (2006) Expression profile analysis in multiple human tumors identifies L1 (CD171) as a molecular marker for differential diagnosis and targeted therapy. Hum Pathol 37(8):1000–1008. doi:10.1016/j.humpath.2006.03.014
Bergmann F, Aulmann S, Wente MN, Penzel R, Esposito I, Kleeff J, Friess H, Schirmacher P (2006) Molecular characterisation of pancreatic ductal adenocarcinoma in patients under 40. J Clin Pathol 59(6):580–584. doi:10.1136/jcp.2005.027292
Warth A, Penzel R, Brandt R, Sers C, Fischer JR, Thomas M, Herth FJ, Dietel M, Schirmacher P, Blaker H (2012) Optimized algorithm for Sanger sequencing-based EGFR mutation analyses in NSCLC biopsies. Virchows Arch 460(4):407–414. doi:10.1007/s00428-012-1219-x
Findeisen P, Kloor M, Merx S, Sutter C, Woerner SM, Dostmann N, Benner A, Dondog B, Pawlita M, Dippold W, Wagner R, Gebert J, von Knebel DM (2005) T25 repeat in the 3′ untranslated region of the CASP2 gene: a sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer Res 65(18):8072–8078. doi:10.1158/0008-5472.CAN-04-4146
Gunawan B, von Heydebreck A, Fritsch T, Huber W, Ringert RH, Jakse G, Fuzesi L (2003) Cytogenetic and morphologic typing of 58 papillary renal cell carcinomas: evidence for a cytogenetic evolution of type 2 from type 1 tumors. Cancer Res 63(19):6200–6205
Gunawan B, von Heydebreck A, Sander B, Schulten HJ, Haller F, Langer C, Armbrust T, Bollmann M, Gasparov S, Kovac D, Fuzesi L (2007) An oncogenetic tree model in gastrointestinal stromal tumours (GISTs) identifies different pathways of cytogenetic evolution with prognostic implications. J Pathol 211(4):463–470. doi:10.1002/path.2128
von Heydebreck A, Gunawan B, Fuzesi L (2004) Maximum likelihood estimation of oncogenetic tree models. Biostatistics 5(4):545–556. doi:10.1093/biostatistics/kxh007
Harada T, Okita K, Shiraishi K, Kusano N, Furuya T, Oga A, Kawauchi S, Kondoh S, Sasaki K (2002) Detection of genetic alterations in pancreatic cancers by comparative genomic hybridization coupled with tissue microdissection and degenerate oligonucleotide primed polymerase chain reaction. Oncology 62(3):251–258
Mahlamaki EH, Hoglund M, Gorunova L, Karhu R, Dawiskiba S, Andren-Sandberg A, Kallioniemi OP, Johansson B (1997) Comparative genomic hybridization reveals frequent gains of 20q, 8q, 11q, 12p, and 17q, and losses of 18q, 9p, and 15q in pancreatic cancer. Gene Chromosome Cancer 20(4):383–391
Schleger C, Arens N, Zentgraf H, Bleyl U, Verbeke C (2000) Identification of frequent chromosomal aberrations in ductal adenocarcinoma of the pancreas by comparative genomic hybridization (CGH). J Pathol 191(1):27–32. doi:10.1002/(sici)1096-9896(200005)191:1<27::aid-path582>3.0.co;2-j
Shiraishi K, Okita K, Kusano N, Harada T, Kondoh S, Okita S, Ryozawa S, Ohmura R, Noguchi T, Iida Y, Akiyama T, Oga A, Fukumoto Y, Furuya T, Kawauchi S, Sasaki K (2001) A comparison of DNA copy number changes detected by comparative genomic hybridization in malignancies of the liver, biliary tract and pancreas. Oncology 60(2):151–161
Fukushige S, Waldman FM, Kimura M, Abe T, Furukawa T, Sunamura M, Kobari M, Horii A (1997) Frequent gain of copy number on the long arm of chromosome 20 in human pancreatic adenocarcinoma. Genes Chromosomes Cancer 19(3):161–169
Zhao J, Moch H, Scheidweiler AF, Baer A, Schaffer AA, Speel EJ, Roth J, Heitz PU, Komminoth P (2001) Genomic imbalances in the progression of endocrine pancreatic tumors. Genes Chromosomes Cancer 32(4):364–372
Speel EJ, Richter J, Moch H, Egenter C, Saremaslani P, Rutimann K, Zhao J, Barghorn A, Roth J, Heitz PU, Komminoth P (1999) Genetic differences in endocrine pancreatic tumor subtypes detected by comparative genomic hybridization. Am J Pathol 155(6):1787–1794
Stumpf E, Aalto Y, Hoog A, Kjellman M, Otonkoski T, Knuutila S, Andersson LC (2000) Chromosomal alterations in human pancreatic endocrine tumors. Genes Chromosomes Cancer 29(1):83–87. doi:10.1002/1098-2264(2000)9999:9999<::AID-GCC1011>3.0.CO;2-Z [pii]
Tonnies H, Toliat MR, Ramel C, Pape UF, Neitzel H, Berger W, Wiedenmann B (2001) Analysis of sporadic neuroendocrine tumours of the enteropancreatic system by comparative genomic hybridisation. Gut 48(4):536–541
Pourani J, Kaserer K, Pfragner R (2002) Cytogenetic and molecular analyses of multiple endocrine neoplasias of the MEN1 syndrome. Int J Oncol 20(5):971–976
Liu XP, Li DY, Liu XL, Xu JD, Furuya T, Kawauchi S, Oga A, Sasaki K (2009) Comparison of chromosomal aberrations between primary tumors and their synchronous lymph-node metastases in intestinal-type gastric carcinoma. Pathol Res Pract 205(2):105–111. doi:10.1016/j.prp.2008.09.003
Buffart TE, van Grieken NC, Tijssen M, Coffa J, Ylstra B, Grabsch HI, van de Velde CJ, Carvalho B, Meijer GA (2009) High resolution analysis of DNA copy-number aberrations of chromosomes 8, 13, and 20 in gastric cancers. Virchows Arch 455(3):213–223. doi:10.1007/s00428-009-0814-y
Pitti RM, Marsters SA, Lawrence DA, Roy M, Kischkel FC, Dowd P, Huang A, Donahue CJ, Sherwood SW, Baldwin DT, Godowski PJ, Wood WI, Gurney AL, Hillan KJ, Cohen RL, Goddard AD, Botstein D, Ashkenazi A (1998) Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 396(6712):699–703. doi:10.1038/25387
Aubele M, Auer G, Braselmann H, Nahrig J, Zitzelsberger H, Quintanilla-Martinez L, Smida J, Walch A, Hofler H, Werner M (2002) Chromosomal imbalances are associated with metastasis-free survival in breast cancer patients. Anal Cell Pathol 24(2–3):77–87
Kimura Y, Noguchi T, Kawahara K, Kashima K, Daa T, Yokoyama S (2004) Genetic alterations in 102 primary gastric cancers by comparative genomic hybridization: gain of 20q and loss of 18q are associated with tumor progression. Mod Pathol 17(11):1328–1337. doi:10.1038/modpathol.3800180
Kang JU, Kang JJ, Kwon KC, Park JW, Jeong TE, Noh SM, Koo SH (2006) Genetic alterations in primary gastric carcinomas correlated with clinicopathological variables by array comparative genomic hybridization. J Korean Med Sci 21(4):656–665
Goeze A, Schluns K, Wolf G, Thasler Z, Petersen S, Petersen I (2002) Chromosomal imbalances of primary and metastatic lung adenocarcinomas. J Pathol 196(1):8–16. doi:10.1002/path.1009
Wrage M, Ruosaari S, Eijk PP, Kaifi JT, Hollmen J, Yekebas EF, Izbicki JR, Brakenhoff RH, Streichert T, Riethdorf S, Glatzel M, Ylstra B, Pantel K, Wikman H (2009) Genomic profiles associated with early micrometastasis in lung cancer: relevance of 4q deletion. Clin Cancer Res 15(5):1566–1574. doi:10.1158/1078-0432.ccr-08-2188
Bardeesy N, DePinho RA (2002) Pancreatic cancer biology and genetics. Nat Rev Cancer 2(12):897–909. doi:10.1038/nrc949
Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, Simons J, Bronson RT, Gordon JI, Tessier-Lavigne M, Weinberg RA (1997) Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature 386(6627):796–804. doi:10.1038/386796a0
Tarafa G, Villanueva A, Farre L, Rodriguez J, Musulen E, Reyes G, Seminago R, Olmedo E, Paules AB, Peinado MA, Bachs O, Capella G (2000) DCC and SMAD4 alterations in human colorectal and pancreatic tumor dissemination. Oncogene 19(4):546–555. doi:10.1038/sj.onc.1203353
Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, Tessier-Lavigne M (1996) Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell 87(2):175–185
Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U (2002) c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol 15(4):462–469. doi:10.1038/modpathol.3880547
Holen KD, Klimstra DS, Hummer A, Gonen M, Conlon K, Brennan M, Saltz LB (2002) Clinical characteristics and outcomes from an institutional series of acinar cell carcinoma of the pancreas and related tumors. J Clin Oncol 20(24):4673–4678
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359(17):1757–1765. doi:10.1056/NEJMoa0804385
Tsuchihashi Z, Khambata-Ford S, Hanna N, Janne PA (2005) Responsiveness to cetuximab without mutations in EGFR. N Engl J Med 353(2):208–209. doi:10.1056/NEJM200507143530218
Kulke MH, Hornick JL, Frauenhoffer C, Hooshmand S, Ryan DP, Enzinger PC, Meyerhardt JA, Clark JW, Stuart K, Fuchs CS, Redston MS (2009) O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res 15(1):338–345. doi:10.1158/1078-0432.CCR-08-1476
Mayer P, Harjung A, Breinig M, Fischer L, Ehemann V, Malz M, Scherubl H, Britsch S, Werner J, Kern MA, Blaker H, Schirmacher P, Bergmann F (2012) Expression and therapeutic relevance of heat-shock protein 90 in pancreatic endocrine tumors. Endocr Relat Cancer 19(3):217–232. doi:10.1530/ERC-11-0227
Song D, Chaerkady R, Tan AC, Garcia-Garcia E, Nalli A, Suarez-Gauthier A, Lopez-Rios F, Zhang XF, Solomon A, Tong J, Read M, Fritz C, Jimeno A, Pandey A, Hidalgo M (2008) Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer. Mol Cancer Ther 7(10):3275–3284. doi:10.1158/1535-7163.MCT-08-0508
Hollingshead M, Alley M, Burger AM, Borgel S, Pacula-Cox C, Fiebig HH, Sausville EA (2005) In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother Pharmacol 56(2):115–125. doi:10.1007/s00280-004-0939-2
Usmani SZ, Bona R, Li Z (2009) 17 AAG for HSP90 inhibition in cancer—from bench to bedside. Curr Mol Med 9(5):654–664
Trepel J, Mollapour M, Giaccone G, Neckers L (2010) Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10(8):537–549. doi:10.1038/nrc2887
Bergmann F, Wandschneider F, Sipos B, Moldenhauer G, Schniewind B, Welsch T, Schirrmacher P, Kloppel G, Altevogt P, Schafer H, Sebens Muerkoster S (2010) Elevated L1CAM expression in precursor lesions and primary and metastastic tissues of pancreatic ductal adenocarcinoma. Oncol Rep 24(4):909–915
Bergmann F, Moldenhauer G, Herpel E, Gaida MM, Strobel O, Werner J, Esposito I, Muerkoster SS, Schirmacher P, Kern MA (2010) Expression of L1CAM, COX-2, EGFR, c-KIT and Her2/neu in anaplastic pancreatic cancer: putative therapeutic targets? Histopathology 56(4):440–448. doi:10.1111/j.1365-2559.2010.03499.x
Kaifi JT, Zinnkann U, Yekebas EF, Schurr PG, Reichelt U, Wachowiak R, Fiegel HC, Petri S, Schachner M, Izbicki JR (2006) L1 is a potential marker for poorly-differentiated pancreatic neuroendocrine carcinoma. World J Gastroenterol 12(1):94–98
Gast D, Riedle S, Riedle S, Schabath H, Schlich S, Schneider A, Issa Y, Stoeck A, Fogel M, Joumaa S, Wenger T, Herr I, Gutwein P, Altevogt P (2005) L1 augments cell migration and tumor growth but not beta3 integrin expression in ovarian carcinomas. Int J Cancer 115(4):658–665. doi:10.1002/ijc.20869
Gavert N, Conacci-Sorrell M, Gast D, Schneider A, Altevogt P, Brabletz T, Ben-Ze’ev A (2005) L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol 168(4):633–642. doi:10.1083/jcb.200408051
Kaifi JT, Strelow A, Schurr PG, Reichelt U, Yekebas EF, Wachowiak R, Quaas A, Strate T, Schaefer H, Sauter G, Schachner M, Izbicki JR (2006) L1 (CD171) is highly expressed in gastrointestinal stromal tumors. Mod Pathol 19(3):399–406. doi:10.1038/modpathol.3800547
Boo YJ, Park JM, Kim J, Chae YS, Min BW, Um JW, Moon HY (2007) L1 expression as a marker for poor prognosis, tumor progression, and short survival in patients with colorectal cancer. Ann Surg Oncol 14(5):1703–1711. doi:10.1245/s10434-006-9281-8
Fogel M, Mechtersheimer S, Huszar M, Smirnov A, Abu-Dahi A, Tilgen W, Reichrath J, Georg T, Altevogt P, Gutwein P (2003) L1 adhesion molecule (CD 171) in development and progression of human malignant melanoma. Cancer Lett 189(2):237–247
Issa Y, Nummer D, Seibel T, Muerkoster SS, Koch M, Schmitz-Winnenthal FH, Galindo L, Weitz J, Beckhove P, Altevogt P (2009) Enhanced L1CAM expression on pancreatic tumor endothelium mediates selective tumor cell transmigration. J Mol Med 87(1):99–112. doi:10.1007/s00109-008-0410-7
Gast D, Riedle S, Issa Y, Pfeifer M, Beckhove P, Sanderson MP, Arlt M, Moldenhauer G, Fogel M, Kruger A, Altevogt P (2008) The cytoplasmic part of L1-CAM controls growth and gene expression in human tumors that is reversed by therapeutic antibodies. Oncogene 27(9):1281–1289. doi:10.1038/sj.onc.1210747
Arlt MJ, Novak-Hofer I, Gast D, Gschwend V, Moldenhauer G, Grunberg J, Honer M, Schubiger PA, Altevogt P, Kruger A (2006) Efficient inhibition of intra-peritoneal tumor growth and dissemination of human ovarian carcinoma cells in nude mice by anti-L1-cell adhesion molecule monoclonal antibody treatment. Cancer Res 66(2):936–943. doi:10.1158/0008-5472.CAN-05-1818
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, Group B-S (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516. doi:10.1056/NEJMoa1103782
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344(11):783–792. doi:10.1056/NEJM200103153441101
Acknowledgments
The authors thank Regine Brandt, Maike Pacena, Anja Bredtmann, Andrea Müller, Stefanie Keller, and the NCT tissue bank Heidelberg for excellent technical support.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 229 kb)
Rights and permissions
About this article

Cite this article
Bergmann, F., Aulmann, S., Sipos, B. et al. Acinar cell carcinomas of the pancreas: a molecular analysis in a series of 57 cases. Virchows Arch 465, 661–672 (2014). https://doi.org/10.1007/s00428-014-1657-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-014-1657-8