
Abstract
The bed nucleus of the stria terminalis (BNST) is a forebrain structure, involved in the modulation of neuroendocrine, cardiovascular and autonomic responses. One of the responses is baroreflex activity, which consists in a neural mechanism responsible for keeping the blood pressure within a narrow range of variation. It has been reported that blockade of BNST α1-adrenoceptors increased the bradycardic component of baroreflex. In addition, such receptors are able to modulate glutamate release in this structure. Interestingly, BNST NMDA receptor antagonism and neuronal nitric oxide synthase (nNOS) inhibition led to the same effect of the α1-adrenoceptors blockade on baroreflex bradycardic response. Therefore, the hypothesis of the present study is that BNST noradrenergic transmission interacts with NMDA/NO pathway through α1 adrenoceptors to modulate the baroreflex activity. Male Wistar rats had stainless steel guide cannulas bilaterally implanted in the BNST. Subsequently, a catheter was inserted into the femoral artery for cardiovascular recordings, and into the femoral vein for assessing baroreflex activation. Injection of the noradrenaline reuptake inhibitor reboxetine in the BNST did not modify the tachycardic, but significantly decreased the bradycardic component of baroreflex. Administration of an α1, but not an α2 antagonist into the BNST prior to reboxetine prevented this effect. Likewise, previous injection of NMDA/NO pathway blockers inhibited the effect of reboxetine on bradycardic response. In conclusion, it was demonstrated for the first time the existence of an interaction between BNST noradrenergic, glutamatergic and nitrergic neurotransmissions in the modulation of bradycardic baroreflex response.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The bed nucleus of the stria terminalis (BNST) was first described by Johnston as the stria terminalis surrounding grey matter. It is a limbic structure able to control emotional responses, most likely because of its reciprocal connections with the centromedial amygdala and incoming terminals from the hippocampus and medial prefrontal cortex (MPFC). These are supramedullary structures involved in behavioural, autonomic and cardiovascular activities [15, 46, 48].
Dunn and Williams were the first to show that electrical stimulation of more rostral sites of BNST-induced pressor responses [17]. Furthermore, the injection of a non-specific synaptic blocker in the BNST potentiated tachycardic response elicited by restraint stress in rats [12]. Additionally, it was described that pharmacological manipulation of BNST decreased tachycardia and blood pressure rise during conditioned emotional response to contextual fear [30]. Such results suggest that BNST could be related to short-term regulation of cardiovascular responses through the autonomic system. One of these responses is the baroreceptor reflex arc, which is a neural mechanism able to influence peripheral resistance and heart rate (HR) to keep blood pressure (BP) within a narrow range of variation. In fact, stressful stimuli can alter baroreflex sensitivity, by displacing HR responses towards higher values of BP [13]. Such response is mainly modulated by brain stem structures, and the BNST is believed to work as an anatomical relay between such areas and those aforementioned supramedullary structures in order to integrate autonomic activity [28, 48].
Indeed, the BNST modulates baroreflex activity, since the pharmacological inhibition of its synapses facilitated bradycardic response, while no alteration was observed in the tachycardic component [9]. However, cobalt chloride inhibits the synaptic release of neurotransmitters, since it precludes calcium entrance in neurons [34], turning impossible to know which area’s neurotransmission system is participating on this response. On the other hand, it is well established that BNST receives extensive noradrenergic projections from A1, A2 and A5 brain stem nuclei, as well as from locus coeruleus, which are the main source of noradrenaline in the brain [5, 37, 51].
It has been shown that noradrenaline injection into the BNST evoked vasopressine-dependent BP rise followed by bradycardia [10]. Furthermore, BP and HR responses were mediated by both α1- and α2-noradrenergic receptors [8]. Posteriorly, Crestani and colleagues demonstrated that BNST α1-, but not α2-receptor antagonism increased the baroreflex bradycardic response with no alteration in tachycardia [11], suggesting that such adrenoceptors are able to modulate the parasympathetic component of the baroreflex activity.
Additionally, electrophysiological and microdyalisis studies revealed that BNST noradrenergic transmission can influence excitatory potential and glutamate release [1, 18, 21, 22]. Interestingly, BNST NMDA glutamatergic receptor blockade led to baroreflex bradycardic response enhancement, the same effect observed after BNST α1 antagonism. Also, NMDA receptor modulation on bradycardic reflex was dependent on neuronal nitric oxide synthase (nNOS) and soluble guanylate cyclase (sGC) activation [2], since such glutamatergic receptors are able to engage nitric oxide (NO) neurotransmission in central nervous system [25].
Therefore, considering that BNST noradrenergic α1-receptor, as well as NMDA receptor/NO/sGC neurotransmissions modulate bradycardic baroreflex activity in the same way, and that noradrenaline is able to modulate glutamatergic release in the area, our hypothesis is there is an interaction between BNST α1-receptor and NMDA receptor/NO/sGC pathway in the modulation of baroreflex activity.
Materials and methods
Ethical approval and animals
Experimental procedures were carried out following protocols approved by the Ethical Review Committee of the School of Medicine of Ribeirão Preto (Protocol number 019/2015), which complies with the Guiding Principles for Research Involving Animals and Human Beings of the American Physiological Society. Male Wistar rats weighing 230–270 g were used. They were provided by the Central Biotery Service of the University of São Paulo. Animals were housed in plastic cages in a temperature-controlled room at 25 °C in the Animal Care Unit of the Department of Pharmacology, School of Medicine of Ribeirão Preto, University of São Paulo. They were kept under a 12:00 h light-dark cycle (lights on between 6:00 h and 18:00 h) and had water and food ad libitum.
Animal preparation
Four days before the experiment, rats were anaesthetized with tribromoethanol (250 mg kg−1, i.p., Sigma, St. Louis, Missouri, USA). After local anaesthesia with 2% lidocaine, the skull was surgically exposed, and stainless steel guide cannulae (26G) were bilaterally implanted into the BNST using a stereotaxic apparatus (Stoelting, Wood Dale, Illinois, USA). Stereotaxic coordinates for guide cannulas directed to BNST were selected from The Rat Brain Atlas of Paxinos and Watson and were antero-posterior = − 0.4 mm from bragma; lateral = 4.0 mm from the medial suture; and vertical = − 5.5 mm from the skull, with a lateral inclination of 23°. Cannulas were fixed to the skull with dental cement and one metal screw. After surgery, animals were treated with a polyantibiotic preparation of streptomycins (30 mg/0.3 mL)/penicillins (72,000 UI/0.3 mL) (i.m., Pentabiotico®, Fort Dodge, Campinas, São Paulo, Brazil) to prevent infection and with the non-steroidal anti-inflammatory flunixine meglumine (0.02 mg/0.3 mL) (s.c., Banamine®, Schering Plough, Cotia, São Paulo, Brazil) for analgesia. One day before the experiment, rats were anaesthetized with tribromoethanol (250 mg kg_1, i.p.), and a catheter (a 4-cm segment of PE-10 that was heat-bound to a 13-cm segment of PE-50, Clay Adams, Parsippany, New Jersey, USA) was inserted into the femoral artery, for recording BP. A second catheter was implanted into the femoral vein for the infusion of vasoactive substances. Both catheters were inserted under the skin and exteriorized on the animal’s dorsum and were fixed to the skin using suture line. After surgery, treatment with anti-inflammatory drugs was repeated.
Measurement of cardiovascular responses
Pulsatile arterial pressure of freely moving animals was recorded using an ML870 preamplifier (LabChart, USA) and an acquisition board (PowerLab, AD Instruments, USA) connected to a computer. Mean arterial pressure (MAP) and heart rate (HR) values were derived from pulsatile recordings and processed on-line.
Drug injection
Needles (33G, Small Parts, Miami Lakes, FL, USA) used for microinjection into the BNST were 1 mm longer than the guide cannulae and were connected to a 1-μL syringe (7002-H, Hamilton Co., Reno, NV, USA) through PE-10 tubing. The needle was carefully inserted into the guide cannula, and drugs were injected in a final volume of 100 nL over a 5-s period. The animals were not handled during drug administration and they stayed under the bedding to avoid animal stress during BNST infusion. After a 30-s period, the needle was removed and inserted into the second guide cannula for microinjection into the contralateral BNST.
Baroreflex assessment
The baroreflex was activated by phenylephrine (α1 adrenoceptor agonist; 50 μg kg−1; 0.34 mL min_1) or sodium nitroprusside (SNP) (NO donor; 50 μg kg_1; 0.8 mL min_1) infusion using an infusion pump (KD Scientific, Holliston, MA, USA). The phenylephrine or SNP infusion lasted 30–40 s and caused, respectively, an increase or decrease in BP.
Method used to evaluate baroreflex activity
Baroreflex curves were constructed, matching MAP variations with HR responses. Paired values for variations in MAP (ΔMAP) and HR (ΔHR) were plotted to create sigmoid curves for each rat, which were used to determine baroreflex activity [45]. To analyse bradycardic and tachycardic responses separately, HR values matching 10, 20, 30 and 40 mmHg of MAP changes were calculated [45]. Values were plotted to create linear regression curves for each rat, and their slopes were compared to determine changes in baroreflex gain.
Drugs
The following drugs were used: the neuronal noradrenaline reuptake inhibitor (reboxetine); Tocris, Westwoods Business Park Ellisville, MO, USA); a selective α1 receptor antagonist (WB4101; Tocris, Westwoods Business Park, Ellisville, MO, USA) and a selective α2 receptor antagonist (RX821002, Tocris, Westwoods Business Park Ellisville, MO, USA). In addition, a NMDA receptor antagonist (AP7, Tocris, Westwoods Business Park Ellisville, MO, USA); a nNOS inhibitor, n-propyl (n-propyl-L-arginine-NPLA, Tocris, Westwoods Business Park, Ellisville, MO, USA); a NO scavenger (c-PTIO Tocris, Westwoods Business Park, Ellisville, MO, USA) and a sGC inhibitor (ODQ, Tocris, Westwoods Business Park, Ellisville, MO, USA) were used. Phenylephrine–HCl (Sigma, St. Louis, MO, USA) and SNP (Sigma, St. Louis, MO, USA) were used for baroreflex assessment. All of these compounds were dissolved in sterile saline (0.9% NaCl). Tribromoethanol (Sigma, St. Louis, MO, USA) and urethane (Sigma, St. Louis, MO, USA) were dissolved in distilled water. The solutions were prepared immediately before use and were kept on ice and protected from light during the experimental sessions.
Experimental protocols
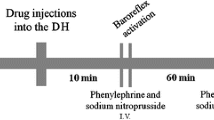
The animals were acclimated in experimental room for at least 30 min before the experimental session. All groups of animals used in our study received three sets of phenylephrine or SNP infusion to determine control values of baroreflex activity. Each animal received one or two microinjections in both BNST hemispheres: the pilot study was composed by an experimental group that received microinjections of 100 nL of reboxetine (2 nmol). In this group, the baroreflex activit was assessed before, 10 min and 60 min after bilateral microinjections into the BNST. Afterwards, other groups of animals were used to find sub-effective doses of the α1-receptor antagonist (WB4101, 5 nmol); α2-receptor antagonist (RX821002, 2 nmol), NMDA receptor antagonist (AP7, 0.1 nmol), nNOS inhibitor (NPLA, 0.04), NO scavenger (c-PTIO, 0.1 nmol) and sGC inhibitor (ODQ, 0.2 nmol). Such doses were found by lowering the effective concentrations that had been used on previous studies [2, 30]. The further experimental groups received a combination of two microinjections in each BNST hemisphere: vehicle of sub-effective doses of such compounds prior to reboxetine (2 nmol). One of these groups received microinjections of 100 nL of reboxetine (2 nmol) and vehicle (saline) or reboxetine (2 nmol) and WB4101 (5 nmol); the third group received microinjections of 100 nL of reboxetine (2 nmol) and vehicle (saline) or reboxetine (2 nmol) and RX821002 (2 nmol); the fourth group received microinjections of 100 nL of reboxetine (2 nmol) combined with either AP7 (0.1 nmol), or NPLA (0.04 nmol), or c-PTIO (0.1 nmol) or ODQ (0.2 nmol) or saline. We used 5 min of interval between the microinjections of sub-effective doses of blockers or vehicle and reboxetine. Phenylephrine and SNP infusion was repeated 10 and 60 min after the last bilateral BNST microinjection. Figure 1 shows the protocols of baroreflex stimulation combined with BNST microinjection times.

Original cardiovascular recording traces of pulsatile arterial pressure (PAP; mmHg), mean arterial pressure (MAP; mmHg) and heart rate (HR; beats per minute bpm) of one animal used in the study. The protocols used for baroreflex stimulation using vasoconstrictor agents (phenilephryne and sodium nitroprusside) combined with BNST microinjections intervals are depicted
Histological procedure
At the end of the experiments, the rats were anaesthetized with urethane (1.25 g kg−1, i.p.), and 100 nL of 1% Evans blue dye was bilaterally injected into the BNST as a marker of injection sites. The chest was surgically opened; the descending aorta occluded; the right atrium severed, and the brain perfused with 10% formalin through the left ventricle. Brains were post-fixed for 24 h at 4 °C, and 40-μm sections were cut with a cryostat (CM-1900, Leica, Wetzlar, Germany). The actual placement of the injection needles was verified in serial sections, according to the Rat Brain Atlas of Paxinos and Watson, 1997.
Data analysis
Baseline cardiovascular values before and after pharmacological treatment in the BNST were compared using Student’s t test. Baroreflex activity was analysed using sigmoid curves which were characterized with five parameters: (i) P1 (beats min−1) lower heart rate plateau, (ii) P2 (beats min−1) upper heart rate plateau; (iii) heart rate range (beats min−1), difference between upper and lower plateau levels (ΔP); and (iv) average baroreflex gain (G, beats min−1 mmHg−1), which is the average slopes of the non-linear curves; mean blood pressure (BP50, mmHg), which is the BP value matching with 50% of variation in HR. Significant differences among sigmoid curves or linear regression parameters were analysed using one-way ANOVA followed by the Dunnett’s post hoc test. The slope of linear regression curves (Δ HR vs. Δ MAP) before, 10 and 60 min after microinjection of each treatment was determined, and results were analysed to detect alterations in cardiac baroreflex gain using one-way ANOVA followed by Dunnett’s post hoc test. Results of statistical tests where P < 0.05 were considered significant.
Results
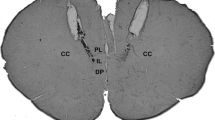
Figure 2 shows a diagrammatic representation of the microinjection sites in BNST of all animals used in the present study. Due to the amount of animals used, some injections sites are overlapped. In addition, all animals had guide cannulas correctly implanted in the area, apart from the groups in which cannulas were targeted to BNST surrounding structures.

a Photomicrography of a positive BNST stereotaxic implantation of one animal used in the study. b Diagramatic representations of the BNST coronal sections showing overlapping microinjection sites of all animals used in this study. The coordinates were based on the rat brain atlas of Paxinos and Watson (2007)
Effects of BNST noradrenaline reuptake inhibition on cardiac baroreflex activity in awake rats
Noradrenaline reuptake inhibitor, reboxetine (2 nmol), was administered into the BNST (n = 7). This dose of reboxetine was based on the study of Hott and colleagues (2012). We observed no alteration either in HR (before = 370 ± 10; after = 370 ± 9 bpm; t = 0.34; P > 0.05) or MAP (before = 99 ± 3.01; after = 100 ± 2.72 mmHg; t = 0.92; P > 0.05) basal levels. On the other hand, blockade of BNST noradrenaline reuptake decreased the slope of the linear regression curve of the bradycardic response (before = − 2.53 ± 0.26; 10 min = − 1.70 ± 0.31; F(2,17) = 13.79; P < 0.01) (Fig. 2; Table 1). The slope of the bradycardic curve returned to the basal levels 60 min after the microinjection (before = − 2.53 ± 0.26; 60 min = − 2.53 ± 0.25; F(2,17) = 12.39; P > 0.05) (Fig. 3; Table 1). Some parameters of the non-linear regression curve (G, P1, ΔP) were also decreased (Fig. 3; Table 1). However, this same pharmacological manipulation was unable to change the slope of the tachycardic linear regression curve (before = − 2.03 ± 0.26; 10 min = − 2.08 ± 0.16; 60 min: − 1.83 ± 0.16; F(2,17) = 11.20; P < 0.05) (Fig. 3).

Linear regression and sigmoidal curves correlating the responses of ΔMAP and ΔHR before, 10 min and 60 min after bilateral microinjection of reboxetine (2 nmol, n = 7) in the BNST—(upper) Correlation r2 values for bradycardic linear regression curves were 0.82 (before); 0.67 (10 min after); 0.83 (60 min). Correlation r2 values for tachycardic linear regression curves were 0.73 (before); 0.90 (10 min); and 0.81 (60 min). (lower) Sigmoid curves r2 correlation values were 0.92 (before); 0.79 (10 min); and 0.94 (60 min). Values are means ± SEM. bpm, beats min1. The symbols on sigmoid curves represent BP50
Characterization of ineffective doses of NMDA/NO pathway inhibitors and α1- and α2-receptors antagonists administered into the BNST on cardiac baroreflex activity in awake rats
In order to determine which BNST noradrenergic receptor is mediating the effects of reboxetine, we sought for sub-effective doses of α1- and α2-receptors antagonists on the baroreflex activity. Such doses were found by reducing the concentrations used in the study of Hott and co-workers (2012) [30]. Injection of the α1 antagonist (WB4101; 5 nmol, n = 5) in the BNST did not alter the baroreflex bradycardic (before = 1.74 ± 0.39; 10 min after = 1.71 ± 0.26; 60 min after = 1.69 ± 0.52; F(2,14) = 1.04; P > 0.05) or tachycardic responses (before = − 1.51 ± 0.49; 10 min after = − 1.55 ± 1.08; 60 min after = − 1.58 ± 1.77; F(2,14) = 0.10; P > 0.05) (graphs not shown). Antagonism of BNST α2 receptors with an equimolar dose (5 pmol; n = 5) did not displace the bradycardic (before = − 1.67 ± 0.53; 10 min after = − 1.71 ± 0.76; 60 min after = − 1.69 ± 0.81; F(2,14) = 0.37; P > 0.05) or the tachycardic (before = − 1.21 ± 1.29; 10 min after = − 1.22 ± 0.87; 60 min after = − 1.24 ± 0.82; F(2,14) = 0.68; P > 0.05) linear regression curves (graphs not shown). Moreover, we looked for sub-effective doses of NMDA/NO pathway inhibitors in order to study a possible interaction with the noradrenergic system inside the BNST. The doses were found by lowering the concentration used in the work of Hott and colleagues [31] Administration of NMDA receptor antagonist (AP7 0.1 nmol, n = 5) into the area did not change the bradycardic (before = − 2.00 ± 0.29; 10 min after = − 1.98 ± 0.42; 60 min after = − 1.99 ± 0.26; F(2,14) = 1.79; P > 0.05) nor tachycardic slopes of regression curves (before = − 1.33 ± 0.09; 10 min after = − 1.32 ± 0.12; 60 min after = − 1.35 ± 0.16; F(2,14) = 0.22; P > 0.05). In addition, nNOS inhibition with NPLA (0.04 nmol, n = 5) in the BNST was unable to modify both bradycardic (before = − 1.71 ± 0.39; 10 min after = − 1.69 ± 0.32; 60 min after = − 1.72 ± 0.26; F(2,14) = 1.43; P > 0.05) and tachycardic regression line slopes (before = − 2.33 ± 0.29; 10 min after = − 2.28 ± 0.34; 60 min after = − 2.12 ± 0.27; F(2,14) = 1.57; P > 0.05). Administration of NO scavenger, c-PTIO (0.1 nmol; n = 5) into the BNST did not affect the bradycardic (before = − 1.11 ± 0.19; 10 min after = − 1.15 ± 0.22; 60 min after = − 1.13 ± 0.16; F(2,14) = 1.99; P > 0.05) or tachycardic (before = − 2.28 ± 0.34; 10 min after = − 2.35 ± 0.20; 60 min after = − 2.23 ± 0.13; F(2,14) = 3.62; P > 0.05) baroreflex responses (data not shown). Likewise, the sGC inhibitor ODQ (0.2 nmol; n = 5) did not affect the bradycardic (before = − 1.86 ± 0.43; 10 min after = − 1.84 ± 0.25; 60 min after = − 1.82 ± 0.38; F(2,14) = 2.89; P > 0.05) or tachycardic responses (before = − 1.97 ± 0.63; 10 min after = − 1.90 ± 0.72; 60 min after = − 2.01 ± 0.64; F(2,14) = 0.99; P > 0.05) responses (graphs not shown). Sigmoid curve parameters (G, P1, P2, ∆P, BP50) were not modified by either of these compounds administered in the BNST at their respective doses (graphs not shown).
Effects of α1-receptor antagonism in the BNST prior to noradrenaline reuptake inhibition on cardiac baroreflex activity in awake rats
In the control group, microinjection of the noradrenaline reuptake inhibitor, reboxetine (2 nmol), preceded by vehicle (saline) (n = 6) in the BNST did not affect the basal levels of HR (before = 351 ± 10; after = 353 ± 11 bpm; t = 1.56; P > 0.05) and MAP (before = 102 ± 2.98; after = 105 ± 2.56 mmHg; t = 3.27; P > 0.05). The slope of the linear regression related to the bradycardic (before = − 2.53 ± 0.17; 10 min = − 1.48 ± 0.23; F(2,17) = 13.67; P < 0.01) was decreased (Fig. 3a). The lower plateau (P1) and the range between the upper and the lower plateaus (ΔP) of the sigmoid curve were also decreased by noradrenaline reuptake inhibition in the area (Fig. 4a; Table 1). On the other hand, the tachycardic linear regression curve slope was not affected by the pharmacological manipulation in the BNST (before = − 2.05 ± 0.17; 10 min = − 1.95 ± 0.12; 60 min = − 2.11 ± 0.24; F(2,17) = 0.21; P > 0.05). The linear regression slope of bradycardic responses (before = − 2.53 ± 0.17; 60 min = − 2.18 ± 0.12; F(2,17) = 13.67; P > 0.05) as well as the sigmoid curve parameters returned to basal levels 60 min after the microinjections (Fig. 4a; Table 1).

Linear regression and sigmoidal curves correlating the responses of ΔMAP and ΔHR before, 10 min and 60 min after bilateral microinjection of the respective combinations depicted in bold into the BNST(n = 6 and n = 5, respectively). (a–b upper) Correlation r2 values for bradycardic linear regression curves were 0.89 and 0.52 (before); 0.55 and 0.78 (10 min); 0.91 and 0.65 (60 min). Correlation r2 values for tachycardic linear regression curves were 0.89 and 0.89 (before); 0.90 and 0.95 (10 min); and 0.81 and 0.89 (60 min). (a–b; lower) Sigmoid curves r2 correlation values were 0.95 and 0.86 (before); 0.92 and 0.93 (10 min); 0.90 and 0.91 (60 min). Values are means ± SEM. bpm, beats.min1. The symbols on sigmoid curves represent BP50
In a second group of animals, injection of the same dose of reboxetine (2 nmol) into the BNST was preceded by the administration of a sub-effective dose of the α1-receptor antagonist, WB4101 (5 nmol, n = 5). The combination of these two compounds did not affect the basal values of HR (before = 363 ± 11; after = 362 ± 12 bpm; t = 0.21; P > 0.05) nor MAP (before = 104 ± 2.42; after = 103 ± 2.01 mmHg; t = 0.69; P > 0.05). Nevertheless, the α1 blockade in the area prevented the effect of reboxetine on both the baroreflex bradycardic response (before = − 2.00 ± 0.28; 10 min = − 2.07 ± 0.16; 60 min = − 2.00 ± 0.27; F(2,17) = 0.24; P > 0.05). The tachycardic response (before = − 1.62 ± 0.16; after = − 1.70 ± 0.11; 60 min = − 1.94 ± 0.24; F(2,17) = 1.21; P > 0.05) was not altered either (Fig. 4b). The modification of the sigmoid parameters (P1, ΔP) was also prevented (Fig. 4b; Table 1).
Effects of α2-receptor antagonism in the BNST prior to noradrenaline reuptake inhibition on cardiac baroreflex activity in awake rats
Microinjection of the noradrenaline reuptake inhibitor, reboxetine (2 nmol), preceded by vehicle (saline) (n = 6) in the BNST did not affect the basal levels of HR (before = 386 ± 5; after = 389 ± 5 bpm; t = 1.90; P > 0.05) and MAP (before = 100 ± 2.52; after = 100 ± 2.42 mmHg; t = 0.16; P > 0.05). The linear regression bradycardic slope (before = − 2.38 ± 0.14; 10 min = − 1.40 ± 0.25; F(2,17) = 14.02; P < 0.01) was decreased (Fig. 5a). The lower plateau (P1), the baroreflex gain (G) and the range between the upper and the lower plateaus (ΔP) of the sigmoid curve were also decreased by noradrenaline reuptake inhibition in the area (Fig. 5a; Table 1). Once again, the tachycardic linear regression curve slope was not affected by the pharmacological manipulation in the BNST (before = − 1.92 ± 0.15; 10 min = − 1.84 ± 0.14; 60 min = − 1.99 ± 0.15; F(2,17) = 2.58; P > 0.05). The linear regression slope of bradycardic responses (before = − 2.38 ± 0.14; 60 min = − 2.41 ± 0.07; F(2,17) = 14.02; P > 0.05) as well as the sigmoid curve parameters (G, P1,∆P) returned to basal levels 60 min after the microinjections (Fig. 5a; Table 1).

Linear regression and sigmoidal curves correlating the responses of ΔMAP and ΔHR before, 10 min and 60 min after bilateral microinjection of the respective combinations depicted in bold (n = 6 and n = 6, rescpetively) into the BNST. (a–b upper) Correlation r2 values for bradycardic linear regression curves were 0.95 and 0.89 (before); 0.64 and 0.50 (10 min); 0.94 and 0.95 (60 min). Correlation r2 values for tachycardic linear regression curves were 0.88 and 0.71 (before); 0.89 and 0.80 (10 min); and 0.91 and 0.71 (60 min). (a–b; lower) Sigmoid curves r2 correlation values were 0.97 and 0.88 (before); 0.93 and 0.88 (10 min); 0.97 and 0.89 (60 min). Values are means ± SEM. bpm, beats min−1. The symbols on sigmoid curves represent BP50
In a second group of animals, injection of reboxetine (2 nmol) into the BNST was preceded by the administration of a sub-effective dose of the α2-receptor antagonist, RX821002 (5 nmol, n = 5). The combination of these two compounds did not affect the basal values of HR (before = 384 ± 6; after = 386 ± 7 bpm; t = 0.87; P > 0.05) nor MAP (before = 104 ± 3.48; after = 105 ± 2.62 mmHg; t = 0.45; P > 0.05). Differently from α1 blockade, the α2 antagonism in the BNST did not prevent the effect of reboxetine on baroreflex bradycardic response (before = − 2.56 ± 0.18; 10 min = − 1.43 ± 0.22; 60 min = − 2.35 ± 0.09; F(2,17) = 16.59; P < 0.001). Likewise, the tachycardic response (before = − 1.96 ± 0.25; 10 min = − 1.76 ± 0.20; 60 min = − 1.97 ± 0.25; F(2,17) = 1.05; P > 0.05) was not changed (Fig. 5b). The sigmoid parameters (P1, ΔP, G) were also decreased (Fig. 5b; Table 1).
Effects of NMDA receptor antagonism in the BNST prior to noradrenaline reuptake inhibition on cardiac baroreflex activity in awake rats
Microinjection of the noradrenaline reuptake inhibitor, reboxetine (2 nmol), preceded by vehicle (saline) (n = 5) in the BNST did not modify the basal levels of HR (before = 360 ± 11; after = 362 ± 11 bpm; t = 1.71; P > 0.05) and MAP (before = 103 ± 2.92; after = 103 ± 2.29 mmHg; t = 0.21; P > 0.05). The linear regression bradycardic slope (before = − 2.59 ± 0.29; 10 min = − 1.52 ± 0.34; F(2,14) = 5.82; P < 0.05) was decreased (Fig. 6a). The lower plateau (P1), the baroreflex gain (G) and the range between the upper and the lower plateaus (ΔP) of the sigmoid curve were also decreased by reboxetine injection in the area (Fig. 6a; Table 1). The tachycardic component of linear regression was not affected by the pharmacological manipulation in the BNST (before = − 2.07 ± 0.04; 10 min = − 2.17 ± 0.08; 60 min = − 1.87 ± 0.04; F(2,14) = 1.13; P > 0.05). The linear regression slope of bradycardic responses (before = − 2.59 ± 0.29; 60 min = − 2.14 ± 0.15; F(2,14) = 5.82; P > 0.05) as well as the sigmoid curve parameters (G, P1, ∆P) returned to basal levels 60 min after the microinjections (Fig. 6a; Table 1).

Linear regression and sigmoidal curves correlating the responses of ΔMAP and ΔHR before, 10 min and 60 min after bilateral microinjection of the respective combinations depicted in bold into the BNST (n = 5 and n = 6, respectively). (a–b; upper) Correlation r2 values for bradycardic linear regression curves were 0.76 and 0.62 (before); 0.64 and 0.70 (10 min); 0.93 and 0.99 (60 min). Correlation r2 values for tachycardic linear regression curves were 0.98 and 0.75 (before); 0.95 and 0.82 (10 min); and 0.96 and 0.92 (60 min). (a–b; lower) Sigmoid curves r2 correlation values were 0.93 and 0.89 (before); 0.93 and 0.97 (10 min); 0.97 and 0.98 (60 min). Values are means ± SEM. bpm, beats min−1. The symbols on sigmoid curves represent BP50
In another group of animals, injection of reboxetine (2 nmol) into the BNST was preceded by the administration of a sub-effective dose of the NMDA receptor antagonist, AP7 (0.1 nmol, n = 6). The combination of these two compounds did not affect the basal values of HR (before = 363 ± 10; after = 366 ± 8 bpm; t = 1.71; P > 0.05) nor MAP (before = 103 ± 2.69; after = 105 ± 2.78 mmHg; t = 2.17; P > 0.05). NMDA-receptor antagonism in the BNST precluded the effect of reboxetine on baroreflex bradycardic response (before = − 1.57 ± 0.22; 10 min = − 1.52 ± 0.22; 60 min = − 1.68 ± 0.05; F(2,17) = 0.21; P > 0.05). Likewise, the tachycardic response (before = − 1.90 ± 0.21; 10 min = − 2.12 ± 0.15; 60 min = − 1.89 ± 0.07; F(2,17) = 1.47; P > 0.05) was not changed (Fig. 6b). None of the sigmoid parameters (P1, P2, ΔP, G) were altered (Fig. 6b; Table 1).
Effects of nNOS inhibition in the BNST prior to noradrenaline reuptake preclusion on cardiac baroreflex activity in awake rats
In the control group, microinjection of the noradrenaline reuptake inhibitor, reboxetine (2 nmol), preceded by vehicle (saline) (n = 6) in the BNST did not change the basal values of HR (before = 354 ± 10; after = 357 ± 9 bpm; t = 1.90; P > 0.05) and MAP (before = 102 ± 1.63; after = 101 ± 1.46 mmHg; t = 0.52; P > 0.05). The slope of the bradycardic linear regression curve (before = − 2.32 ± 0.14; 10 min = − 1.19 ± 0.15; F(2,17) = 21.04; P < 0.001) was reduced (Fig. 7a). The lower plateau (P1), the baroreflex gain (G) and the range between the upper and the lower plateaus (ΔP) of the non-linear regression were also decreased by reboxetine injection in the area (Fig. 7a; Table 1). The tachycardic component of linear regression was not affected by the pharmacological manipulation in the BNST (before = − 1.89 ± 0.14; 10 min = − 1.78 ± 0.08; 60 min = − 2.03 ± 0.04; F(2,17) = 2.52; P > 0.05). The linear regression slope of bradycardic responses (before = − 2.32 ± 0.14; 60 min = − 2.21 ± 0.15; F(2,17) = 21.04; P > 0.05) as well as the sigmoid curve parameters (G, P1,∆P) returned to basal levels 60 min after the microinjections (Fig. 7a; Table 1).

Linear regression and sigmoidal curves correlating the responses of ΔMAP and ΔHR before, 10 min and 60 min after bilateral microinjection of the respective combinations depicted in bold into the BNST (n = 6 and n = 7, respectively). (a–b; upper) Correlation r2 values for bradycardic linear regression curves were 0.78 and 0.81 (before); 0.70 and 0.57 (10 min); 0.91 and 0.90 (60 min). Correlation r2 values for tachycardic linear regression curves were 0.85 and 0.92 (before); 0.95 and 0.89 (10 min); and 0.80 and 0.77 (60 min). (a–b; lower) Sigmoid curves r2 correlation values were 0.93 and 0.94 (before); 0.95 and 0.89 (10 min); 0.95 and 0.94 (60 min). Values are means ± SEM. bpm, beats min−1. The symbols on sigmoid curves represent BP50
In a parallel group, injection of reboxetine (2 nmol) into the BNST was preceded by the administration of a sub-effective dose of the nNOS inhibitor, NPLA (0.4 nmol, n = 7). The basal values of HR (before = 360 ± 11; after = 361 ± 10 bpm; t = 0.24; P > 0.05) nor MAP (before = 105 ± 1.73; after = 107 ± 1.32 mmHg; t = 1.62; P > 0.05) were not altered by these two compounds. nNOS inhibition in the BNST precluded the effect of reboxetine on baroreflex bradycardic response (before = − 1.98 ± 0.21; 10 min = − 2.29 ± 0.36; 60 min = − 1.85 ± 0.18; F(2,20) = 1.17; P > 0.05). In the same way, the tachycardic response (before = − 1.78 ± 0.08; 10 min = − 1.83 ± 0.12; 60 min = −1.88 ± 0.15; F(2,20) = 0.12; P > 0.05) was not changed (Fig. 7b). The alteration in the sigmoid parameters (P1, ΔP, G) was also prevented (Fig. 7b; Table 1).
Effects of NO scavenging in the BNST prior to noradrenaline reuptake preclusion on cardiac baroreflex activity in awake rats
Microinjection of the noradrenaline reuptake inhibitor, reboxetine (2 nmol), preceded by vehicle (saline) (n = 6) in the BNST did not change the basal values of HR (before = 367 ± 11; after = 371 ± 10 bpm; t = 2.35; P > 0.05) and MAP (before = 106 ± 2.08; after = 105 ± 2.26 mmHg; t = 0.22; P > 0.05). The slope of the bradycardic linear regression curve (before = − 2.47 ± 0.18; 10 min = − 1.26 ± 0.13; F(2,17) = 22.25; P < 0.001) was reduced (Fig. 8a). The lower plateau (P1) and the range between the upper and the lower plateaus (ΔP) of the non-linear regression were also decreased by reboxetine injection in the area (Fig. 8a; Table 1). The tachycardic component of linear regression was not affected by the pharmacological manipulation in the BNST (before = − 1.92 ± 0.08; 10 min = − 2.10 ± 0.07; 60 min = − 1.95 ± 0.05; F(2,17) = 3.25; P > 0.05). The linear regression slope of bradycardic responses (before = − 2.47 ± 0.18; 60 min = − 2.11 ± 0.13; F(2,17) = 22.25; P > 0.05) as well as the sigmoid curve parameters (P1, ∆P) returned to basal levels 60 min after the microinjections (Fig. 8a; Table 1).

Linear regression and sigmoidal curves correlating the responses of ΔMAP and ΔHR before, 10 min and 60 min after bilateral microinjection of the respective combinations depicted in bold into the BNST (n = 6 and n = 5, respectively). (a-b; upper) Correlation r2 values for bradycardic linear regression curves were 0.87 and 0.74 (before); 0.63 and 0.88 (10 min); 0.90 and 0.81 (60 min). Correlation r2 values for tachycardic linear regression curves were 0.94 and 0.86 (before); 0.98 and 0.59 (10 min); and 0.92 and 0.81 (60 min). (a–b; lower) Sigmoid curves r2 correlation values were 0.95 and 0.95 (before); 0.95 and 0.92 (10 min); 0.91 and 0.94 (60 min). Values are means ± SEM. bpm, beats min−1. The symbols on sigmoid curves represent BP50
In a parallel group, injection of reboxetine (2 nmol) into the BNST was preceded by the administration of a sub-effective dose of NO scavenger, c-ptio (0.1 nmol, n = 5). The basal values of HR (before = 383 ± 8; after = 383 ± 9 bpm; t = 0.31; P > 0.05) nor MAP (before = 105 ± 1.78; after = 104 ± 0.87 mmHg; t = 0.17; P > 0.05) were not altered by these two compounds. NO scavenging in the BNST inhibited the effect of reboxetine on baroreflex bradycardic response (before = −1.52 ± 0.18; 10 min = − 2.00 ± 0.13; 60 min = − 1.81 ± 0.26; F(2,14) = 3.28; P > 0.05). In the same way, the tachycardic response (before = − 2.99 ± 0.33; 10 min = − 2.66 ± 0.41; 60 min = − 2.93 ± 0.20; F(2,14) = 0.36; P > 0.05) was not changed (Fig. 8b). The alteration in the sigmoid parameters (P1, ΔP) was also prevented (Fig. 8b; Table 1).
Effects of sGC inhibition in the BNST prior to noradrenaline reuptake preclusion on cardiac baroreflex activity in awake rats
Microinjection of the noradrenaline reuptake inhibitor, reboxetine (2 nmol), preceded by vehicle (saline) (n = 5) in the BNST did not change the basal levels of neither HR (before = 374 ± 10; after = 375 ± 11 bpm; t = 0.69; P > 0.05) nor MAP (before = 102 ± 3.08; after = 103 ± 2.87 mmHg; t = 1.05; P > 0.05). The slope of the bradycardic linear regression curve (before = − 2.47 ± 0.32; 10 min = − 1.60 ± 0.31; F(2,14) = 5.60; P < 0.05) was reduced (Fig. 9a). The lower plateau (P1), the range between the upper and the lower plateaus (ΔP) and the average baroreflex gain (G) of the non-linear regression were also decreased by reboxetine injection in the area (Fig. 9a; Table 1). The tachycardic component of linear regression was not significantly affected by the pharmacological manipulation in the BNST (before = − 1.97 ± 0.08; 10 min = − 2.15 ± 0.09; 60 min = − 1.87 ± 0.04; F(2,14) = 5.48; P > 0.05). The linear regression slope of bradycardic responses (before = − 2.47 ± 0.32; 60 min = − 2.01 ± 0.08; F(2,14) = 5.60; P > 0.05) as well as the sigmoid curve parameters (G, P1, ∆P) returned to basal levels 60 min after the microinjections (Fig. 9a; Table 1).

Linear regression and sigmoidal curves correlating the responses of ΔMAP and ΔHR before, 10 min and 60 min after bilateral microinjection of the respective combinations depicted in bold into the BNST (n = 5 and n = 5, respectively). (a–b; upper) Correlation r2 values for bradycardic linear regression curves were 0.77 and 0.71 (before); 0.83 and 0.83 (10 min); 0.96 and 0.89 (60 min). Correlation r2 values for tachycardic linear regression curves were 0.97 and 0.68 (before); 0.97 and 0.69 (10 min); and 0.99 and 0.96 (60 min). (a–b; lower) Sigmoid curves r2 correlation values were 0.93 and 0.90 (before); 0.94 and 0.92 (10 min); 0.98 and 0.92 (60 min). Values are means ± SEM. bpm, beats min−. The symbols on sigmoid curves represent BP50
Treatment of BNST with the soluble guanylate cyclase (sGC) inhibitor, ODQ (0.2 nmol) prior to reboxetine (2 nmol, n = 5) did not modify the basal levels of HR (before = 364 ± 8; after = 366 ± 7 bpm; t = 1.77; P > 0.05) nor MAP (before = 107 ± 2.47; after = 108 ± 2.78 mmHg; t = 1.00; P > 0.05) were not altered by the injections. sGC inhibition in the BNST blocked the effect of reboxetine on baroreflex bradycardic response (before = − 2.16 ± 0.34; 10 min = − 1.97 ± 0.26; 60 min = − 2.19 ± 0.16; F(2,14) = 0.58; P > 0.05). Such as it happened in the control group, the tachycardic response (before = − 2.39 ± 0.40; 10 min = − 2.36 ± 0.35; 60 min = − 2.63 ± 0.09; F(2,14) = 0.19; P > 0.05) was not altered (Fig. 9b). The alteration in the sigmoid parameters (P1, ΔP, G) was also prevented (Fig. 9b; Table 1).
Effects of vehicle (saline 0.9%) injection in the BNST as well as of reboxetine in BNST surrounding structures on cardiac baroreflex activity in awake rats
Injection of saline 0.9% in the BNST (n = 5) was unable to change the basal levels of HR (before = 372 ± 12; after = 366 ± 14 bpm; t = 2.69; P > 0.05) and MAP (before = 98 ± 1.38; after = 101 ± 2.09 mmHg; t = 1.48; P > 0.05). Neither bradycardic (before = − 1.97 ± 0.46; 10 min = − 1.89 ± 0.35; 60 min = − 1.99 ± 0.58; F(2,14) = 1.22; P > 0.05) nor tachycardic (before = − 2.34 ± 0.27; 10 min = − 2.19 ± 0.46; 60 min = − 2.29 ± 0.39; F(2,14) = 0.78; P > 0.05) slopes were affected by its microinjection. In addition none of the sigmoid curve parameters (G, P1, P2, ∆P, BP50) were displaced (graphs not shown).
Moreover, reboxetine (2 nmol, n = 6) was injected in BNST surrounding structures. Such areas include a range of at least 1 mm outside (upper, lower, left or right) the region that assembles BNST showed in Fig. 2. The volume used was not enough to reach BNST and were, therefore, consider outside the aimed structure. It did not modify HR (before = 347 ± 6; after = 343 ± 9 bpm; t = 1.88; P > 0.05) or MAP (before = 101 ± 0.59; after = 103 ± 2.44 mmHg; t = 2.66; P > 0.05) basal levels. In the same way, it was not capable of altering bradycardic (before = − 1.88 ± 0.09; 10 min = − 1.83 ± 0.05; 60 min = − 1.86 ± 0.04; F(2,17) = 1.44; P > 0.05), tachycardic slopes (before = − 2.00 ± 0.21; 10 min = − 1.99 ± 0.29; 60 min = − 2.13 ± 0.34; F(2,17) = 2.47; P > 0.05) of linear regression curves or even the sigmoid parameters (G, P1,P2, ∆P, BP50) (graphs not shown).
Discussion
In the present study, we observed that potentiation of BNST noradrenergic transmission reduced baroreflex bradycardic response with no alteration in tachycardic reflex (Fig. 3). In addition, α1-receptor antagonism in BNST prevented the effect of reboxetine (Fig. 4). However, previous microinjection of a α2-receptor antagonist into the area was unable to do it (Fig. 5). Such result is in line with the study of Crestani and co-workers, in which BNST α1 blockade increased the magnitude of bradycardia, while α2-receptor antagonism had no effect [11]. In this same study, the authors observed that systemic treatment with a muscarinic receptor antagonist inhibited bradycardia enhancement, suggesting that such effect is mediated by parasympathetic activation [11]. Consequently, we highlight that BNST α1, but not α2-adrenoceptors negatively modulate the baroreflex bradycardic response by reducing parasympathetic output. Noradrenaline can also bind to β-adrenoceptors. However, injection of non-selective β-receptors into BNST did not displace neither bradycardic nor tachycardic baroreflex curves [11]. Also, the return of baroreflex activity to basal levels 60 min after BNST administration suggests that reboxetine half-life does not exceed this period of time.
Interestingly, peripheral administration of a muscarinic receptor antagonist prevented the bradycardic potentiating effect of an α1-receptor antagonist injected into the BNST [11]. It strongly suggests that BNST noradrenergic system mediates baroreflex parasympathetic activity during BP increase, through α1-receptors. Additionally, BNST projects to the nucleus of the solitary tract (NTS) and nucleus ambiguous [26, 29], which are potential neural relays for BNST α1-receptors containing neurons for the modulation of parasympathetic activity. Furthermore, the connections between BNST and brain stem seem to be reciprocal, since the majority of noradrenergic terminals found in BNST arise from NTS [23]. Therefore, peripheral inputs could be involved in the engagement of NTS noradrenergic neurons targeted to BNST during baroreflex stimulation. Moreover, BNST receives projections from the hippocampus, amygdaloid nuclei and medial prefrontal cortex, which are also engaged in baroreflex modulation [19, 24, 35]. Thus, BNST could work as an integrator between those areas and the brain stem to modulate the cardiovascular system. Specifically, pharmacological ablation of medial amygdaloid nucleus induced a similar effect of BNST α1 antagonism on baroreflex activity [11, 24]. Therefore, we suggest that connections between these two structures may be key mediators of such integration. Additionally, the BNST circuitry highlighted in the present study is not involved in the modulation of HR and MAP basal levels, since drug administration in the area was unable to change such parameters (“Results” section).
Furthermore, BNST is activated during stressful conditions [32, 36]. Such conditions may stimulate the central noradrenergic system [38]. Interestingly, under stressful situations, the extracellular level of noradrenaline inside the BNST is increased [42]. Hott and colleagues showed that antagonism of BNST α1-receptors precluded the HR and BP enhancement during the contextual fear conditioning task [30]. Such HR and BP responses are discriminative of aversive situations, in which the baroreflex tachycardic activity is potentiated, while the bradycardic reflex is suppressed [13]. As we can see through our results, activation of BNST α1-receptors decreases bradycardic response. Therefore, we believe that during acute stress, BNST noradrenaline release is increased, which in turn activates α1-adrenoceptors to suppress baroreflex bradycardic activity, allowing HR and BP increase in the aversive environment.
It has been demonstrated that glutamate release can be modulated by noradrenergic neurotransmission inside BNST [21,22,23, 49, 50]. Egli and colleagues strongly suggested that the receptor involved in such release was alpha-2 receptor, but not alpha-1 [18]. In our study, we observed that BNST alpha-2 antagonism lacks to inhibit reboxetine effect on bradycardic reduction (Fig. 5). Instead, alpha-1 antagonism blocks the effect of reboxetine (Fig. 4). It is still not clear if α1 adrenoceptor is postsynaptic or presynaptic in BNST. Considering that α1-receptor may modulate glutamate release in BNST and that this release necessarily occurs on presynaptic membrane [21,22,23, 49, 50], we suggest that such receptors are more likely to modulate glutamate exocytosis acting on presynaptic level. Additionally, it has been suggested that alpha-1 receptors could be located presynaptically in BNST [4]. Moreover, NMDA glutamatergic receptors are present in BNST. Interestingly, BNST NMDA and α1-receptors antagonism increase baroreflex bradycardic response in the same way [2, 11]. Therefore, one of our matters was to determine if there is an interaction between α1 and NMDA receptors in BNST to modulate the bradycardic response of the baroreflex. Our results show that administration of NMDA antagonist in BNST prevented the effect of reboxetine, demonstrating a link between noradrenergic and glutamatergic neurotransmissions in the modulation of bradycardic baroreflex response.
Additionally, NMDA receptors enhance calcium influx to neurons, which may stimulate NO synthesis through activation of neuronal nitric oxide synthase (nNOS), in the central nervous system [25]. As previously shown, inhibition of this enzyme in BNST also increased bradycardic baroreflex gain [2]. These results suggest that NMDA activation and NO production in the area may play a role in baroreflex control. Since stimulation of BNST α1 receptors activates NMDA channels to reduce the bradycardic response, we sought to evaluate if this effect would engage nNOS. In fact, nNOS inhibition was able to preclude effect of reboxetine on bradycardic reflex (Fig. 7, Table 1). It demonstrates that the effect of noradrenergic stimulation depends not only on NMDA channels but also NO production by nNOS.
After its synthesis, NO may be released from the postsynaptic neuron, acting as a retrograde messenger, by diffusing in presynaptic terminals membranes [39]. In order to evaluate the possible site of NO action, we employed a pharmacological agent called “carboxy-PTIO”. Such compound is a NO scavenger, unable to cross the lipid membrane, binding exclusively to NO molecules outside the cells [33]. Our results show that the reduction in bradycardic response induced by reboxetine leads to NO release, since carboxy-PTIO injection in BNST inhibited this effect (Fig. 8, Table 1). Thus, we suggest that NO may be acting as a retrograde messenger. Moreover, one of the molecular targets of NO at presynaptic level is the soluble guanylate cyclase (sGC) [3]. This enzyme is responsible for converting GTP in cGMP, a second messenger that can influence ion channels, and consequently, facilitate glutamate release [3]. Inhibition of this enzyme in BNST was able to preclude the effect of noradrenergic stimulation on bradycardic response (Fig. 9, Table 1).
In sum, our results demonstrate for the first time an interaction between noradrenergic glutamatergic and nitrergic systems in BNST in the modulation of the baroreflex bradycardic response. We believe that during bradycardic stimulation, noradrenaline is released from terminals in the BNST. Noradrenaline, then, activates presynaptic α1 receptors, which in turn, stimulate glutamate release and NMDA triggering. This receptor engages production of NO by nNOS. NO diffuses to the presynaptic terminal to activate sGC that is able to influence ion channels to facilitate the glutamate release. This whole neurocircuitry acts to decrease the magnitude of bradycardic reflex during BP increase. A schematic representation of the suggested BNST neurotransmission is depicted in Fig. 10.

BNST schematic neurotransmission proposed to be involved in the modulation of cardiac baroreflex response: (1) during baroreflex stimulation, noradrenaline is released in the BNST due to neuronal inputs. Noradrenaline, in turn, activates presynaptic α1-adrenoceptors in BNST neurons; (2) once activated, α1 receptors engages calcium release from endoplasmic reticulum on account of IP3 mobilization. (3) Calcium ions stimulate L-glutamate exocytosis. (4) L-glutamate engages postsynaptic NMDA receptors, leading to calcium influx. (5) Calcium depolarizes postsynaptic cells. (6) Depolarization stimulates BNST outputs to brain stem nuclei involved in parasympathetic control, leading to (7) bradycardic inhibition. (6′) In parallel, calcium entrance through NMDA receptors triggers NO synthesis by nNOS. (7′) NO is released from postsynaptic neuron, diffusing to presynaptic membrane, as a retrograde messenger. (8′) In presynaptic cell, NO activates sGC, enhancing kinases activity that facilitates calcium influx through ion channels. Calcium potentiates L-glutamate exocytosis (9′) and, therefore, depolarization of BNST outputs. The facilitation of this process leads to reduction in bradycardic activity
On the other hand, it is important to mention that GABAergic circuitry was identified in BNST and it has also been linked to noradrenergic, nitrergic and glutamatergic systems in cardiovascular control, especially during stress response [4, 43]. Therefore, we cannot exclude the role of such neurotransmitter in the area and its possible involvement on baroreflex activity. However, its physiological contribution for the modulation of this response needs further investigation.
Similarly to α1 antagonism, blockade of NMDA receptors, nNOS inhibition and NO scavenging in BNST also decreased the HR and BP rise evoked by conditioned emotional response [31]. Moreover, there was a positive correlation between concentration of NO metabolites (NOx) in BNST and time spent in freezing in conditioned animals. Additionally, injection of NMDA antagonist in the area was able to decrease NOx level [31]. Recently, Barreto-de-Souza and colleagues (2018) showed that BNST α1-receptor and NMDA receptor antagonism prevented the effect of a NO donor on HR rise during restraint stress. Moreover, the authors demonstrated that nNOS activation in BNST modulated the same HR response due to NMDA injection [4]. In fact, restraint stress induced baroreflex shifting towards higher BP values, allowing tachycardia along with BP rise [13]. Taking these findings together with our results, it is possible to assume that noradrenergic, glutamatergic and nitrergic neurotransmissions inside BNST interact to modulate baroreflex adjustments needed during stress response.
Another possibility is that this BNST neurotransmission may be activated by physical exercise [47]. Camargo and co-workers observed a significant NOx formation in BNST of rats submitted to treadmill running and that this increase was blocked by peripheral administration of a NMDA antagonist [6]. Also, a nNOS inhibitor injected intraperitoneally was able reduce HR during running [6]. Taking together with the results of the present study, such data suggest that BNST NMDA/NO pathway is engaged during baroreflex adjustments required in physical exercise.
In addition to neural control of baroreflex, the BNST seems to be involved in the neuroendocrine changes elicited during stressful situations [7]. In such situations, the hypothalamic-pituitary axis is engaged, and the existence of connections between BNST and hypothalamic nuclei has been shown [16, 27, 28]. Inside BNST, corticotropin releasing factor (CRF) neurotransmission has been associated with cardiovascular adaptation induced by conditioned stressors [40]. In addition, Oliveira and colleagues demonstrated that CRF microinjection in BNST enhanced BP and HR increases induced by acute stress, which were completely abolished by previous treatment with NMDA/nNOS/sGC inhibitors [41]. Moreover, plasma adrenocorticotropic hormone levels are blunted by BNST α1 blockade before immobilization stress in rats [7]. Therefore, it can be suggested that interaction between BNST noradrenergic, glutamatergic and nitrergic systems may have a cross-link with hypothalamic-pituitary axis for the modulation of baroreflex acitivity during a stress response. Additionally, stimulation of α1 adrenoceptor inside the area induced BP rises and reflex bradycardia on account of vasopressin release in blood stream [10]. Thus, BNST noradrenergic neurotransmission may be important for other baroreflex hormonal mechanisms during aversive situations.
BNST is also related to cardiovascular dysfunction induced by chronic exposure to drugs of abuse, such as cocaine [14]. This compound, similarly to reboxetine, inhibits noradrenaline reuptake, facilitating noradrenergic transmission [44]. Cruz and colleagues showed that repeated administration with cocaine increased the gain of both bradycardic and tachycardic components of baroreflex in rats [14]. Interestingly, pharmacological ablation of BNST prevented such baroreflex alterations [14]. Therefore, it is possible to assume that BNST noradrenergic, and consequently, glutamtergic/nitrergic stimulation, could be a part of the autonomic and baroreflex toxic effects of cocaine. It seems controversial that facilitation of BNST noradrenergic transmission would decrease bradycardic response with no alteration in tachycardia, while cocaine administration enhanced both responses [14]. However, this compound is likely to evoke functional changes in other brain areas involved in baroreflex control, such as MPFC, hippocampus and amygdaloid nuclei [19, 24, 45]. Therefore, its action on those structures may also account for the effect observed on baroreflex response.
Perspectives and significance
The present study adds an important observation in the field of integrative physiology, since it describes how different neurotransmitters in BNST may modulate the main short-term cardiovascular adjustment response. In addition, the present work could serve as a background for further studies relating BNST neurotransmission in the alteration of baroreflex, once that such area takes part in stress response, in which baroreflex reseting is necessary. Moreover, chronic stress displaces baroreflex response [20], but the mechanisms by which it is altered is not establish yet. Once that BNST is sensitive to stress stimuli, the present work could be one of the firsts steps to elucidate baroreflex dysfunctions after chronic stress states.
Conclusion
In conclusion, our results show an interaction between BNST noradrenergic neurotransmission and NMDA/nNOS/sGC pathway in the modulation of the bradycardic response of baroreflex activity.
Data availability
The data presented in the present work is original and the paper is being under consideration in any other journal.
References
Aliaga E, Bustos G, Gysling K (1995) Release of endogenous catecholamines from the striatum and bed nucleus of stria terminalis evoked by potassium and N-methyl-D-aspartate: in vitro microdialysis studies. J Neurosci Res 40:89–98. https://doi.org/10.1002/jnr.490400110
Alves FHF, Crestani CC, Resstel LBM, Correa FMA (2009) Bed nucleus of the stria terminalis N-methyl-D-aspartate receptors and nitric oxide modulate the baroreflex cardiac component in unanesthetized rats. J Neurosci Res 87:1703–1711. https://doi.org/10.1002/jnr.21974
Arnold WP, Mittal CK, Katsuki S, Murad F (1977) Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A 74:3203–3207
Barretto-de-Souza L, Adami MB, Benini R, Crestani CC (2018) Dual role of nitrergic neurotransmission in the bed nucleus of the stria terminalis in controlling cardiovascular responses to emotional stress in rats. Br J Pharmacol 175:3773–3783. https://doi.org/10.1111/bph.14447
Byrum CE, Guyenet PG (1987) Afferent and efferent connections of the A5 noradrenergic cell group in the rat. J Comp Neurol 261:529–542. https://doi.org/10.1002/cne.902610406
Camargo LHA, Alves FHF, Biojone C, Correa FMA, Resstel LBM, Crestani CC (2013) Involvement of N-methyl-D-aspartate glutamate receptor and nitric oxide in cardiovascular responses to dynamic exercise in rats. Eur J Pharmacol 713:16–24. https://doi.org/10.1016/j.ejphar.2013.04.046
Cecchi M, Khoshbouei H, Javors M, Morilak DA (2002) Modulatory effects of norepinephrine in the lateral bed nucleus of the stria terminalis on behavioral and neuroendocrine responses to acute stress. Neuroscience 112:13–21
Crestani CC, Alves FHF, Resstel LB, Corrêa FMA (2008) Both alpha1 and alpha2-adrenoceptors mediate the cardiovascular responses to noradrenaline microinjected into the bed nucleus of the stria terminal of rats. Br J Pharmacol 153:583–590. https://doi.org/10.1038/sj.bjp.0707591
Crestani CC, Alves FHF, Resstel LBM, de Corrêa FMA (2006) The bed nucleus of the stria terminalis modulates baroreflex in rats. Neuroreport 17:1531–1535. https://doi.org/10.1097/01.wnr.0000236854.40221.40
Crestani CC, Alves FHF, Resstel LBM, Corrêa FMA (2007) Cardiovascular effects of noradrenaline microinjection in the bed nucleus of the stria terminalis of the rat brain. J Neurosci Res 85:1592–1599. https://doi.org/10.1002/jnr.21250
Crestani CC, Alves FHF, Resstel LBM, Correa FMA (2008) Bed nucleus of the stria terminalis alpha(1)-adrenoceptor modulates baroreflex cardiac component in unanesthetized rats. Brain Res 1245:108–115. https://doi.org/10.1016/j.brainres.2008.09.082
Crestani CC, Alves FHF, Tavares RF, Corrêa FMA (2009) Role of the bed nucleus of the stria terminalis in the cardiovascular responses to acute restraint stress in rats. Stress 12:268–278. https://doi.org/10.1080/10253890802331477
Crestani CC, Tavares RF, Alves FHF, Resstel LBM, Correa FMA (2010) Effect of acute restraint stress on the tachycardiac and bradycardiac responses of the baroreflex in rats. Stress Amst Neth 13:61–72. https://doi.org/10.3109/10253890902927950
Cruz FC, Alves FHF, Leão RM, Planeta CS, Crestani CC (2013) Role of the bed nucleus of the stria terminalis in cardiovascular changes following chronic treatment with cocaine and testosterone: a role beyond drug seeking in addiction? Neuroscience 253:29–39. https://doi.org/10.1016/j.neuroscience.2013.08.034
Dong HW, Petrovich GD, Swanson LW (2001) Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res Brain Res Rev 38:192–246
Dunn JD (1987) Plasma corticosterone responses to electrical stimulation of the bed nucleus of the stria terminalis. Brain Res 407:327–331. https://doi.org/10.1016/0006-8993(87)91111-5
Dunn JD, Williams TJ (1995) Cardiovascular responses to electrical stimulation of the bed nucleus of the stria terminalis. J Comp Neurol 352:227–234. https://doi.org/10.1002/cne.903520206
Egli RE, Kash TL, Choo K, Savchenko V, Matthews RT, Blakely RD, Winder DG (2005) Norepinephrine modulates glutamatergic transmission in the bed nucleus of the stria terminalis. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 30:657–668. https://doi.org/10.1038/sj.npp.1300639
Ferreira-Junior NC, Lagatta DC, Fabri DR, Alves FHF, Corrêa FMA, Resstel LBM (2017) Hippocampal subareas arranged in the dorsoventral axis modulate cardiac baroreflex function in a site-dependent manner in rats. Exp Physiol 102:14–24. https://doi.org/10.1113/EP085827
Firmino EMS, Kuntze LB, Lagatta DC, Dias DPM, Resstel LBM (2019) Effect of chronic stress on cardiovascular and ventilatory responses activated by both chemoreflex and baroreflex in rats. J Exp Biol 222:1. https://doi.org/10.1242/jeb.204883
Forray MI, Andrés ME, Bustos G, Gysling K (1995) Regulation of endogenous noradrenaline release from the bed nucleus of stria terminalis. Biochem Pharmacol 49:687–692
Forray MI, Bustos G, Gysling K (1999) Noradrenaline inhibits glutamate release in the rat bed nucleus of the stria terminalis: in vivo microdialysis studies. J Neurosci Res 55:311–320. https://doi.org/10.1002/(SICI)1097-4547(19990201)55:3<311::AID-JNR6>3.0.CO;2-E
Forray MI, Gysling K (2004) Role of noradrenergic projections to the bed nucleus of the stria terminalis in the regulation of the hypothalamic-pituitary-adrenal axis. Brain Res Brain Res Rev 47:145–160. https://doi.org/10.1016/j.brainresrev.2004.07.011
Fortaleza EAT, Ferreira-Junior NC, Lagatta DC, Resstel LBM, Corrêa FMA (2015) The medial amygdaloid nucleus modulates the baroreflex activity in conscious rats. Auton Neurosci Basic Clin 193:44–50. https://doi.org/10.1016/j.autneu.2015.07.003
Garthwaite J, Garthwaite G, Palmer RM, Moncada S (1989) NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur J Pharmacol 172:413–416
Gray TS, Magnuson DJ (1987) Neuropeptide neuronal efferents from the bed nucleus of the stria terminalis and central amygdaloid nucleus to the dorsal vagal complex in the rat. J Comp Neurol 262:365–374. https://doi.org/10.1002/cne.902620304
Herman JP, Cullinan WE, Watson SJ (1994) Involvement of the bed nucleus of the stria terminalis in tonic regulation of paraventricular hypothalamic CRH and AVP mRNA expression. J Neuroendocrinol 6:433–442
Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE (2003) Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol 24:151–180
Holstege G, Meiners L, Tan K (1985) Projections of the bed nucleus of the stria terminalis to the mesencephalon, pons, and medulla oblongata in the cat. Exp Brain Res 58:379–391
Hott SC, Gomes FV, Fabri DRS, Reis DG, Crestani CC, Côrrea FMA, Resstel LBM (2012) Both α1- and β1-adrenoceptors in the bed nucleus of the stria terminalis are involved in the expression of conditioned contextual fear. Br J Pharmacol 167:207–221. https://doi.org/10.1111/j.1476-5381.2012.01985.x
Hott SC, Gomes FV, Uliana DL, Vale GT, Tirapelli CR, Resstel LBM (2017) Bed nucleus of the stria terminalis NMDA receptors and nitric oxide modulate contextual fear conditioning in rats. Neuropharmacology 112:135–143. https://doi.org/10.1016/j.neuropharm.2016.05.022
Hubert GW, Muly EC (2014) Distribution of AMPA receptor subunit glur1 in the bed nucleus of the stria terminalis and effect of stress. Synap N Y N 68:194–201. https://doi.org/10.1002/syn.21729
Ko GY, Kelly PT (1999) Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J Neurosci 19:6784–6794
Kretz R (1984) Local cobalt injection: a method to discriminate presynaptic axonal from postsynaptic neuronal activity. J Neurosci Methods 11:129–135
Lagatta DC, Ferreira-Junior NC, Resstel LBM (2015) Medial prefrontal cortex TRPV1 channels modulate the baroreflex cardiac activity in rats. Br J Pharmacol 172:5377–5389. https://doi.org/10.1111/bph.13327
Micioni Di Bonaventura MV, Ciccocioppo R, Romano A, Bossert JM, Rice KC, Ubaldi M, St Laurent R, Gaetani S, Massi M, Shaham Y, Cifani C (2014) Role of bed nucleus of the stria terminalis corticotrophin-releasing factor receptors in frustration stress-induced binge-like palatable food consumption in female rats with a history of food restriction. J Neurosci 34:11316–11324. https://doi.org/10.1523/JNEUROSCI.1854-14.2014
Moore RY, Bloom FE (1979) Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci 2:113–168. https://doi.org/10.1146/annurev.ne.02.030179.000553
Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, Petre CO (2005) Role of brain norepinephrine in the behavioral response to stress. Prog Neuro-Psychopharmacol Biol Psychiatry 29:1214–1224. https://doi.org/10.1016/j.pnpbp.2005.08.007
Musleh WY, Shahi K, Baudry M (1993) Further studies concerning the role of nitric oxide in LTP induction and maintenance. Synap N Y N 13:370–375. https://doi.org/10.1002/syn.890130409
Nijsen M (2001) CRH Signalling in the bed nucleus of the stria terminalis is involved in stress-induced cardiac vagal activation in conscious rats. Neuropsychopharmacology 24:1–10. https://doi.org/10.1016/S0893-133X(00)00167-6
Oliveira LA, Gomes-de-Souza L, Benini R, Crestani CC (2018) Control of cardiovascular responses to stress by CRF in the bed nucleus of stria terminalis is mediated by local NMDA/nNOS/sGC/PKG signaling. Psychoneuroendocrinology 89:168–176. https://doi.org/10.1016/j.psyneuen.2018.01.010
Pacak K, McCarty R, Palkovits M, Kopin IJ, Goldstein DS (1995) Effects of immobilization on in vivo release of norepinephrine in the bed nucleus of the stria terminalis in conscious rats. Brain Res 688:242–246
Pati D, Marcinkiewcz CA, DiBerto JF, Cogan ES, McElligott ZA, Kash TL (2020) Chronic intermittent ethanol exposure dysregulates a GABAergic microcircuit in the bed nucleus of the stria terminalis. Neuropharmacology 168:107759. https://doi.org/10.1016/j.neuropharm.2019.107759
Ravna AW, Sylte I, Dahl SG (2003) Molecular mechanism of citalopram and cocaine interactions with neurotransmitter transporters. J Pharmacol Exp Ther 307:34–41. https://doi.org/10.1124/jpet.103.054593
Resstel LBM, Fernandes KBP, Corrêa FMA (2004) Medial prefrontal cortex modulation of the baroreflex parasympathetic component in the rat. Brain Res 1015:136–144. https://doi.org/10.1016/j.brainres.2004.04.065
Spencer SJ, Buller KM, Day TA (2005) Medial prefrontal cortex control of the paraventricular hypothalamic nucleus response to psychological stress: possible role of the bed nucleus of the stria terminalis. J Comp Neurol 481:363–376. https://doi.org/10.1002/cne.20376
Tarasova OS, Borzykh AA, Kuz’min IV, Borovik AS, Lukoshkova EV, Sharova AP, Vinogradova OL, Grigor’ev AI (2012) Dynamics of heart rate changes in rats following stepwise change of treadmill running speed. Ross Fiziol Zh Im I M Sechenova 98:1372–1379
Ulrich-Lai YM, Herman JP (2009) Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10:397–409. https://doi.org/10.1038/nrn2647
Walker DL, Davis M (1997) Double dissociation between the involvement of the bed nucleus of the stria terminalis and the central nucleus of the amygdala in startle increases produced by conditioned versus unconditioned fear. J Neurosci 17:9375–9383
Weitlauf C, Egli RE, Grueter BA, Winder DG (2004) High-frequency stimulation induces ethanol-sensitive long-term potentiation at glutamatergic synapses in the dorsolateral bed nucleus of the stria terminalis. J Neurosci 24:5741–5747. https://doi.org/10.1523/JNEUROSCI.1181-04.2004
Woulfe JM, Hrycyshyn AW, Flumerfelt BA (1988) Collateral axonal projections from the A1 noradrenergic cell group to the paraventricular nucleus and bed nucleus of the stria terminalis in the rat. Exp Neurol 102:121–124
Acknowledgements
The authors wish to thank Camargo, L.H.A. and Mesquita O. for technical support.
Code availability
The authors permit the use of software to screen for plagiarism.
Funding
This study was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Protocol number 461/2009); the Conselho Nacional para o Desenvolvimento Científico e Tecnológico (CNPq) (Protocol number 156718/2012-0); the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) fellowship and the Fundação de Apoio ao Ensino, Pesquisa e Assistência do Hospital das Clínicas da FMRP-USP (FAEPA).
Author information
Authors and Affiliations
Contributions
Davi C. Lagatta. and Leonardo Resstel conceived and designed this research; experiments were performed by Davi, Campos Lagatta., Luciana Bärg Kuntze, Daniela.Lescano Uliana and Anna Bárbara Borges Asis. Data were analysed by Davi Campos Lagatta., Luciana Bärg Kuntze and Leonardo Resstel. Results of experiments were interpreted by Davi Campos Lagatta., Luciana Bärg Kuntze and Leonardo Rresstel. Figures were prepared and manuscript was firstly drafted by Davi Campos Lagatta. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
They authors declare that there are no conflicts of interests.
Ethical approval
Experimental procedures were carried out following protocols approved by the Ethical Review Committee of the School of Medicine of Ribeirão Preto (Protocol number 019/2015), which complies with the Guiding Principles for Research Involving Animals and Human Beings of the American Physiological Society.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lagatta, D.C., Kuntze, L.B., Uliana, D.L. et al. Bed nucleus of the stria terminalis modulates baroreflex cardiac activity: an interaction between alpha-1 receptors and NMDA/nitric oxide pathway. Pflugers Arch - Eur J Physiol 473, 253–271 (2021). https://doi.org/10.1007/s00424-020-02475-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-020-02475-1