
Abstract
Histone methylation is a key epigenetic mechanism and plays a major role in regulating gene expression during oocyte maturation and early embryogenesis. This mechanism can be briefly defined as the process by which methyl groups are transferred to lysine and arginine residues of histone tails extending from nucleosomes. While methylation of the lysine residues is catalyzed by histone lysine methyltransferases (KMTs), protein arginine methyltransferases (PRMTs) add methyl groups to the arginine residues. When necessary, the added methyl groups can be reversibly removed by histone demethylases (HDMs) by a process called histone demethylation. The spatiotemporal regulation of methylation and demethylation in histones contributes to modulating the expression of genes required for proper oocyte maturation and early embryonic development. In this review, we comprehensively evaluate and discuss the functional importance of dynamic histone methylation in mammalian oocytes and early embryos, regulated by KMTs, PRMTs, and HDMs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term “epigenetics” was first introduced by Conrad Waddington in the early 1940s, and he defined it as “the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being" (Waddington 1942). It is now described as the functional changes in genes without any alteration in their DNA sequences, which can be inherited by the next generations (Bird 2007). Indeed, epigenetic mechanisms can lead to conformational changes in the chromatin structure in order to regulate the corresponding gene expression (Saksouk et al. 2015; Li and Reinberg 2011). As is known, chromatin is a highly organized complex of DNA and histone proteins, both of which are tightly packaged to form the fundamental units, nucleosomes. In nucleosomes, a DNA fragment of approximately 147 base pairs wraps around an octamer core composed of unique histone proteins, two copies each of H2A, H2B, H3, and H4 (Tessarz and Kouzarides 2014). The fifth type of histone, linker histone protein H1, binds to the entry and exit sites of the wrapped DNA to maintain nucleosome integrity, and also covers internucleosomal DNA for protection from any potential nuclease attack (Woodcock et al. 2006).
The histone tails extending out of nucleosomes can be subjected to a set of modifications involving acetylation, phosphorylation, ubiquitination, isomerization, ADP-ribosylation, and methylation for chromatin remodeling (Kadoch et al. 2016). Acetylation (addition of acetyl groups) and deacetylation (removal of acetyl groups) of the lysine residues are dynamic processes, catalyzed by two main enzyme families: histone/lysine acetyltransferases [HATs; also referred to as lysine acetyltransferases (KATs)] and histone deacetylases (HDACs), respectively (Allis et al. 2007; DesJarlais and Tummino 2016). In general, acetylation of lysine residues involves transcriptional activation of target genes by permitting chromatin accessibility to related factors, while deacetylation of these sites results in transcriptional suppression (Rajan et al. 2020). On the other hand, phosphorylation is a process in which a phosphate group is added to histones based on intracellular signaling pathways by protein kinases, and these phosphate groups can be removed by phosphatases, which is called histone dephosphorylation (Cao and Dang 2018). It is worth noting that phosphorylation commonly occurs on the serine, threonine, and tyrosine residues of histones (Rossetto et al. 2012), and is involved in modulating different cellular events such as DNA damage response (Rogakou et al. 1998), mitosis, and meiosis (de la Barre et al. 2000; Wei et al. 1998), apoptosis (Ajiro 2000), and transcriptional regulation (Metzger et al. 2008).
Ubiquitination of lysine residues in histones such as H2A and H2B by ubiquitin ligases contributes to modulation of DNA damage repair and transcription (Cao and Yan 2012). Ubiquitinated histones can be transformed into a non-modified form by deubiquitinating enzymes. Another histone modification, isomerization, is a non-covalent histone modification and takes place primarily on the proline amino acids of histone H3. It not only controls transcriptional activity but is also involved in the methylation of lysine residues on histone tails (Nelson et al. 2006). ADP-ribosylation, catalyzed by ADP-ribose transferases, occurs in the tails of all histone types for the purpose of mediating DNA damage response and other basic cellular processes (Messner et al. 2010).
In the following sections, we will focus exclusively on the spatiotemporal modulation and functions of histone methylation by specific histone methyltransferases during oocyte maturation and early embryonic development.
Histone methylation
Histone methylation was first described by Allfrey and Mirsky in the early 1960s (Allfrey and Mirsky 1964). This modification commonly takes place in the arginine and lysine residues localized in the N-terminal tails of histones H3 and H4 (Dambacher et al. 2010; Ng et al. 2009).
Methylation of arginine residues
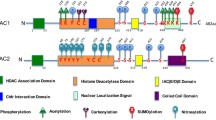
In the arginine residues, methylation can occur as mono- (me1) or dimethylation (me2) [appearing symmetrically (me2s) or asymmetrically (me2a)] (Greer and Shi 2012). This methylation is catalyzed by the family of protein arginine methyltransferases (PRMTs), which transfer methyl groups from S-adenosyl-L-methionine (SAM) to target sites (Bedford and Clarke 2009). Notably, nine members of the PRMT family (type I enzymes, PRMT1, 2, 3, 4, 6, and 8; type II enzymes, PRMT5 and PRMT9; type III enzyme, PRMT7) have been identified in mammals (Yang and Bedford 2013). As can be seen in Fig. 1a, PRMTs contain a widely conserved methyltransferase (MTase) domain, and some of them have extra domain(s) such as Src-homology 3 (SH3), zinc-finger (Zn), myristoylation (Myr), and F-box motifs (Bachand 2007). The MTase domain further includes subdomains for interacting with the methyl donor and substrate proteins. While the SH3 domain in the PRMT3 and PRMT9 proteins provides binding to the proteins with proline-rich motifs (Cura et al. 2017), the N-terminal Myr domain appearing in the PRMT8 protein facilitates plasma membrane targeting (Lee et al. 2005a). The zinc-finger domain of PRMT3 seems to contribute to the arginine methylation process (Frankel and Clarke 2000), while the F-box motifs play a role in substrate recognition (Mason and Laman 2020). Methylation of arginine residues at histone tails through cooperation of these domains functions in activation or suppression of gene expression based on cellular requirement and in chromatin reorganization and localization of transcription factors (Fulton et al. 2018; Di Lorenzo and Bedford 2011).

Schematic structures of arginine and lysine protein methyltransferases and lysine-specific demethylases. a The basic domains of protein arginine methyltransferases (PRMTs). PRMTs contain a conserved methyltransferase (MTase) domain in all members, and some of them have additional domain(s) such as Src-homology 3 (SH3), zinc-finger (Zn), myristoylation (Myr), and F-box motifs. Each domain contributes to methylation of arginine residues. b The basic domains of histone lysine methyltransferases (KMTs). Most KMT families have an evolutionarily conserved SET [su(var)3–9, enhancer of zeste and trithorax] domain. It is noteworthy that the disruptor of the telomeric silencing1-like family does not contain a SET domain. The SET domain-containing lysine methyltransferases are classified into seven subfamilies: SUV39, SET1, EZ, RIZ, SET2, SUV4-20, and SMYD. c The basic domains of lysine-specific demethylases (LSDs or KDMs). LSDs include highly conserved three domains: N-terminal Swi3p/Rsc8p/Moira (SWIRM), amine oxidase-like (AOL), and TOWER. This figure was created utilizing the BioRender program (BioRender; Toronto, Canada)
Methylation of lysine residues
Methylation of histone lysine residues is specifically performed by histone lysine methyltransferases (KMTs) (Cui et al. 2019). It is worth noting that me1, me2, and trimethyl (me3) groups can be added by transferring methyl groups from the methyl donor, SAM, to target regions (Martin and Zhang 2005). Most KMT families have evolutionarily conserved su(var)3–9, enhancer of the zeste and trithorax domain (SET; approximately 130 amino acids in length) responsible for catalytic activity (Jenuwein et al. 1998; Dillon et al. 2005) (Fig. 1b). The SET domain is composed of a conserved anti-parallel beta-barrel and a variable inset that enclose a knot-like structure displaying the enzyme activity (Qian and Zhou 2006). The pre-SET and post-SET domains surrounding the SET domain contain nine cysteines and three zinc conserved sites (Dillon et al. 2005). While the exact function of the cysteine-rich region remains unknown (Scheer and Zaph 2017), the plant homeodomain (PHD) contributes to binding to methylated lysine residues (Hyun et al. 2017). The C2H2 motif serves as zinc-finger binding domain (Dillon et al. 2005), and the lysine-rich motif is an important site for establishing an interaction with ubiquitin and for binding to the nucleosome core (Oh et al. 2010). Basically, SET domain-containing KMTs are classified into seven subfamilies (SUV39, SET1, E2, RIZ, SET2, SUV4-20, and SMYD), and some methyltransferases such as SET7/9 and SET8 even including the SET domain are not included in this classification as they do not share common domains (Dillon et al. 2005).
Another KMT family, disruptor of telomeric silencing 1-like (DOT1L), which does not contain a SET domain, is involved in H3K79 methylation exclusively through its catalytic domain (Feng et al. 2002). Importantly, several KMTs are capable of catalyzing methylation of the same lysine residues. For example, H3K36me2 is carried out by lysine methyltransferases including nuclear receptor binding SET domain protein 1 (NSD1), NSD2, NSD3, and absent, small, or homeotic 1-like (ASH1L), whereas SET domain-containing 2 (SETD2) is the only enzyme performing H3K36me3 in somatic cells (Husmann and Gozani 2019). Also, the six KMTs including SET domain-containing 1A (SETD1A), SETD1B, mixed lineage leukemia protein-1 (MLL1), MLL2, MLL3, and MLL4 can methylate H3K4 (Eissenberg and Shilatifard 2010).
In general, histone lysine methylation leads to changes in the chromatin states, resulting in activation or repression of gene expression (Black et al. 2012). In most genes, while methylation of H3K9, H3K27, and H4K20 represses the transcriptional activity, methylation of H3K4, H3K36, and H3K79 stimulates this process (Black et al. 2012). It is worth noting that there is also crosstalk among histone modifications including lysine methylation, acetylation, phosphorylation, and isomerization (Kouzarides 2007). Regarding this issue, Clements et al. (2003) documented that phosphorylation on histone H3 at serine 10 (H3S10) mediates acetylation of H3K14 by the histone acetyltransferase GCN5 (Clements et al. 2003). Moreover, the isomerization of histone 3 proline 38 (H3P38) is essential for methylation of H3K36 (Nelson et al. 2006).
Histone demethylation
Histone lysine methylation is a reversible process in which methyl groups added by KMTs can be removed by histone lysine demethylases (KDMs) (Black et al. 2012). KDMs are flavin adenine dinucleotide (FAD)-dependent amine oxidases and catalyze demethylation by generating FADH2 (hydroquinone form) and hydrogen peroxide (H2O2) (DesJarlais and Tummino 2016). One of the KDMs, a lysine-specific histone demethylase 1 (LSD1; also known as KDM1A), is part of the co-repressor complex C-terminal binding protein 1 (CtBP1) and performs demethylation of H3K4me1 and H3K4me2 (Shi et al. 2003, 2004). KDM1A includes the Swi3p/Rsc8p/Moira (SWIRM), N-terminal amine oxidase-like (AOL-N), TOWER, and C-terminal AOL (AOL-C) domains (Anand and Marmorstein 2007). The SWIRM domain found in many chromatin-associated proteins has a compact helix-turn-helix-related fold and several shorter helices surrounding the central helix (Yoneyama et al. 2007). Although SWIRM’s function within KDM1A is not clearly revealed, it is known to play a role in binding to DNA and in establishing an association between protein complex and nucleosomal substrates (Qian et al. 2005). The AOL domain is composed of two subdomains, FAD-binding and substrate-binding, both of which comprise a large cavity center for the catalytic process (Yoneyama et al. 2007). Another KDM1A domain, TOWER, which divides the AOL domain, includes a helix-turn-helix formation, and forms a binding site for the partner proteins such as corepressor element silencing factor CoREST (Lee et al. 2005b).
The histone lysine demethylase KDM1B further possesses a zinc-finger domain next to the SWIRM domain at its N-terminus, but there is no TOWER domain (Ismail et al. 2018). The zinc-finger domain contributes to binding the FAD cofactor and forming a structural scaffolding (Zhang et al. 2013). The JmjC proteins also perform demethylation by utilizing the amine oxidase and Jumonji C (JmjC) domains (Black et al. 2012). The JmjC domain interacts with Fe(II) cofactor and α-ketoglutarate in order to provide hydroxylation of methylated substrates (Klose et al. 2006). Notably, one of the JmjC proteins, arginine demethylase, JMJD6, facilitates demethylation of arginine residues (Chang et al. 2007; Walport et al. 2016).
As a result, these lysine and arginine methyltransferases and demethylases are responsible for dynamic changes in histone methylation for regulating gene expression during oocyte maturation, maternal-to-zygotic transition, and early embryogenesis in order to enable proper early development. In the following sections, the spatiotemporal modulation of histone (de)methylation in mammalian oocytes and early embryos will be explained.
Oogenesis from primordial germ cells to mature oocytes
In mice, the precursors of primordial germ cells (PGCs) first appear among epiblast cells at about 6.5 days of embryonic development (E6.5), and then migrate toward the extraembryonic mesoderm at the base of the allantois, where they differentiate into PGCs (McLaren and Lawson 2005). The PGCs migrate to the genital ridges between E9.5 and E11.5 along with the dorsal mesentery of the hindgut (Molyneaux et al. 2001; McLaren and Buehr 1990). In the genital ridges, they undergo a number of mitotic divisions and then differentiate into oogonial stem cells (OSCs). Once OSCs enter the meiotic division at E14.5 following many mitotic divisions, they are called primordial oocytes arrested at the prophase of the first meiotic division. As is well known, these oocytes are surrounded by squamous follicular cells (also known as pre-granulosa cells) to create primordial follicles during the fetal and postnatal early periods (Pepling and Spradling 2001). The primordial follicles serve as ovarian reserve and remain in a dormant state until puberty (Epifano and Dean 2002; Zheng et al. 2014). A subset of primordial follicles can be stimulated based on the balance between activator and inhibitor factors (Oktem and Urman 2010), and begin to grow in a gonadotropin-independent manner at puberty. The primary follicles arising from primordial follicles are composed of a layer of cuboidal granulosa cells enclosing primary oocytes, and then secondary follicles including two or more layers of granulosa cells emerge in a gonadotropin-dependent manner. When small spaces occur between granulosa cells, these follicles are defined as preantral follicles with centrally localized germinal vesicle (GV) oocytes. After formation of a single large antrum by coalescing the small spaces, antral follicles appear. Meanwhile, GV oocytes in the antral follicles undergo nuclear maturation to complete the first round of meiotic division dependent on a luteinizing hormone (LH) surge. The ovulated cumulus–oocyte complexes (COCs) include the secondary oocytes arrested at the metaphase II (MII) phase of the second meiotic division until fertilization, and these oocytes also have a first polar body persistent in the perivitelline space (Sánchez and Smitz 2012).
Dynamic histone methylation in oocytes
As is known, transcription is in an active state in growing GV oocytes during folliculogenesis, but its silencing occurs in the fully grown GV oocytes, having a surrounded nucleolus, progressively condensed chromatin, and alteration of several transcription factors including the TATA binding protein at the time of meiotic resumption (De La Fuente and Eppig 2001; Albertini et al. 2003). Thus, various types of histone methylation, especially on histone H3, play key roles in regulating several cellular events for successful oocyte maturation from the GV to MII stages (Fig. 2).

The main functions of methylation on histone H3 lysine (K) residues in different developmental and intracellular events. The rectangular boxes with different colors on the lysine residues depict the events. This figure was created utilizing the BioRender program (BioRender; Toronto, Canada)
Lysine methylation
An evolutionarily conserved histone modification, H3K4 methylation in the oocyte genome is established in both promoters and bodies of genes to regulate transcriptional activity (Sha et al. 2020). Therefore, H3K4me1, H3K4me2, and H3K4me3 are generally observed in actively transcribing genes (Barski et al. 2007). In mouse oocytes, Dahl et al. (2016) reported that H3K4me3 was not only present in the promoters but was also accumulated in the intergenic regions independent of transcription (Dahl et al. 2016). In the promoters, H3K4me3 is also involved in a time-dependent transcriptional repression of certain genes required for meiotic resumption and embryonic genome activation (EGA) (Dahl et al. 2016; Zhang et al. 2016a). Although the general consensus about H3K4me3 is that it leads to transcriptional activation, its contribution to transcriptional repression during meiotic resumption and EGA in mice may be performed by exclusive cooperation with transcriptional repressors and/or other epigenetic mechanisms such as DNA methylation. Consistent with this suggestion, it was found that MLL2 involved in the establishment of non-canonical H3K4me3 marks interacts with CpG islands (Denissov et al. 2014; Hu et al. 2013).
In human oocytes, H3K4me3 levels were found to decrease from the GV to the MII stage, but it was not significant (Zhang et al. 2012) (Table 1). Hanna et al. (2018) revealed that H3K4me3 is limited to active promoters in non-growing oocytes, and it is also deposited in the intergenic regions, putative enhancers, and silent promoters at the later stages of oogenesis in mice (Hanna et al. 2018). In bovine oocytes, H3K4me3 remains at stable levels from the GV to MII stages (Wu et al. 2020). It is worth noting that the transcript levels of the three KDM5 family demethylases involved in demethylation of H3K4, KDM5A, KDM5B (also known as JARID1B), and KDM5C, were analyzed in porcine oocytes, but no significant differences were found between GV and MII oocytes (Huang et al. 2015) (Table 2). The protein levels of these methyltransferases should be analyzed in the same samples. Together, these results indicate that although there are small differences in H3K4me3 levels among species, the H3K4me1, H3K4me2, and H3K4me3 modifications seem to have important roles in controlling expression of the chromatin modifiers and transcriptional regulators including transcription factors, coactivators, enhancers, and silencers. Thus, they contribute to regulating transcription and chromatin remodeling during oocyte development.
H3K9me2 was found at similar levels during meiotic maturation in human oocytes at the stages of germinal vesicle breakdown (GVBD), MI, and MII (Qiao et al. 2010). This means that H3K9me2 begins to be established in the GV oocytes. Similarly, the genome of mouse oocytes had H3K9me2 and H3K9me3 at the GV and MII stages during meiotic maturation (Liu et al. 2004). Russo et al. (2013) also noted that the genome of sheep GV oocytes underwent H3K9me3 (Russo et al. 2013). The expression of the SUV39H1 and SUV39H2 genes, both of which catalyze trimethylation of H3K9, showed dynamic changes in bovine oocytes produced by in vitro maturation (IVM) (Zhang et al. 2016b). The accumulated H3K9me3 level in the GV oocytes disappeared in the MII oocytes. Consistently, SUV39H1 and SUV39H2 mRNA levels in the MII oocytes were dramatically lower than in the GV oocytes (Table 2).
The H3K79me2 and H3K79me3 signals were also analyzed in mouse oocytes, and they were detected in the GV, GVBD, and MII stages (Ooga et al. 2008). These modifications may be regulated by specific nuclear factors such as SMYD3 and DOT1L present in the GV oocytes during folliculogenesis (Phillips et al. 2016). As is known, H3K27 methylation is catalyzed by polycomb repressive complex 2 (PRC2) and represses gene transcription (Wiles and Selker 2017). Trimethylation of H3K27 at unmethylated genomic regions leads to transcriptional repression, and this modification is removed during oocyte maturation for facilitating transcription of the related genes (Zheng et al. 2016). Analysis of H3K27 methylation levels in porcine oocytes during maturation showed that H3K27me1, H3K27me2, and H3K27me3 signals were intensively present in the nuclei of the GV and MII stages (Marinho et al. 2017) (Table 1).
The enhancer of zeste homolog 2 (Ezh2; a catalytic subunit of the PRC2 complex performing trimethylation of H3K27) gene expression at the mRNA and protein levels exhibited a similar pattern as the H3K27me3 levels during oocyte maturation in mice (Qu et al. 2016). Specifically, the EZH2 protein level gradually increased from the GVBD to MII stages, but was found at a low level in the GV oocytes. The KDM6B protein [also known as Jumonji domain-containing protein 3 (JMJD3)] carries out demethylation of H3K27me3 to permit gene transcription, and the KDM6B transcript was found at high levels in bovine GV and MII oocytes (Canovas et al. 2012). In future work, the KDM6B protein levels should be evaluated in the same oocytes to more reliably compare the expression distribution with histone methylation levels during oocyte maturation. As there are a limited number of studies that have examined dynamic changes in histone methylation-related gene expression, further investigations are required to elucidate the (de)methyltransferases specifically functioning in methylation and demethylation of target histones, and their potential roles during oocyte maturation.
Arginine methylation
The methylation of H3 arginine 17 (H3R17) and H4R3 also affects the transcriptional activation in mouse oocytes during maturation (Sarmento et al. 2004). Indeed, it was shown that H3R17 and H4R3 methylation contribute to forming a complex with coactivators to facilitate the binding of histone acetyltransferases so that gene transcription can occur (Wang et al. 2001; Bauer et al. 2002; Li et al. 2010b). Sarmento et al. (2004) revealed that H3R17 and H4R3 methylation existed in the chromatin of GV oocytes, whereas these modifications were almost absent in the chromosomes of MII oocytes (Sarmento et al. 2004). The dimethylation of H4R3 was observed in human oocytes at the GV, GVBD, MI, and MII stages (Qiao et al. 2010). The similar distribution of methylation on the arginine and lysine amino acids of histones H3 and H4 in the oocytes at different stages suggests that they may be localized in the same or nearby nucleosomes to control transcriptional activity and genomic reorganization during maturation.
Early embryonic development from one-cell to blastocyst
When a competent sperm cell fertilizes a mature oocyte, the second meiotic division is completed, and a one-cell embryo (also known as a zygote) having male and female pronuclei is formed. The pronuclei replicate their genomes as they approach each other, and then fuse within 24 h of fertilization (Li et al. 2010a). The first cleavage division occurs 1 day after fertilization whereby a two-cell embryo including two even blastomeres is produced (Fujimori 2010). The four-cell embryo, 12 h after first cleavage, and then the eight-cell embryo emerge at the end of the second and third mitotic divisions, respectively. The eight-cell embryo experiences a compaction process, and subsequently a morula composed of 12–16 embryonic cells which are tightly aggregated is formed. Eventually, a blastocyst arises, containing two distinct cell types, inner cell mass (ICM) and outer cells (also known as trophectoderm), with different polarization (Grobstein 1985).
The early embryonic development is mainly regulated by epigenetic mechanisms including histone methylation. The genome of early embryos developing from one-cell to blastocyst stages undergoes methylation, especially on histone H3, to regulate molecular events such as EGA, X-chromosome inactivation, genomic imprinting, and chromatin reorganization required for proper early development (Li 2002) (Fig. 2).
Dynamic histone methylation in early embryos
H3K4 methylation
Among the methylation on histone H3, the H3K4 methylation profile was analyzed by Shao et al. (2014) in mouse early embryos from one-cell to blastocyst stages (Shao et al. 2014). Although H3K4me1 was at a higher level in the two-cell embryos when compared to the others, H3K4me2 at low levels in the four-cell and eight-cell embryos increased in the one-cell embryo, morula, and blastocyst, and was further enhanced in the two-cell embryos. On the other hand, the lowest H3K4me3 level in the one-cell embryos increased in the four-cell and eight-cell embryos, was further enhanced in the four-cell embryos, and reached the highest levels in the morula- and blastocyst-stage embryos (Table 1). In the bovine early embryos, H3K4me3 at low levels in the eight-cell and morula-stage embryos increased in the blastocysts, and was further enhanced in the two-cell and four-cell embryos (Wu et al. 2020). Zhang et al. (2012) evaluated the H3K4me3 distribution in human early embryos, and found that it was at high levels in the one-cell and four-cell embryos, decreased in the blastocysts, and further reduced in the eight-cell embryos (Zhang et al. 2012) (Table 1). Taken together, these reports show that H3K4me3 levels exhibit remarkable differences during early embryonic development among these species, and the fluctuation in H3K4me3 levels in the early embryos indicates that it may function in regulating temporal gene expression. The mRNA levels of H3K4 demethylases including KDM5A, KDM5B, and KDM5C were evaluated in porcine embryos, and revealed that although there were no differences in the one-cell, two-cell, four-cell, and blastocyst-stage embryos for KDM5A and KDM5C gene expression, the KDM5B transcript levels increased in the two-cell embryos, and were further enhanced in the blastocysts (Huang et al. 2015) (Table 2). Although there seems to be a correlation between H3K4 methylation levels and the KDM transcript levels, their protein levels should be explored in the same species to reach a consensus on the relationship between the histone methylation and demethylase levels in early embryos.
As EGA facilitates successful embryonic development, its inhibition by 5,6-dichloro-1-β-d-ribofuranosyl-benzimidazole blocking RNA polymerase II results in reduced H3K4me3 levels (Abe et al. 2018). A study by Aoshima et al. (2015) reported that H3K4me1, H3K4me2, and H3K4me3 marks involved in active transcription are associated with EGA minimally appearing in one-cell embryos, and the loss of H3K4 marks led to decreased EGA in mice (Aoshima et al. 2015). In the same study, it was also revealed that knockdown of Mll3/4 gene (encoding an enzyme carrying out H3K4 methylation) expression in the one-cell embryos caused embryonic developmental arrest (Aoshima et al. 2015). These findings indicate that EGA as a crucial process for early developmental progression seems to be regulated by changes in the H3K4 methylation marks and the corresponding enzymes.
During fertilization, the male pronucleus was found to undergo a major exchange of protamines with maternal histones (Derijck et al. 2006; Svoboda 2018). Although H3K4me2 and H3K4me3 modifications were not initially observed in the male pronucleus, both pronuclei had H3K4me1 at similar levels in the mouse zygote (Lepikhov and Walter 2004). Later in development, the male pronucleus acquired H3K4me2 and H3K4me3 in the one-cell embryos, and these modifications were maintained during early cleavage divisions in mice (Aoshima et al. 2015). Therefore, maternal H3K4me3 undergoes active demethylation by the demethylases KDM1A and KDM5B to establish proper H3K4 methylation marks for normal EGA and early development (Zhang et al. 2016a; Dahl et al. 2016).
H3K27 methylation
The trimethylation of H3K27 plays a crucial role in establishing genomic imprinting and transcriptional repression in early embryos (Inoue et al. 2017; Zheng et al. 2016). It was found that in mice, the H3K27me3 signal in the morula was more intense than that in the blastocysts (Wu et al. 2014). This modification at low levels in the eight-cell and blastocyst-stage embryos increased in the one-cell stage, further increased in the four-cell stage, and reached the highest level in the two-cell embryos, which showed significant differences from H3K27me1 and H3K27me2 levels analyzed in the same embryos in a porcine model (Marinho et al. 2017). The H3K27me1 and H3K27me2 at high levels in the GV and MII oocytes decreased in the early embryos at the one-cell, two-cell, four-cell, and eight-cell stages, and increased again in the blastocysts. Fu et al. (2017) also evaluated H3K27me3 levels in bovine embryos and found that levels were low in the eight-cell embryos, then increased in the blastocysts and reached high levels in the two-cell and four-cell embryos (Table 1). It is important to note that H3K27me3 exhibits similar distributions in the early embryos of porcine and bovine models, suggesting that this mark may perform the same function in these species during early embryonic development.
The gene expression of a H3K27me3 histone methyltransferase Ezh2 in mice was found at the highest level in the one-cell embryos and was reduced in the two-cell, four-cell, and eight-cell early embryos having the same levels as the MII oocytes, further decreased in the morula, and reached the lowest level in the blastocysts (Wu et al. 2014). Huang et al. (2014) also demonstrated that EZH2 was intensively localized in the nuclei of early embryos from one-cell to blastocyst stages (Huang et al. 2014). The transcript level of H3K27me3 demethylase KDM6B in bovines decreased significantly from one-cell to morula-stage embryos, and there a level of higher expression was found in blastocysts than in the early cleavage embryos (Canovas et al. 2012) (Table 2). Although there are similar and different expression distributions of the Ezh2 and KDM6B genes during early development, their protein levels should be analyzed in the early embryos of these species to address their potential relationship with H3K27me3 accumulation.
H3K9 methylation
H3K9me2 was found to be absent or weakly present in the male pronucleus, whereas it was strongly detected in the female pronucleus of mouse one-cell embryos (Liu et al. 2004). The same study revealed that the H3K9me2 levels in the one-cell and two-cell embryos at the G2 stage were reduced by almost half of those observed at their G1 stage. In bovine preimplantation embryos obtained by in vitro fertilization (IVF) procedure, although there were no significant differences for the H3K9me3 levels during early development, high levels were found in the early embryos from two-cell to blastocyst stages (Zhang et al. 2016b). In contrast, the expression of histone H3K9me3 methyltransferase SUV39H1 in bovines was at high levels in the two-cell embryos but significantly decreased in the four-cell embryos, and no expression was found in the eight-cell embryos and blastocysts (Zhang et al. 2016b). The expression of another histone H3K9me3 methyltransferase, SUV39H2, was found at low levels in the two-cell embryos, increased significantly in the four-cell embryos, and then predominantly decreased in the eight-cell embryos and blastocysts (Zhang et al. 2016b). Although only mRNA levels of these genes were analyzed in this study, they did not exhibit comparable expression patterns in the early bovine embryos. This suggests that other methyltransferases such as G9a and PRDM family members may participate in H3K9 methylation, or that levels of the demethylases including JHDM2, JHMD3, and PHF8 are increased in the early embryonic stages. It is worth noting that the dynamic changes in SUV39H1 and SUV39H2 gene expression in early embryos might be associated with their potential roles in chromosome segregation, cell division, and DNA repair (Peters et al. 2001; Sidler et al. 2014).
H3K79 methylation
H3K79me2 and H3K79me3 modifications were found to accumulate in mouse oocytes during meiotic maturation, and a weak signal was present in the female pronucleus following fertilization, but there was no signal in the male pronucleus (Ooga et al. 2008). The weak signal in the two-cell, four-cell, and morula-stage embryos significantly increased in the blastocysts. The expression of the Dot1L gene performing H3K79 methylation was also evaluated in mouse early embryos by the same group (Ooga et al. 2013). The study showed that Dot1L mRNA at a high level in the one-cell embryos decreased significantly in the two-cell and four-cell embryos, and increased at the blastocyst stage. Analogously, the DOT1L protein intensity at high levels in the one-cell and two-cell embryos decreased to a low level in the four-cell embryos, and then increased dramatically in the blastocysts (Table 2). When H3K79 methylation marks were compared with the Dot1L gene expression, the H3K79me2 and H3K79me3 levels partially overlapped with the DOT1L protein levels in the early embryos from one-cell to blastocyst stages (Ooga et al. 2013).
DOTL1-mediated H3K79 methylation was found to function in meiotic progression in mouse oocytes because its suppression using siRNA technology resulted in markedly reduced H3K79 methylation levels, which blocked the division at the first meiotic metaphase (Wang et al. 2014). Consistent with the known roles of DOT1L, its loss led to developmental defects including growth impairment, abnormal angiogenesis in the yolk sac, and cardiac dilation, and these embryos died between 9.5 and 10.5 days post coitum (Jones et al. 2008). Moreover, Dot1L-deficient embryonic stem cells displayed global loss of H3K79 as well as decreased heterochromatic marks at the centromeres and telomeres. A recent article reported that maternal DOT1L accumulation is not required for successful early mouse development (Liao and Szabo 2020). Most likely, DOT1L expression and establishment of H3K79 methylation are mainly regulated by the embryonic genome following EGA. All these findings indicate that in concert with other histone methylation, regulation of both H3K79 methylation and spatiotemporal expression of the Dot1L gene is a crucial process for generating competent early embryos.
Lack of histone methyltransferases in oocytes and early embryos
Many studies have examined the functional impacts of the depletion or lack of genes involved in histone methylation and demethylation processes during oocyte maturation and early embryonic development. One of these studies was on the histone-lysine N-methyltransferase SETD1B (also referred to as H3K4 methyltransferase) involved in regulating gene expression during oocyte maturation (Sha et al. 2020). The conditional knockout (cKO) of Setd1b in mouse oocytes led to downregulation of the expression of oogenic genes such as oocyte maturation, alpha (Omt2a), oocyte maturation, beta (Omt2b), oocyte-secreted protein 1 (Oosp1), and oogenesin 3 (Oog3), having roles in transcriptional regulation and cell cycle reorganization (Brici et al. 2017). Zona pellucida defects, abnormal cytoplasmic maturation, and polyspermy have also been found to occur in the oocytes with SETD1B deficiency, but there is no effect on the primordial germ cell migration. Additionally, loss of SETD1B was observed to cause a developmental defect in oocyte-to-embryo transition based on morphological evaluation, impaired chromatin configuration from non-surrounded nucleolus (NSN; type of chromatin configuration showing transcriptional activity) to surrounded nucleolus (SN; displaying transcriptional silencing), formation of smaller meiotic spindles, arrest at the one-cell embryonic stage, and eventually female infertility (Brici et al. 2017) (Table 3). In a recently published study, GV oocyte-specific ablation of Setd1b resulted in an impaired oogenic gene expression program in mouse GV and MII oocytes (Hanna et al. 2021). Furthermore, in the Setd1b cKO oocytes, H3K4me3 levels decreased in the promoters of transcriptionally active and inactive genes, but some genes having a CpG-rich region gained H3K4me3 marks and subsequently underwent hypomethylation. The resulting impairments in the oocytes and early embryos due to the loss of the Setd1b gene suggest that H3K4 methylation acts as an orchestral conductor in modulating early development-related gene expression.
Loss of the Setdb1 gene
The loss of the H3K9 methyltransferase Setdb1 (performing di- and trimethylation) in mice using cKO technology caused severe defects in meiotic arrest and meiotic resumption during oocyte maturation (Kim et al. 2016). Moreover, it led to increased double-strand breaks in the oocytes, defective mitotic cell cycle progression in the one-cell embryos, and progressive developmental delay in the early embryos, which were therefore unable to reach the blastocyst stage (Table 3). The same study further revealed that SETDB1 is required not only for modulating global H3K9me2 levels but also for silencing of retrotransposons such as long terminal repeats (LTRs) in the growing oocytes. Restoring SETDB1 activity in the Setdb1 cKO GV oocytes could completely rescue the defects related to meiosis and early embryonic development (Kim et al. 2016).
Eymery et al. (2016) also created cKO of the Setdb1 gene in growing mouse oocytes to explore its function during meiotic maturation and early embryonic development (Eymery et al. 2016). Although SETDB1-deficient oocytes were able to undergo GVBD (the first step of meiotic maturation) with delayed time, they were unable to complete meiotic maturation, most likely due to altered expression of the genes involved in cell cycle and chromosome condensation. The loss of Setdb1 also caused aneuploidy owing to defective kinetochore–microtubule interactions, and impaired protein kinase A signaling in the mouse oocytes (Eymery et al. 2016). Histone H3K9 methyltransferase Eset (involved in heterochromatin formation and gene silencing during early embryogenesis) mutant mice were generated to investigate its role during preimplantation embryonic development (Dodge et al. 2004). In this study, it was shown that although maternal Eset transcripts existed in oocytes and were maintained during preimplantation development, embryonic Eset began to be expressed from the blastocyst stage and persisted throughout postimplantation development. The lack of the Eset gene caused abnormalities in development of the ICM in the blastocysts, establishment of embryonic stem cells, and peri-implantation lethality at 9.5–16.5 days post coitum (Table 3). However, there was no change observed in the global H3K9me3 level or DNA methylation in the Eset−/− blastocysts. Therefore, the researchers suggested that maternally stored ESET likely provides for maintenance of H3K9me3 levels during early embryonic development. Importantly, ESET may have further functions in addition to its primary role in H3K9 methylation, since different phenotypic effects emerge upon lack of this protein during the pre-, peri-, and postimplantation periods. Overall, these studies by Kim et al. (2016) and Eymery et al. (2016) on the Setdb1-knockout mice models reached comparable results, including reduced global H3K9me2 levels in the oocytes, defective meiotic maturation, abnormal kinetochore–microtubule interactions and spindle organization, defects in cell cycle progression and chromosome segregation, and impaired early embryonic development (Table 3).
Loss of the Setd2 gene
The histone-lysine N-methyltransferase SETD2 was found to exhibit strong intensity in the nucleus of NSN-type porcine oocytes, whereas there was no expression in the nuclei of the SN, MI, or MII oocytes (Diao et al. 2018). A weak cytoplasmic SETD2 distribution was observed in the SN, MI, and MII oocytes. SETD2 catalyzes H3K36me3 for transcriptional activation, and its knockdown using siRNA led to impairment in both meiotic maturation and first polar body extrusion in mouse oocytes (Li et al. 2018). The immunostaining of Setd2-knockdown oocytes for the tubulin protein showed that there was a kinetochore–microtubule mis-attachment, incorrect chromosome alignment, and an increased rate of aneuploidy. A later study by Xu et al. (2019) reported that maternal SETD2 deficiency led to reduced H3K36me3 levels, decreased MII oocyte number, and abnormal DNA methylome in mouse oocytes (Xu et al. 2019). Moreover, it caused the loss of maternal imprints and ectopic H3K4me3 formation at the imprinting control regions, arrest at the one-cell-stage embryos, failure of DNA replication and EGA, and abnormal epigenome creation in the early embryos derived from aberrant parental epigenomic reprogramming, as well as inhibited preimplantation and postimplantation development. These phenotypes (Table 3) that appeared due to maternal depletion of SETD2 indicate that SETD2 is required for properly establishing a maternal epigenome, which is essential for successful early embryonic development.
Loss of other histone methyltransferases
Another histone lysine methyltransferase, MLL2, contributes to regulating gene expression by way of catalyzing trimethylation of H3K4 in oocytes and early embryos (Andreu-Vieyra et al. 2010). The cKO of Mll2 using Gdf9 promoter in mouse oocytes caused several defects involving ovarian follicle loss, impaired ovulation, increased oocyte death, failure of transcriptional repression, and abnormal histone modifications (Andreu-Vieyra et al. 2010) (Table 3). As a result, the loss of MII2 led to female sterility due to the emerging phenotypes.
Depletion of the lysine methyltransferase Ezh2 by Morpholino antisense oligos resulted in chromosome misalignment, disrupted kinetochore–microtubule interactions, abnormal spindle formation, aneuploidy, and accelerated first polar body extrusion in mouse oocytes (Qu et al. 2016). The overexpression of Ezh2 in the same study by injecting its mRNA into oocytes led to chromosome misalignment, aneuploidy, and impaired first polar body extrusion. The researchers also documented that although methyltransferase activity of EZH2 was not required for meiotic maturation in the oocytes, it was essential for maintaining the stability of the spindle assembly checkpoint protein BUBR1. In a previous study, one-cell embryos were treated with Ezh2 siRNA (Huang et al. 2014). Severe growth retardation in the early embryos and reduced rates of blastocyst formation, along with an increased number of cracked (having morphologically abnormal blastomeres and zona pellucida) and dead early embryos, were determined in the siRNA-treated group. In addition, expression of the pluripotency-associated transcription factor genes (Oct4, Sox2, and Nanog) and H3K27me2 and H3K27me3 levels decreased markedly in the early embryos injected with Ezh2 siRNA (Huang et al. 2014) (Table 3). These investigations suggest that the EZH2 protein at required levels plays key roles in oocyte maturation and early embryonic development via methylation of the target genes in a spatiotemporal manner.
The functional importance of the G9a gene [also known as euchromatic histone lysine methyltransferase 2 (EHMT2), a H3K9 methyltransferase] was also examined in mice (Yeung et al. 2019). The oocyte-specific cKO of G9a led to various types of defects, including impaired chromatin reorganization, decreased H3K9me2 levels, developmental defects, reduced CG and non-CG methylation levels, changes in the expression of the genes and endogenous retrovirus, abnormal chromosome segregation and arrest in the oocytes or early embryos, and reduced female fertility. Additionally, the proportions of the transcriptionally inactive and SN-type oocytes decreased in the G9a cKO ovaries (Yeung et al. 2019). Similarly, it was reported in a previous study that G9a-deficient embryos showed markedly decreased H3K9 methylation profiles, mainly in euchromatic regions, severe growth retardation, and early lethality at E9.5–E12.5 (Tachibana et al. 2002) (Table 3).
In a recently published study, another histone lysine methyltransferase, ASH1L (making H3K36 methylation for stimulating gene expression), was knocked down using siRNA in bovine cumulus cells (Cui et al. 2021). The decrease in ASH1L resulted in reduced H3K36me1, H3K36me2, and H3K36me3 levels, increased apoptosis rate, and reduced proliferation in cultured bovine cumulus cells. The authors concluded that establishment of histone methylation in the granulosa cells during folliculogenesis may be an important process for generating good-quality oocytes and early embryos.
As revealed in the abovementioned studies (Table 3), establishing methylation on target histones is very important for proper progression of oocyte maturation and early embryonic development. However, further studies are needed to identify the molecular determinants and mechanisms underlying the emergence of those phenotypic defects that derive from the loss of histone methyltransferases and histone methylation. Also, it remains unknown whether potential functions of histone methyltransferases other than their primary role may have any impact on the emergence of those phenotypes.
Loss of the histone demethylases in oocytes and early embryos
The loss of H3K4 demethylases
In addition to examining the effects of histone methyltransferases loss, the potential impacts of the lack of histone demethylases in oocytes and early embryos were also evaluated. Although there was no effect of the disrupted histone H3K4 demethylase Kdm1b gene on early mouse development and oogenesis, oocytes from KDM1B-deficient females showed markedly increased H3K4 methylation and defects in establishing methylation marks in the maternally imprinting genes Mest, Grb10, Zac1, and Impact (Ciccone et al. 2009). Moreover, the early embryo produced from these oocytes exhibited biallelic expression or biallelic suppression of the affected genes, and died before mid-gestation (Table 4). Another H3K4 demethylase, the LSD1 gene, was observed at higher levels in MII oocytes compared to GV oocytes during IVM in goats (Liu et al. 2020). Additionally, the inhibition of LSD1 by its specific inhibitor, GSK-LSD1, led to a reduced first polar body extrusion rate, abnormal spindle assembly, misaligned chromosomes, and an increase in H3K4me2 levels in oocytes.
The demethylation of H3K4me2 and H3K4me3 performed by KDM5B is a crucial process for proper early embryonic development (Han et al. 2017). The lack of Kdm5b in mice resulted in increased H3K4me3 levels in the early embryos and postnatal lethality in the majority of pups (Albert et al. 2013). Furthermore, functional defects in the respiratory system, disorganized cranial nerves, aberrant eye development, and homeotic skeletal transformations were observed. By contrast, Zou et al. (2014) revealed that the majority of Kdm5b knockout mouse embryos survived beyond the neonatal stage (Zou et al. 2014). These mice exhibited reduced body weight and female fertility rates, increased mortality, and delayed mammary gland development (Table 4). The differences between the two studies may derive from the use of different strategies to create the knockout mouse models.
The altered expression of the H3K27, H3K9, and H3K36 demethylases
The knockdown of the KDM5B gene expression by Huang et al. (2015) in porcine MII oocytes led to increased abundance of H3K4me3 in four-cell and blastocyst-stage embryos, but a decrease in H3K27me3 levels in blastocysts (Huang et al. 2015). Also, impaired early embryonic development and enhanced expression of the homeobox (HOX, undergoing silencing during early development) and ten-eleven translocation (TET, having a role in DNA methylation) genes were observed in the knockdown group (Table 4). Notably, changes in the TET genes (TET1, TET2, and TET3) expression indicated an association between the establishment of histone methylation and DNA methylation. The researchers also suggested based on their results that a decreased H3K27me3 profile in the blastocysts may arise from the upregulation of histone demethylase KDM6A.
The H3K27me3 demethylase KDM6B (also called JMJD3) gene expression was knocked down in bovine MII oocytes using siRNA injection in order to investigate its roles during early embryonic development (Canovas et al. 2012). The H3K27me3 levels that normally decrease in the preimplantation embryos did not change in the cleavage-stage embryos produced from KDM6B-knockdown oocytes, and similarly there was no remarkable change observed in the early embryos derived from the parthenogenetically activated MII oocytes. Additionally, the rate of blastocyst formation decreased in the KDM6B knockdown group when compared to their control counterparts (Canovas et al. 2012). In parallel with the previous study, knockdown of the histone demethylase JMJD1C (involved in demethylation of H3K9me1 and H3K9me2) gene expression using siRNA technology in bovine MII oocytes caused reduced rates of both blastocyst formation and cleavage of one-cell embryos after IVF (Li et al. 2015) (Table 4). These studies suggest that these demethylases contribute to temporally establishing the H3K9 and H3K27 methylation states for proper early embryonic development in bovines.
To investigate the functional importance of KDM1A catalyzing H3K9me2 as well as H3K4me1 and H3K4me2 demethylation during early embryogenesis, the Kdm1a gene was maternally deleted in a mouse model (Ancelin et al. 2016). The early embryos developed from Kdm1a-knockout oocytes were arrested at the two-cell stage. In the same study (Ancelin et al. 2016), the one-cell embryos from wild-type mice were treated with pargyline, an inhibitor of KDM1A enzymatic activity, to evaluate its importance for preimplantation embryonic development. Most one-cell embryos treated with pargyline showed arrested development at the two-cell stage, and some of them failed to develop beyond the four-cell stage, as was observed in the Kdm1a knockout models. On the other hand, maternal absence of KDM1A did not affect H3K4me1, H3K4me2, H3K4me3, H3K9me1, or H3K9me2 levels in the one-cell embryos but caused an abnormal increase in H3K9me3. In the two-cell embryos, there were significant increases in the H3K4 and H3K9 methylation states (-me1, -me2, and -me3), impairment of the normal changes of transcriptome, and enhanced LINE-1 protein levels and γH2AX foci in the Kdm1a-depleted group. Another study by Wasson et al. (2016) reported that deficiency of maternally expressing Kdm1a led to early developmental arrest at the one-cell or two-cell stage and a failure of maternal-to-zygotic transition in mice (Wasson et al. 2016). Also, the hypomorphic phenotypes including decreased litter size, increased perinatal lethality, enhanced fragmented and multicellular embryos, and abnormal behavior and imprinting defects in the progeny were detected in the Kdm1a knockout group (Table 4). Taken together, these studies indicate that KDM1A is a key player in regulating early and postnatal embryonic development. Importantly, other factors involved in modulating Kdm1a gene expression in oocytes and early embryos should be determined to understand the background of the phenotypes that emerge in the loss of KDM1A.
As KDM4A is involved in the demethylation of H3K9me2 and H3K9me3, as well as H3K36me2 and H3K36me3, maternal expression of the Kdm4a gene is critical for embryo survival and female fertility in mice (Sankar et al. 2017). Expectedly, Kdm4a knockout mice exhibited a lower implantation rate, decreased developmental potential, and female infertility when compared to wild-type mice (Table 4). It is worth noting that no global change in H3K9me3 methylation level was observed in the absence of KDM4A. This may reflect the compensating for the loss of KDM4A by other demethylases such as KDM4B and KDM4C. In a newly published study, it was reported that KDM4A-mediated H3K9me3 demethylation in mouse oocytes is required for EGA and normal early embryonic development (Sankar et al. 2020). In the impairment of H3K9me3 demethylation, most genes expressed during EGA undergo repression in the two-cell embryos.
Overall, the studies evaluating the potential roles of the histone demethylases suggest that KDMs play crucial roles in temporally regulating the expression of the genes required for oocyte maturation, early embryonic development, and postnatal development. It is worth noting that histone methyltransferases and demethylases work in balance for reorganizing chromatin. Further studies are needed to uncover the possible relationship between histone methyltransferases and demethylases, as well as their interaction with other epigenetic mechanisms, for example, DNA methylation.
The potential effect of aging on histone methylation
Reduced reproductive features accompanied by biological aging are closely associated with decreased oocyte quality and defective embryonic development (Qiao et al. 2014; Cui et al. 2013). These unfavorable outcomes may arise in part from failure to establish proper histone methylation. The potential effects of the early postnatal development periods on histone methylation were evaluated in growing mouse GV oocytes (Kageyama et al. 2007). The levels of H3K4me2, H3K4me3, H3K9me2, and H3K9me3 were higher in the growing oocytes from a 15-day-old mouse compared with 5- to 10-day-old mice. Expectedly, the Set7 (performing H3K4me2), Mll (performing H3K4me3), and G9a (performing H3K9me2) genes exhibited higher expression levels in the oocytes of a 15-day-old mouse than in the 5- and 10-day-old groups. The researchers suggested that the increase in H3K4 methylation profiles may be associated with the change in chromatin configuration independent of transcription; however, the altered H3K9 methylation levels seem to be related to the suppression of gene expression and the formation of a heterochromatin structure during oocyte growth.
A study by Manosalva and Gonzalez (2010) analyzed the changes in histone methylation levels in the GV and MII oocytes obtained from young (2-month-old) to old (11-month-old) mice (Manosalva and Gonzalez 2010). The H3K9me3, H3K36me2, H3K79me2, and H4K20me2 levels decreased significantly in the GV and MII oocytes of the old mice, and lower H3K4me2 and H3K9me2 levels were found in the MII oocytes from the old mice when compared with the GV and MII oocytes of young mice (Manosalva and Gonzalez 2010). Furthermore, the number of oocytes with NSN was lower in the old mice than young ones. These findings indicate that changes in histone methylation profiles in oocytes with aging may alter the chromatin configuration, influencing temporal transcriptional activity. Another study on this subject revealed that H3K4me2 and H3K4me3 levels were reduced in the GV oocytes of old mice (42–44 weeks old), but the H3K4me2 level increased significantly in the MII oocytes of old mice when compared with their young (6–8 weeks old) counterparts (Shao et al. 2015). In a recently published study, Petri et al. (2020) reported that in vitro postovulatory aging enhanced H3K9me3 levels in mouse MII oocytes (Petri et al. 2020). As a result, histone methylation exhibited dynamic changes from GV to MII oocytes during maturation, and biological aging seemed to be a prominent factor affecting methylation levels. More work is required to determine the short- and long-term influences of histone methylation changes due to biological aging on oocyte and early embryo quality.
Conclusion
In conclusion, correctly establishing methylation of histones in a spatiotemporal manner contributes to proper completion of oocyte maturation and pre- and postimplantation development. For this purpose, dynamic changes in the expression levels of KMTs and HDMs take place in oocytes and early embryos. The lack or overproduction of these enzymes leads to altered histone methylation profiles that cause impaired oogenic and/or embryonic gene expression, abnormal EGA, arrest in early embryos, and even embryonic lethality. Unsuitable changes in histone methylation patterns, as well as other epigenetic modifications, may be one of the main reasons for decreased reproductive functions and ultimately female infertility. Further studies are needed to elucidate the mechanisms modulating histone methylation and demethylation processes during oocyte maturation and early embryogenesis. Findings from these studies would aid in determining the molecular background of female infertility deriving from inappropriate changes in histone methylation, and in the discovery of new treatment strategies to maintain female fertility for a longer period.
References
Abe KI, Funaya S, Tsukioka D, Kawamura M, Suzuki Y, Suzuki MG, Schultz RM, Aoki F (2018) Minor zygotic gene activation is essential for mouse preimplantation development. Proc Natl Acad Sci USA 115(29):E6780–E6788. https://doi.org/10.1073/pnas.1804309115
Ajiro K (2000) Histone H2B phosphorylation in mammalian apoptotic cells. An association with DNA fragmentation. J Biol Chem 275(1):439–443. https://doi.org/10.1074/jbc.275.1.439
Albert M, Schmitz SU, Kooistra SM, Malatesta M, Morales Torres C, Rekling JC, Johansen JV, Abarrategui I, Helin K (2013) The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet 9(4):e1003461. https://doi.org/10.1371/journal.pgen.1003461
Albertini DF, Sanfins A, Combelles CM (2003) Origins and manifestations of oocyte maturation competencies. Reprod Biomed Online 6(4):410–415. https://doi.org/10.1016/s1472-6483(10)62159-1
Allfrey VG, Mirsky AE (1964) Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science 144(3618):559. https://doi.org/10.1126/science.144.3618.559
Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y (2007) New nomenclature for chromatin-modifying enzymes. Cell 131(4):633–636. https://doi.org/10.1016/j.cell.2007.10.039
Anand R, Marmorstein R (2007) Structure and mechanism of lysine-specific demethylase enzymes. J Biol Chem 282(49):35425–35429. https://doi.org/10.1074/jbc.R700027200
Ancelin K, Syx L, Borensztein M, Ranisavljevic N, Vassilev I, Briseno-Roa L, Liu T, Metzger E, Servant N, Barillot E, Chen CJ, Schule R, Heard E (2016) Maternal LSD1/KDM1A is an essential regulator of chromatin and transcription landscapes during zygotic genome activation. Elife. https://doi.org/10.7554/eLife.08851
Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K, Stewart AF, Matzuk MM (2010) MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. https://doi.org/10.1371/journal.pbio.1000453
Aoshima K, Inoue E, Sawa H, Okada Y (2015) Paternal H3K4 methylation is required for minor zygotic gene activation and early mouse embryonic development. EMBO Rep 16(7):803–812. https://doi.org/10.15252/embr.201439700
Bachand F (2007) Protein arginine methyltransferases: from unicellular eukaryotes to humans. Eukaryot Cell 6(6):889–898. https://doi.org/10.1128/EC.00099-07
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (2007) High-resolution profiling of histone methylations in the human genome. Cell 129(4):823–837. https://doi.org/10.1016/j.cell.2007.05.009
Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T (2002) Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep 3(1):39–44. https://doi.org/10.1093/embo-reports/kvf013
Bedford MT, Clarke SG (2009) Protein arginine methylation in mammals: who, what, and why. Mol Cell 33(1):1–13. https://doi.org/10.1016/j.molcel.2008.12.013
Bird A (2007) Perceptions of epigenetics. Nature 447(7143):396–398. https://doi.org/10.1038/nature05913
Black JC, Van Rechem C, Whetstine JR (2012) Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 48(4):491–507. https://doi.org/10.1016/j.molcel.2012.11.006
Brici D, Zhang Q, Reinhardt S, Dahl A, Hartmann H, Schmidt K, Goveas N, Huang J, Gahurova L, Kelsey G, Anastassiadis K, Stewart AF, Kranz A (2017) Setd1b, encoding a histone 3 lysine 4 methyltransferase, is a maternal effect gene required for the oogenic gene expression program. Development 144(14):2606–2617. https://doi.org/10.1242/dev.143347
Canovas S, Cibelli JB, Ross PJ (2012) Jumonji domain-containing protein 3 regulates histone 3 lysine 27 methylation during bovine preimplantation development. Proc Natl Acad Sci USA 109(7):2400–2405. https://doi.org/10.1073/pnas.1119112109
Cao X, Dang W (2018) Histone modification changes during aging: cause or consequence?—what we have learned about epigenetic regulation of aging from model organisms. Epigenetics of aging and longevity. Elsevier, pp 309–328
Cao J, Yan Q (2012) Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front Oncol 2:26. https://doi.org/10.3389/fonc.2012.00026
Chang B, Chen Y, Zhao Y, Bruick RK (2007) JMJD6 is a histone arginine demethylase. Science 318(5849):444–447. https://doi.org/10.1126/science.1145801
Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T (2009) KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461(7262):415–418. https://doi.org/10.1038/nature08315
Clements A, Poux AN, Lo WS, Pillus L, Berger SL, Marmorstein R (2003) Structural basis for histone and phosphohistone binding by the GCN5 histone acetyltransferase. Mol Cell 12(2):461–473. https://doi.org/10.1016/s1097-2765(03)00288-0
Cui LB, Zhou XY, Zhao ZJ, Li Q, Huang XY, Sun FZ (2013) The Kunming mouse: as a model for age-related decline in female fertility in human. Zygote 21(4):367–376. https://doi.org/10.1017/S0967199412000123
Cui JY, Fu ZD, Dempsey J (2019) The role of histone methylation and methyltransferases in gene regulation. Toxicoepigenetics. Elsevier, pp 31–84
Cui LX, Tian YQ, Hao HS, Zou HY, Pang YW, Zhao SJ, Zhao XM, Zhu HB, Du WH (2021) Knockdown of ASH1L methyltransferase induced apoptosis inhibiting proliferation and H3K36 methylation in bovine cumulus cells. Theriogenology 161:65–73. https://doi.org/10.1016/j.theriogenology.2020.11.007
Cura V, Marechal N, Troffer-Charlier N, Strub JM, van Haren MJ, Martin NI, Cianferani S, Bonnefond L, Cavarelli J (2017) Structural studies of protein arginine methyltransferase 2 reveal its interactions with potential substrates and inhibitors. FEBS J 284(1):77–96. https://doi.org/10.1111/febs.13953
Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY, Preissl S, Jermstad I, Haugen MH, Suganthan R, Bjoras M, Hansen K, Dalen KT, Fedorcsak P, Ren B, Klungland A (2016) Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537(7621):548–552. https://doi.org/10.1038/nature19360
Dambacher S, Hahn M, Schotta G (2010) Epigenetic regulation of development by histone lysine methylation. Heredity (edinb) 105(1):24–37. https://doi.org/10.1038/hdy.2010.49
de la Barre AE, Gerson V, Gout S, Creaven M, Allis CD, Dimitrov S (2000) Core histone N-termini play an essential role in mitotic chromosome condensation. EMBO J 19(3):379–391
De La Fuente R, Eppig JJ (2001) Transcriptional activity of the mouse oocyte genome: companion granulosa cells modulate transcription and chromatin remodeling. Dev Biol 229(1):224–236. https://doi.org/10.1006/dbio.2000.9947
Denissov S, Hofemeister H, Marks H, Kranz A, Ciotta G, Singh S, Anastassiadis K, Stunnenberg HG, Stewart AF (2014) Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 141(3):526–537. https://doi.org/10.1242/dev.102681
Derijck AA, van der Heijden GW, Giele M, Philippens ME, van Bavel CC, de Boer P (2006) gammaH2AX signalling during sperm chromatin remodelling in the mouse zygote. DNA Repair (amst) 5(8):959–971. https://doi.org/10.1016/j.dnarep.2006.05.043
DesJarlais R, Tummino PJ (2016) Role of histone-modifying enzymes and their complexes in regulation of chromatin biology. Biochemistry 55(11):1584–1599. https://doi.org/10.1021/acs.biochem.5b01210
Di Lorenzo A, Bedford MT (2011) Histone arginine methylation. FEBS Lett 585(13):2024–2031. https://doi.org/10.1016/j.febslet.2010.11.010
Diao YF, Lin T, Li X, Oqani RK, Lee JE, Kim SY, Jin DI (2018) Dynamic changes of SETD2, a histone H3K36 methyltransferase, in porcine oocytes, IVF and SCNT embryos. PLoS ONE 13(2):e0191816. https://doi.org/10.1371/journal.pone.0191816
Dillon SC, Zhang X, Trievel RC, Cheng X (2005) The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 6(8):227. https://doi.org/10.1186/gb-2005-6-8-227
Dodge JE, Kang YK, Beppu H, Lei H, Li E (2004) Histone H3–K9 methyltransferase ESET is essential for early development. Mol Cell Biol 24(6):2478–2486. https://doi.org/10.1128/mcb.24.6.2478-2486.2004
Eissenberg JC, Shilatifard A (2010) Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol 339(2):240–249. https://doi.org/10.1016/j.ydbio.2009.08.017
Epifano O, Dean J (2002) Genetic control of early folliculogenesis in mice. Trends Endocrinol Metab 13(4):169–173. https://doi.org/10.1016/s1043-2760(02)00576-3
Eymery A, Liu Z, Ozonov EA, Stadler MB, Peters AH (2016) The methyltransferase Setdb1 is essential for meiosis and mitosis in mouse oocytes and early embryos. Development 143(15):2767–2779. https://doi.org/10.1242/dev.132746
Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol 12(12):1052–1058. https://doi.org/10.1016/s0960-9822(02)00901-6
Frankel A, Clarke S (2000) PRMT3 is a distinct member of the protein arginine N-methyltransferase family. Conferral of substrate specificity by a zinc-finger domain. J Biol Chem 275(42):32974–32982. https://doi.org/10.1074/jbc.M006445200
Fu Y, Xu JJ, Sun XL, Jiang H, Han DX, Liu C, Gao Y, Yuan B, Zhang JB (2017) Function of JARID2 in bovines during early embryonic development. Peer J 5:e4189. https://doi.org/10.7717/peerj.4189
Fujimori T (2010) Preimplantation development of mouse: a view from cellular behavior. Dev Growth Differ 52(3):253–262. https://doi.org/10.1111/j.1440-169X.2010.01172.x
Fulton MD, Brown T, Zheng YG (2018) Mechanisms and inhibitors of histone arginine methylation. Chem Rec 18(12):1792–1807. https://doi.org/10.1002/tcr.201800082
Greer EL, Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13(5):343–357. https://doi.org/10.1038/nrg3173
Grobstein C (1985) The early development of human embryos. J Med Philos 10(3):213–236. https://doi.org/10.1093/jmp/10.3.213
Han M, Xu W, Cheng P, Jin H, Wang X (2017) Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget 8(5):8980–8991. https://doi.org/10.18632/oncotarget.13858
Hanna CW, Taudt A, Huang J, Gahurova L, Kranz A, Andrews S, Dean W, Stewart AF, Colome-Tatche M, Kelsey G (2018) MLL2 conveys transcription-independent H3K4 trimethylation in oocytes. Nat Struct Mol Biol 25(1):73–82. https://doi.org/10.1038/s41594-017-0013-5
Hanna CW, Huang J, Belton C, Reinhardt S, Dahl A, Andrews S, Stewart AF, Kranz A, Kelsey G (2021) Loss of SETD1B results in the redistribution of genomic H3K4me3 in the oocyte. bioRxiv. https://doi.org/10.1101/2021.03.11.434836
Hu D, Garruss AS, Gao X, Morgan MA, Cook M, Smith ER, Shilatifard A (2013) The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat Struct Mol Biol 20(9):1093–1097. https://doi.org/10.1038/nsmb.2653
Huang XJ, Wang X, Ma X, Sun SC, Zhou X, Zhu C, Liu H (2014) EZH2 is essential for development of mouse preimplantation embryos. Reprod Fertil Dev 26(8):1166–1175. https://doi.org/10.1071/RD13169
Huang J, Zhang H, Wang X, Dobbs KB, Yao J, Qin G, Whitworth K, Walters EM, Prather RS, Zhao J (2015) Impairment of preimplantation porcine embryo development by histone demethylase KDM5B knockdown through disturbance of bivalent H3K4me3-H3K27me3 modifications. Biol Reprod 92(3):72. https://doi.org/10.1095/biolreprod.114.122762
Husmann D, Gozani O (2019) Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol 26(10):880–889. https://doi.org/10.1038/s41594-019-0298-7
Hyun K, Jeon J, Park K, Kim J (2017) Writing, erasing and reading histone lysine methylations. Exp Mol Med 49(4):e324. https://doi.org/10.1038/emm.2017.11
Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y (2017) Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547(7664):419–424. https://doi.org/10.1038/nature23262
Ismail T, Lee HK, Kim C, Kwon T, Park TJ, Lee HS (2018) KDM1A microenvironment, its oncogenic potential, and therapeutic significance. Epigenetics Chromatin 11(1):33. https://doi.org/10.1186/s13072-018-0203-3
Jenuwein T, Laible G, Dorn R, Reuter G (1998) SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell Mol Life Sci 54(1):80–93. https://doi.org/10.1007/s000180050127
Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, Baltus GA, Kadam S, Zhai H, Valdez R, Gonzalo S, Zhang Y, Li E, Chen T (2008) The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 4(9):e1000190. https://doi.org/10.1371/journal.pgen.1000190
Kadoch C, Copeland RA, Keilhack H (2016) PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry 55(11):1600–1614. https://doi.org/10.1021/acs.biochem.5b01191
Kageyama S, Liu H, Kaneko N, Ooga M, Nagata M, Aoki F (2007) Alterations in epigenetic modifications during oocyte growth in mice. Reproduction 133(1):85–94. https://doi.org/10.1530/REP-06-0025
Kim J, Zhao H, Dan J, Kim S, Hardikar S, Hollowell D, Lin K, Lu Y, Takata Y, Shen J, Chen T (2016) Maternal Setdb1 is required for meiotic progression and preimplantation development in mouse. PLoS Genet 12(4):e1005970. https://doi.org/10.1371/journal.pgen.1005970
Klose RJ, Kallin EM, Zhang Y (2006) JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 7(9):715–727. https://doi.org/10.1038/nrg1945
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705. https://doi.org/10.1016/j.cell.2007.02.005
Lee J, Sayegh J, Daniel J, Clarke S, Bedford MT (2005a) PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J Biol Chem 280(38):32890–32896. https://doi.org/10.1074/jbc.M506944200
Lee MG, Wynder C, Cooch N, Shiekhattar R (2005b) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437(7057):432–435. https://doi.org/10.1038/nature04021
Lepikhov K, Walter J (2004) Differential dynamics of histone H3 methylation at positions K4 and K9 in the mouse zygote. BMC Dev Biol 4:12. https://doi.org/10.1186/1471-213X-4-12
Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3(9):662–673. https://doi.org/10.1038/nrg887
Li G, Reinberg D (2011) Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev 21(2):175–186. https://doi.org/10.1016/j.gde.2011.01.022
Li L, Zheng P, Dean J (2010a) Maternal control of early mouse development. Development 137(6):859–870. https://doi.org/10.1242/dev.039487
Li X, Hu X, Patel B, Zhou Z, Liang S, Ybarra R, Qiu Y, Felsenfeld G, Bungert J, Huang S (2010b) H4R3 methylation facilitates beta-globin transcription by regulating histone acetyltransferase binding and H3 acetylation. Blood 115(10):2028–2037. https://doi.org/10.1182/blood-2009-07-236059
Li C, Gao Y, Wang S, Xu F, Dai L, Jiang H, Yu X, Chen C, Yuan B, Zhang JB (2015) Expression pattern of JMJD1C in oocytes and its impact on early embryonic development. Genet Mol Res 14(4):18249–18258
Li C, Diao F, Qiu D, Jiang M, Li X, Han L, Li L, Hou X, Ge J, Ou X, Liu J, Wang Q (2018) Histone methyltransferase SETD2 is required for meiotic maturation in mouse oocyte. J Cell Physiol 234(1):661–668. https://doi.org/10.1002/jcp.26836
Liao J, Szabo PE (2020) Maternal DOT1L is dispensable for mouse development. Sci Rep 10(1):20636. https://doi.org/10.1038/s41598-020-77545-6
Liu H, Kim JM, Aoki F (2004) Regulation of histone H3 lysine 9 methylation in oocytes and early pre-implantation embryos. Development 131(10):2269–2280. https://doi.org/10.1242/dev.01116
Liu Z, Zhang G, Deng M, Yang H, Pang J, Cai Y, Wan Y, Wang F (2020) Inhibition of lysine-specific histone demethylase 1A results in meiotic aberration during oocyte maturation in vitro in goats. Theriogenology 143:168–178. https://doi.org/10.1016/j.theriogenology.2019.12.011
Manosalva I, Gonzalez A (2010) Aging changes the chromatin configuration and histone methylation of mouse oocytes at germinal vesicle stage. Theriogenology 74(9):1539–1547. https://doi.org/10.1016/j.theriogenology.2010.06.024
Marinho LSR, Rissi VB, Lindquist AG, Seneda MM, Bordignon V (2017) Acetylation and methylation profiles of H3K27 in porcine embryos cultured in vitro. Zygote 25(5):575–582. https://doi.org/10.1017/S0967199417000405
Martin C, Zhang Y (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6(11):838–849. https://doi.org/10.1038/nrm1761
Mason B, Laman H (2020) The FBXL family of F-box proteins: variations on a theme. Open Biol 10(11):200319. https://doi.org/10.1098/rsob.200319
McLaren A, Buehr M (1990) Development of mouse germ cells in cultures of fetal gonads. Cell Differ Dev 31(3):185–195. https://doi.org/10.1016/0922-3371(90)90131-f
McLaren A, Lawson KA (2005) How is the mouse germ-cell lineage established? Differentiation 73(9–10):435–437. https://doi.org/10.1111/j.1432-0436.2005.00049.x
Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, Rutishauser D, Huang D, Caflisch A, Hottiger MO (2010) PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res 38(19):6350–6362. https://doi.org/10.1093/nar/gkq463
Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, Patnaik D, Higgins JM, Potier N, Scheidtmann KH, Buettner R, Schule R (2008) Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol 10(1):53–60. https://doi.org/10.1038/ncb1668
Molyneaux KA, Stallock J, Schaible K, Wylie C (2001) Time-lapse analysis of living mouse germ cell migration. Dev Biol 240(2):488–498. https://doi.org/10.1006/dbio.2001.0436
Nelson CJ, Santos-Rosa H, Kouzarides T (2006) Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 126(5):905–916. https://doi.org/10.1016/j.cell.2006.07.026
Ng SS, Yue WW, Oppermann U, Klose RJ (2009) Dynamic protein methylation in chromatin biology. Cell Mol Life Sci 66(3):407–422. https://doi.org/10.1007/s00018-008-8303-z
Oh S, Jeong K, Kim H, Kwon CS, Lee D (2010) A lysine-rich region in Dot1p is crucial for direct interaction with H2B ubiquitylation and high level methylation of H3K79. Biochem Biophys Res Commun 399(4):512–517. https://doi.org/10.1016/j.bbrc.2010.07.100
Oktem O, Urman B (2010) Understanding follicle growth in vivo. Hum Reprod 25(12):2944–2954. https://doi.org/10.1093/humrep/deq275
Ooga M, Inoue A, Kageyama S, Akiyama T, Nagata M, Aoki F (2008) Changes in H3K79 methylation during preimplantation development in mice. Biol Reprod 78(3):413–424. https://doi.org/10.1095/biolreprod.107.063453
Ooga M, Suzuki MG, Aoki F (2013) Involvement of DOT1L in the remodeling of heterochromatin configuration during early preimplantation development in mice. Biol Reprod 89(6):145. https://doi.org/10.1095/biolreprod.113.113258
Pepling ME, Spradling AC (2001) Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev Biol 234(2):339–351. https://doi.org/10.1006/dbio.2001.0269
Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107(3):323–337. https://doi.org/10.1016/s0092-8674(01)00542-6
Petri T, Dankert D, Demond H, Wennemuth G, Horsthemke B, Grummer R (2020) In vitro postovulatory oocyte aging affects H3K9 trimethylation in two-cell embryos after IVF. Ann Anat 227:151424. https://doi.org/10.1016/j.aanat.2019.151424
Phillips TC, Wildt DE, Comizzoli P (2016) Incidence of methylated histones H3K4 and H3K79 in cat germinal vesicles is regulated by specific nuclear factors at the acquisition of developmental competence during the folliculogenesis. J Assist Reprod Genet 33(6):783–794. https://doi.org/10.1007/s10815-016-0706-4
Qian C, Zhou MM (2006) SET domain protein lysine methyltransferases: structure, specificity and catalysis. Cell Mol Life Sci 63(23):2755–2763. https://doi.org/10.1007/s00018-006-6274-5
Qian C, Zhang Q, Li S, Zeng L, Walsh MJ, Zhou MM (2005) Structure and chromosomal DNA binding of the SWIRM domain. Nat Struct Mol Biol 12(12):1078–1085. https://doi.org/10.1038/nsmb1022
Qiao J, Chen Y, Yan LY, Yan J, Liu P, Sun QY (2010) Changes in histone methylation during human oocyte maturation and IVF- or ICSI-derived embryo development. Fertil Steril 93(5):1628–1636. https://doi.org/10.1016/j.fertnstert.2009.03.002
Qiao J, Wang ZB, Feng HL, Miao YL, Wang Q, Yu Y, Wei YC, Yan J, Wang WH, Shen W, Sun SC, Schatten H, Sun QY (2014) The root of reduced fertility in aged women and possible therapentic options: current status and future perspects. Mol Aspects Med 38:54–85. https://doi.org/10.1016/j.mam.2013.06.001
Qu Y, Lu D, Jiang H, Chi X, Zhang H (2016) EZH2 is required for mouse oocyte meiotic maturation by interacting with and stabilizing spindle assembly checkpoint protein BubRI. Nucleic Acids Res 44(16):7659–7672. https://doi.org/10.1093/nar/gkw463
Rajan PK, Udoh UA, Sanabria JD, Banerjee M, Smith G, Schade MS, Sanabria J, Sodhi K, Pierre S, Xie Z, Shapiro JI, Sanabria J (2020) The role of histone acetylation-/methylation-mediated apoptotic gene regulation in hepatocellular carcinoma. Int J Mol Sci. https://doi.org/10.3390/ijms21238894
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273(10):5858–5868. https://doi.org/10.1074/jbc.273.10.5858
Rossetto D, Avvakumov N, Cote J (2012) Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 7(10):1098–1108. https://doi.org/10.4161/epi.21975
Russo V, Bernabo N, Di Giacinto O, Martelli A, Mauro A, Berardinelli P, Curini V, Nardinocchi D, Mattioli M, Barboni B (2013) H3K9 trimethylation precedes DNA methylation during sheep oogenesis: HDAC1, SUV39H1, G9a, HP1, and Dnmts are involved in these epigenetic events. J Histochem Cytochem 61(1):75–89. https://doi.org/10.1369/0022155412463923
Saksouk N, Simboeck E, Dejardin J (2015) Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 8:3. https://doi.org/10.1186/1756-8935-8-3
Sánchez F, Smitz J (2012) Molecular control of oogenesis. Biochim Biophys Acta 1822(12):1896–1912. https://doi.org/10.1016/j.bbadis.2012.05.013
Sankar A, Kooistra SM, Gonzalez JM, Ohlsson C, Poutanen M, Helin K (2017) Maternal expression of the histone demethylase Kdm4a is crucial for pre-implantation development. Development 144(18):3264–3277. https://doi.org/10.1242/dev.155473
Sankar A, Lerdrup M, Manaf A, Johansen JV, Gonzalez JM, Borup R, Blanshard R, Klungland A, Hansen K, Andersen CY, Dahl JA, Helin K, Hoffmann ER (2020) KDM4A regulates the maternal-to-zygotic transition by protecting broad H3K4me3 domains from H3K9me3 invasion in oocytes. Nat Cell Biol 22(4):380–388. https://doi.org/10.1038/s41556-020-0494-z
Sarmento OF, Digilio LC, Wang Y, Perlin J, Herr JC, Allis CD, Coonrod SA (2004) Dynamic alterations of specific histone modifications during early murine development. J Cell Sci 117(Pt 19):4449–4459. https://doi.org/10.1242/jcs.01328
Scheer S, Zaph C (2017) The lysine methyltransferase G9a in immune cell differentiation and function. Front Immunol 8:429. https://doi.org/10.3389/fimmu.2017.00429
Sha QQ, Zhang J, Fan HY (2020) Function and regulation of histone H3 lysine-4 methylation during oocyte meiosis and maternal-to-zygotic transition. Front Cell Dev Biol 8:597498. https://doi.org/10.3389/fcell.2020.597498
Shao GB, Chen JC, Zhang LP, Huang P, Lu HY, Jin J, Gong AH, Sang JR (2014) Dynamic patterns of histone H3 lysine 4 methyltransferases and demethylases during mouse preimplantation development. In Vitro Cell Dev Biol Anim 50(7):603–613
Shao G-B, Wang J, Zhang L-P, Wu C-Y, Jin J, Sang J-R, Lu H-Y, Gong A-H, Du F-Y, Peng W-X (2015) Aging alters histone H3 lysine 4 methylation in mouse germinal vesicle stage oocytes. Reprod Fertil Dev 27(2):419–426
Shi Y, Sawada J, Sui G, el Affar B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422(6933):735–738. https://doi.org/10.1038/nature01550
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119(7):941–953. https://doi.org/10.1016/j.cell.2004.12.012
Sidler C, Li D, Wang B, Kovalchuk I, Kovalchuk O (2014) SUV39H1 downregulation induces deheterochromatinization of satellite regions and senescence after exposure to ionizing radiation. Front Genet 5:411. https://doi.org/10.3389/fgene.2014.00411
Svoboda P (2018) Mammalian zygotic genome activation. Semin Cell Dev Biol 84:118–126. https://doi.org/10.1016/j.semcdb.2017.12.006
Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16(14):1779–1791. https://doi.org/10.1101/gad.989402
Tessarz P, Kouzarides T (2014) Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15(11):703–708. https://doi.org/10.1038/nrm3890
Waddington CHJE (1942) The Epigenotype. Endeavour 1:18–20
Walport LJ, Hopkinson RJ, Chowdhury R, Schiller R, Ge W, Kawamura A, Schofield CJ (2016) Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat Commun 7:11974. https://doi.org/10.1038/ncomms11974
Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, Briggs SD, Allis CD, Wong J, Tempst P, Zhang Y (2001) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293(5531):853–857. https://doi.org/10.1126/science.1060781
Wang X, Gao W, Ma X, Wang X, Song C, Huang X, Liu H (2014) Dot1L mediated histone H3 lysine79 methylation is essential to meiosis progression in mouse oocytes. Neuro Endocrinol Lett 35(6):523–530
Wasson JA, Simon AK, Myrick DA, Wolf G, Driscoll S, Pfaff SL, Macfarlan TS, Katz DJ (2016) Maternally provided LSD1/KDM1A enables the maternal-to-zygotic transition and prevents defects that manifest postnatally. Elife. https://doi.org/10.7554/eLife.08848
Wei Y, Mizzen CA, Cook RG, Gorovsky MA, Allis CD (1998) Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc Natl Acad Sci USA 95(13):7480–7484. https://doi.org/10.1073/pnas.95.13.7480
Wiles ET, Selker EU (2017) H3K27 methylation: a promiscuous repressive chromatin mark. Curr Opin Genet Dev 43:31–37. https://doi.org/10.1016/j.gde.2016.11.001
Woodcock CL, Skoultchi AI, Fan Y (2006) Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res 14(1):17–25. https://doi.org/10.1007/s10577-005-1024-3
Wu FR, Zhang Y, Ding B, Lei XH, Huang JC, Wang CH, Liu Y, Wang R, Li WY (2014) H3K27me3 may be associated with Oct4 and Sox2 in mouse preimplantation embryos. Genet Mol Res 13(4):10121–10129. https://doi.org/10.4238/2014.December.4.6
Wu X, Hu S, Wang L, Li Y, Yu H (2020) Dynamic changes of histone acetylation and methylation in bovine oocytes, zygotes, and preimplantation embryos. J Exp Zool B Mol Dev Evol 334(4):245–256. https://doi.org/10.1002/jez.b.22943
Xu Q, Xiang Y, Wang Q, Wang L, Brind’Amour J, Bogutz AB, Zhang Y, Zhang B, Yu G, Xia W, Du Z, Huang C, Ma J, Zheng H, Li Y, Liu C, Walker CL, Jonasch E, Lefebvre L, Wu M, Lorincz MC, Li W, Li L, Xie W (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 51(5):844–856. https://doi.org/10.1038/s41588-019-0398-7
Yang Y, Bedford MT (2013) Protein arginine methyltransferases and cancer. Nat Rev Cancer 13(1):37–50. https://doi.org/10.1038/nrc3409
Yeung WKA, Brind’Amour J, Hatano Y, Yamagata K, Feil R, Lorincz MC, Tachibana M, Shinkai Y, Sasaki H (2019) Histone H3K9 methyltransferase G9a in oocytes is essential for preimplantation development but dispensable for CG methylation protection. Cell Rep 27(1):282–293, e284. https://doi.org/10.1016/j.celrep.2019.03.002
Yoneyama M, Tochio N, Umehara T, Koshiba S, Inoue M, Yabuki T, Aoki M, Seki E, Matsuda T, Watanabe S, Tomo Y, Nishimura Y, Harada T, Terada T, Shirouzu M, Hayashizaki Y, Ohara O, Tanaka A, Kigawa T, Yokoyama S (2007) Structural and functional differences of SWIRM domain subtypes. J Mol Biol 369(1):222–238. https://doi.org/10.1016/j.jmb.2007.03.027
Zhang A, Xu B, Sun Y, Lu X, Gu R, Wu L, Feng Y, Xu C (2012) Dynamic changes of histone H3 trimethylated at positions K4 and K27 in human oocytes and preimplantation embryos. Fertil Steril 98(4):1009–1016. https://doi.org/10.1016/j.fertnstert.2012.06.034
Zhang Q, Qi S, Xu M, Yu L, Tao Y, Deng Z, Wu W, Li J, Chen Z, Wong J (2013) Structure-function analysis reveals a novel mechanism for regulation of histone demethylase LSD2/AOF1/KDM1b. Cell Res 23(2):225–241. https://doi.org/10.1038/cr.2012.177
Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, Ming J, Wu X, Zhang Y, Xu Q, Liu W, Kou X, Zhao Y, He W, Li C, Chen B, Li Y, Wang Q, Ma J, Yin Q, Kee K, Meng A, Gao S, Xu F, Na J, Xie W (2016a) Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537(7621):553–557. https://doi.org/10.1038/nature19361
Zhang S, Wang F, Fan C, Tang B, Zhang X, Li Z (2016b) Dynamic changes of histone H3 lysine 9 following trimethylation in bovine oocytes and pre-implantation embryos. Biotechnol Lett 38(3):395–402. https://doi.org/10.1007/s10529-015-2001-3
Zheng W, Zhang H, Gorre N, Risal S, Shen Y, Liu K (2014) Two classes of ovarian primordial follicles exhibit distinct developmental dynamics and physiological functions. Hum Mol Genet 23(4):920–928. https://doi.org/10.1093/hmg/ddt486
Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, Li Y, Wang Q, Ma J, Peng X, Xu F, Xie W (2016) Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol Cell 63(6):1066–1079. https://doi.org/10.1016/j.molcel.2016.08.032
Zou MR, Cao J, Liu Z, Huh SJ, Polyak K, Yan Q (2014) Histone demethylase Jumonji AT-rich interactive domain 1B (JARID1B) controls mammary gland development by regulating key developmental and lineage specification genes. J Biol Chem 289(25):17620–17633. https://doi.org/10.1074/jbc.M114.570853
Funding
This research did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bilmez, Y., Talibova, G. & Ozturk, S. Dynamic changes of histone methylation in mammalian oocytes and early embryos. Histochem Cell Biol 157, 7–25 (2022). https://doi.org/10.1007/s00418-021-02036-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-021-02036-2