
Abstract
To analyze the frequency and clinical phenotype of neurosarcoidosis (NS) in one of the largest nationwide cohorts of patients with sarcoidosis reported from southern Europe. NS was evaluated according to the Diagnostic Criteria for Central Nervous System and Peripheral Nervous System Sarcoidosis recently proposed by Stern et al. Pathologic confirmation of granulomatous disease was used to subclassify NS into definite (confirmation in neurological tissue), probable (confirmation in extraneurological tissue) and possible (no histopathological confirmation of the disease). Of the 1532 patients included in the cohort, 85 (5.5%) fulfilled the Stern criteria for NS (49 women, mean age at diagnosis of NS of 47.6 years, 91% White). These patients developed 103 neurological conditions involving the brain (38%), cranial nerves (36%), the meninges (3%), the spinal cord (10%) and the peripheral nerves (14%); no patient had concomitant central and peripheral nerve involvements. In 59 (69%) patients, neurological involvement preceded or was present at the time of diagnosis of the disease. According to the classification proposed by Stern et al., 11 (13%) were classified as a definite NS, 61 (72%) as a probable NS and the remaining 13 (15%) as a possible NS. In comparison with the systemic phenotype of patients without NS, patients with CNS involvement presented a lower frequency of thoracic involvement (82% vs 93%, q = 0.018), a higher frequency of ocular (27% vs 10%, q < 0.001) and salivary gland (15% vs 4%, q = 0.002) WASOG involvements. In contrast, patients with PNS involvement showed a higher frequency of liver involvement (36% vs 12%, p = 0.02) in comparison with patients without NS. Neurosarcoidosis was identified in 5.5% of patients. CNS involvement prevails significantly over PNS involvement, and both conditions do not overlap in any patient. The systemic phenotype associated to each involvement was clearly differentiated, and can be helpful not only in the early identification of neurological involvement, but also in the systemic evaluation of patients diagnosed with neurosarcoidosis.
Similar content being viewed by others

Introduction
Sarcoidosis is an immune-mediated granulomatous systemic disease1 that affects adults with a slight predominance in women at a mean age around 40 years2, with an estimated prevalence ranging between 1 and 40 cases per 100,000 individuals2,3. Sarcoidosis may mimic a wide variety of processes, and the diagnosis is established when specific clinical and imaging findings are supported by histologic evidence of non-caseating granulomas4. Spontaneous remission may occur in up to two thirds of patients, while the remaining cases may follow a chronic course5.
The lungs and the thoracic lymph nodes are involved in nearly 90% of patients with sarcoidosis6. However, extrathoracic involvement is a key characteristic of the disease, and involvement of the nervous system (neurosarcoidosis) is one of the most characteristic systemic features involving around 3–9% of patients7,8. The clinical picture is heterogeneous since any part of the nervous system (brain, meninges, cranial nerves, spinal cord, peripheral nerves) can be affected9. Given the key role of histopathological examination for supporting the diagnosis of sarcoidosis, a major challenge regarding neurosarcoidosis is the difficulty of obtaining neural tissue, especially in those cases with involvement of the central nervous system. The diverse clinical presentation and the complex approach for obtaining neurological tissue make to consider neurosarcoidosis as one of the extrathoracic involvements more complex to diagnose and manage10.
Despite numerous publications on neurosarcoidosis, there is no international consensus about their definition and classification. A variety of diagnostic criteria have been proposed in the last 20 years with a wide methodological approach among them and therefore with significant limitations for being applied. A recent study from the Neurosarcoidosis Consortium Consensus Group has launched a consensus diagnostic set of criteria providing definitions for possible, probable and definite neurosarcoidosis10. In this study, we have analyzed the frequency and clinical phenotype of neurosarcoidosis in one of the largest nationwide cohorts of patients with sarcoidosis reported from southern Europe following these new diagnostic criteria.
Methods
Patients
The SARCOGEAS-Study Group was founded in 2015 with the aim of collecting a large series of patients with sarcoidosis from Spanish hospitals with substantial experience in the management of systemic autoimmune diseases6. Both incident and prevalent cases were included. By January 2020, the multicenter database included 1532 consecutive patients diagnosed with sarcoidosis in 40 centers according to the criteria proposed by the American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) 1999 statement on sarcoidosis11: (a) clinical or radiologic findings consistent with sarcoidosis, such as pulmonary disease, uveitis, mediastinal bilateral hilar lymphadenopathy (BHL), or erythema nodosum; (b) tissue biopsy with histologic evidence of non-caseating granulomas; (c) absence of other causes of granulomatous disease. Patients lacking the histopathological criteria (b) were included if they presented at least one of the following features: elevated serum angiotensin-converting enzyme, organ-specific abnormal uptake on gallium-67 citrate scintigraphy, elevated lymphocyte count or elevated CD4/CD8 ratio in bronchoalveolar lavage fluid, or active extrathoracic involvement classified as highly probable according to the WASOG extrathoracic classification12,13. Extrathoracic involvement at diagnosis was defined according to the 2014 WASOG organ assessment instrument, including only the clinical scenarios classified as highly probable or at least probable13. The study was conducted in accordance with the amended Declaration of Helsinki. The Ethics Committee of the coordinating center (Clinical Research Ethics Committee of the Hospital Clinic, Barcelona, Spain, code HCB2016/0181) approved the protocol, and written informed consent was obtained from patients with current follow-up.
Variables
Patients included in the Registry as having neurosarcoidosis according to the 2014 WASOG definition were selected. These cases were re-evaluated according to the Proposed Diagnostic Criteria for Central Nervous System and Peripheral Nervous System Sarcoidosis recently reported by Stern et al.10. The mandatory inclusion criteria consisted of a clinical presentation and diagnostic evaluation suggesting neurosarcoidosis, as defined by the clinical manifestations and magnetic resonance imaging (MRI), cerebrospinal fluid (CSF), and/or electromyogram/nerve conduction study (EMG/NCS) findings suggestive of granulomatous inflammation of the nervous system. Exclusion criteria included a rigorous exclusion of other causes of neurological disease. Pathologic confirmation of granulomatous disease was used to subclassify neurosarcoidosis into definite (non-caseating granulomas demonstrated in neurological tissue), probable (non-caseating granulomas demonstrated in extraneurological tissue) and possible (no histopathological confirmation of the disease)10 (Sup. Figure 1).
The following variables were retrospectively collected at the time of diagnosis of neurosarcoidosis, defined as the date of fulfilment of the Stern criteria: age, sex, signs and symptoms at neurological presentation, ACE serum levels, Scadding radiographic stages14, neurological diagnostic tests (MRI, CSF, EMG/NCS, PET), histopathological study, treatment and outcomes. The neurological diagnosis was achieved in each center after a multidisciplinary evaluation including neurologists, internists and radiologists, and was independently re-evaluated by MRC and YB, who applied the classification criteria and the subclassification into the 5 anatomical clusters proposed by Stern et al.10 (Sup. Table 1), requiring to the participant centers additional information in case was necessary to confirm the fulfilment of the Stern criteria.
Statistical analysis
Descriptive data are presented as mean and standard deviation (SD) for continuous variables and numbers and percentages (%) for categorical variables. Phenotyping comparisons were made considering the presence or absence of neurosarcoidosis, and among the two main subsets of neurosarcoidosis (peripheral and central nervous system involvement). The Chi-square test was used to study the association between Scadding subsets with the main epidemiological, clinical, extrathoracic and therapeutic variables. One-way ANOVA tests were used to compare continuous variables. All significance tests were two-tailed and values of p < 0.05 were considered significant. P-values were adjusted for multiple comparisons using the false discovery rate (FDR) correction. All analyses were conducted using the R V.3.2.3 for Windows statistical software package (R: A Language and Environment for Statistical, R Core Team, R Foundation for Statistical Computing, Vienna, Austria, 2015, https://www.R-project.org/).
Results
Of the 1532 patients included in the cohort, 102 were reported as having neurosarcoidosis according to the WASOG criteria. Among them, 17 patients were excluded after applying the Stern criteria due to lack of enough information for applying the criteria (n = 11), lack of abnormal neurological diagnostic tests (n = 4) or other etiologies unrelated to sarcoidosis (n = 2). Therefore, the remaining 85 (5.5%) of patients fulfilled the Stern criteria for neurosarcoidosis and were specifically analysed. There were 49 (58%) women and 36 (42%) men, with a mean age at diagnosis of neurosarcoidosis of 47.6 years (range 22–76); all patients except 8 were classified as White. Scadding radiologic stage consisted of stage 0 in 15 (18%) patients, stage I in 28 (33%), stage II in 31 (36%), stage III in 9 (11%) and stage IV in 2 (2%) patients. Histopathological demonstration of non-caseating granulomas was reported in 72 (85%) patients. In addition to the neurological involvement, 59 (69%) patients presented other extrathoracic manifestations and 21 (25%) presented only with thoracic manifestations; in the remaining 5 (6%) patients, the nervous system was the unique organ involved by sarcoidosis at the time of diagnosis (Table 1).
Anatomical involvements
The 85 patients with neurosarcoidosis developed 103 neurological conditions involving the brain (n = 39, 38%), cranial nerves (n = 37, 36%), the meninges (n = 3, 3%), the spinal cord (n = 10, 10%) and the peripheral nerves (n = 14, 14%). In 59 (69%) patients, neurological involvement preceded or was present at the time of diagnosis of the disease. Raised serum ECA levels were reported in 41/74 (55%) of cases. Forty-two patients had cerebrospinal fluid (CSF) analysis. Eight had completely normal CSF constituents. The most consistent abnormality was an elevated CSF protein (33/42 patients, 79%), followed by CSF pleocytosis (21/42 patients, 50%, with a range of 11–110 white cells, overwhelmingly monocytes). There were two cases of mild hypoglycorrhachia, in whom extensive microbiological studies were negative. Oligoclonal bands were studied in 8 patients (negative in 5, matched positive in 2 and positive with intrathecal synthesis alone in one). The CSF ACE level was measured in only one patient (normal levels).
Table 2 summarizes the demographics and therapeutic information of patients with neurosarcoidosis according to the different anatomical phenotypes.
According to the classification proposed by Stern et al., 11 (13%) were classified as a definite neurosarcoidosis, 61 (72%) as a probable neurosarcoidosis and the remaining 13 (15%) as a possible neurosarcoidosis (Table 1).
Brain involvement
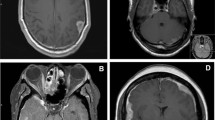
Brain involvement was reported in 39 patients (25 women, mean age at diagnosis of neurosarcoidosis of 46.1 years). Neurological involvement preceded or was present at the time of diagnosis of sarcoidosis in 27 (69%) cases, and the nervous system was the only organ involved in 4 (10%). Clinical neurological presentation consisted of focal motor deficits (n = 14), headache (n = 12), clinical involvement of cranial nerves (n = 8), confusion/cognitive impairment (n = 7), seizures (n = 6), polyuria/polydipsia (n = 3), signs and symptoms of ICH (n = 2) and fever (n = 1); the neurological signs and symptoms often overlapped. Neurological imaging studies disclosed intraparenchymal brain lesions in 23 cases consisting of small subcortical or periventricular white matter lesions (n = 20) (Fig. 1a) or 1–2 large isolated lesions (n = 3) (Fig. 1b,c), leptomeningeal involvement in 18 (especially along the skull base, including the involvement of the pituitary tract in 6 patients) (Fig. 2a), dural involvement in 5 (Fig. 2b) and hydrocephalus in 2 cases; no patient showed stroke or intracerebral haemorrhage. Other diagnostic tests included abnormal PET uptake of the nervous system in 2/11 (18%) and abnormal CSF results in 22/25 (88%). There were 15 patients that presented with other concomitant anatomical neurological involvements (cranial nerves in 11, myelitis in 5, meningitis in one). According to the classification proposed by Stern et al., 8 (21%) were classified as a definite neurosarcoidosis, 25 (64%) as a probable neurosarcoidosis and the remaining 6 (15%) as a possible neurosarcoidosis. Frontline therapeutic approach included the use of corticosteroids in all patients except 2, including intravenous administration in 10 (methylprednisolone pulses in 8, dexamethasone in 2) and oral administration in the remaining 25 (with a dose higher than 30 mg/day in 22 cases). Immunosuppressants were used in 13 patients (methotrexate in 7, azathioprine in 3, mycophenolate in 2, cyclophosphamide in 2) and biological therapies in 5 (infliximab in 4, adalimumab in 1, rituximab in 1); infliximab was stopped in 2 cases due to multiple episodes of infections (n = 1) and tuberculosis (n = 1). Data from therapeutic response was available in 36 cases showing a complete response in 21 (58%) cases, partial response in 13 (36%) and no response in 2 (6%). A relapse was reported in 6 patients, including 2 asymptomatic patients in neuroradiological studies disclosed disease progression.

Intraparenchymal lesions. Pontine right lateral lesion that enhances with gadolinium: (a) Flair sequence and (b) T1 sequence with gadolinium; (c) Subcortical white matter lesions, Flair sequence.

Leptomeningeal and dural involvement. (a) Diffuse leptomeningeal involvement and involvement of the pituitary tract; (b) Dural involvement.
Cranial nerve involvement
Cranial nerve involvement was reported in 37 patients (25 women, mean age at diagnosis of neurosarcoidosis of 47.1 years). Neurological involvement preceded or was present at the time of diagnosis of sarcoidosis in 26 (70%) cases, and the nervous system was the only organ involved in only 1 (3%) case. Clinical neurological presentation consisted of signs and symptoms suggestive of involvement of the VII (n = 24), II (n = 5), V (n = 2) and VI (n = 1) cranial nerves; the remaining 5 cases presented with multiple cranial nerve involvement (3 cases involving the ocular cranial nerves, one case involving the VII and XII and one involving the IX and X). Neurological imaging studies disclosed involvement of the optic nerves (Fig. 3), together with other neurological involvements in 12 cases, mainly brain involvement in 11. Other diagnostic tests included abnormal PET uptake of the nervous system in 1/8 (12%) cases and abnormal CSF results in 10/15 (67%).

Involvement of optic chiasm and hypophysis; (a) T1 sequence; (b) T1 sequence after gadolinium.
According to the classification proposed by Stern et al., 2 (5%) were classified as a definite neurosarcoidosis, 27 (73%) as a probable neurosarcoidosis and the remaining 8 (22%) as a possible neurosarcoidosis. Frontline therapeutic approach included the use of corticosteroids in all patients except 3, including methylprednisolone pulses in 5, and oral administration in 29 (with a dose higher than 30 mg/day in all cases). Immunosuppressants were used in 11 patients (methotrexate in 6, azathioprine in 2, mycophenolate in 2, cyclophosphamide in 2) and biological therapies in 2 (infliximab and rituximab, respectively). Data from therapeutic response was available in 35 cases in included a complete response in 23 (66%), partial improvement in 9 (26%) and no response in 3 (8%). Only 1 patient relapsed during the follow-up.
Meningeal involvement
Three patients presented with lymphocytic meningitis (1 woman, mean age at diagnosis of neurosarcoidosis of 50 years); in one case, neurological involvement preceded or was present at the time of diagnosis confirmation of the disease. All patients presented signs and symptoms of meningism (one together with paraparesia) and abnormal CSF studies, with normal neuroradiological studies. There were 2 patients that showed other concomitant anatomical neurological involvements. According to the classification proposed by Stern et al., all cases were classified as probable neurosarcoidosis. Frontline therapeutic approach included the use of corticosteroids with a dose higher than 30 mg/day in all patients, and azathioprine was added in one case. A complete response was reported in 2 cases, while the remaining patient showed a partial response. One patient developed a haemorrhagic stroke during the follow-up.
Spinal cord involvement
Spinal cord involvement was reported in 10 patients (5 women, mean age at diagnosis of neurosarcoidosis of 45.3 years). In all cases, neurological involvement preceded or was present at the time of diagnosis confirmation of the disease, and the nervous system was the only organ involved in only 1 (10%) case. Clinical neurological presentation consisted of focal motor deficits (n = 9), headache (n = 3), meningism (n = 1) and cranial nerve involvement (n = 1). Neurological imaging studies disclosed intramedullary (Fig. 4a) or extramedullary involvement (Fig. 4b) (with dural enhancement on 4 cases) located in the cervical region (n = 3), the cervical and dorsal regions (n = 2), the dorsal region (n = 2) and the conus medullaris/cauda equina region (n = 2) (Fig. 4c); enhancement extended for more than 3 segments in 5 patients. Abnormal CSF results were reported in 7/8 (88%). There were 5 patients that showed concomitant brain lesions. According to the classification proposed by Stern et al., 8 (80%) were classified as a probable neurosarcoidosis and the remaining 2 (20%) as a possible neurosarcoidosis. Frontline therapeutic approach included the use of corticosteroids with a dose higher than 30 mg/day in all patients (including methylprednisolone pulses in one case). Immunosuppressants were used in 5 patients (methotrexate in 3, mycophenolate in 2, cyclophosphamide in 1) and biological therapies in 1 (infliximab). Data from therapeutic response showed a complete response in 3 (30%), partial improvement in 6 (60%) and no response in 1 (10%, no response to MMF, IFX started but stopped for infections). One patient relapsed at 6-months of follow-up during corticosteroid tapering.

Medullary involvement: (a) Intramedullary involvement; (b) Extramedullary involvement; (c) Gadolinium enhancement at cauda equina (T1 sequence and T1 sequence after gadolinium).
Neuropathy
Peripheral nerve involvement was reported in 14 patients (5 women, mean age at diagnosis of neurosarcoidosis of 50.4 years). In 11 cases, neurological involvement preceded or was present at the time of diagnosis confirmation of the disease. Clinical neurological presentation consisted of signs and symptoms suggestive of sensory neuropathy in 12 cases and radicular paresthesia/paraparesis in the remaining 2. Electromyogram/nerve conduction studies disclosed axonal polyneuropathy (n = 7), pure sensitive neuropathy/small fiber neuropathy (n = 4), polyradiculopathy (n = 2) and inconclusive, non-classified polyneuropathy (n = 1). Neurological imaging studies were normal in 7/7. Other diagnostic tests included abnormal CSF results in 6/7 (86%). No patients showed concomitant involvement of the central nervous system. According to the classification proposed by Stern et al., 2 (14%) were classified as a definite neurosarcoidosis, 11 (79%) as a probable neurosarcoidosis and the remaining one (7%) as a possible neurosarcoidosis. Frontline therapeutic approach included the use of corticosteroids in 10 (71%) cases, including methylprednisolone pulses in 2, and oral administration in 8 (with a dose higher than 30 mg/day in 6 cases). Immunosuppressants were used in 2 patients (methotrexate) and IVIG in 1. Data from therapeutic response was available in 11 cases and included a complete response in 5 (45.5%), partial improvement in 1 (9%) and no response in 5 (45.5%).
Comparison studies
Differences according to anatomical involvement
No significant differences were found between patients with CNS involvement and those with PNS involvement except for a trend for a higher frequency of women in patients with CNS involvement and a higher frequency of renal involvement in those with PNS involvement (14% vs 0%, p = 0.025) (Table 3). In patients with CNS involvement, no significant differences were found between those with brain involvement and those with CNS involvement out of the brain except for a higher frequency of multiple anatomical involvements in patients with brain involvement (38% vs 3%, p < 0.001) (Sup. Table 2). Patients presenting with a unique CNS neurological involvement showed a higher frequency of raised ECA levels (62% vs 29%, p = 0.036) and a lower frequency of salivary gland involvement (10% vs 31%, p = 0.044) in comparison with those with multiple CNS sites involved (Sup. Table 3).
Comparison with patients without neurosarcoidosis
Patients with CNS neurosarcoidosis presented a higher frequency of radiological Scadding Stage 0 (18% vs 7%, q = 0.018), a higher frequency of ocular (27% vs 10%, q < 0.001) and salivary gland (15% vs 4%, q = 0.002) WASOG involvements, and a higher frequency of use of corticosteroids (96% vs 47%, q < 0.001) and immunosuppressive/biological agents (35% vs 8%, q < 0.001) in comparison with patients without neurosarcoidosis, while those with PNS neurosarcoidosis showed a higher frequency of liver involvement (36% vs 12%, p = 0.02) and a trend for a higher frequency of spleen involvement (21% vs 7%, p = 0.08) that were not statistically significant in the corrected statistical analysis (Table 3).
Discussion
Neurosarcoidosis is a potentially life-threatening presentation of sarcoidosis that has been reported in 3–9% of patients with sarcoidosis7,8,15,16,17,18. There are few large studies focusing on the characterization of this extrathoracic presentation of sarcoidosis, with only 7 series including more than 30 cases that have been published in the last 20 years, mainly from the US19,20,21,22,23 and Northern Europe9,24 (Sup. Table 4). In this study, we have characterized the largest series of patients with neurosarcoidosis from Southern Europe, and the second one reported worldwide, using for the first time the new diagnostic criteria proposed by Stern et al.10. Neurosarcoidosis was identified in 5.5% of our patients with sarcoidosis, a similar figure than that reported in previous cohorts of patients7,8,15,16,17,18.
According to previous studies (Sup. Table 4), patients with neurosarcoidosis are women in 44–77% of cases, with a frequency of non-White people ranging between 40 and 96%, and a mean age at diagnosis of the neurological disease ranging from 40 to 48 years; neurosarcoidosis is present at the time of diagnosis of sarcoidosis in 60–72% of cases, and is the single organ involved in 3–20% of reported cases. The epidemiological profile in our series was quite similar (59% women, mean age at diagnosis around 48 years), and also the timing of presentation (neurosarcoidosis was present at the time of diagnosis in 59% of cases being the single organ involved in 6%). We report one of the oldest mean age at diagnosis of neurosarcoidosis among the large reported series, a fact that could be linked with having the reported series with the highest frequency of White people (92% of our cases, in comparison with only 4–60% of patients included in previous studies) (Sup. Table 4), since sarcoidosis is diagnosed at older ages in White patients in comparison with other ethnicities2.
Among patients with CNS involvement, we found a clear predominance of brain (55%) and cranial nerve (52%) involvements, and a low frequency of spinal cord (14%) and meningeal (4%) involvements. The main studies show that brain (33–52%) and cranial nerve (31–76%) involvements also are the predominant presentation of neurosarcoidosis18,19,22,24,25,26,27,28,29,30, as has also been confirmed in a recent systematic review analysing 652 patients with neurosarcoidosis (parenchymal brain lesions were reported in 51% of cases and cranial neuropathies in 55%)31. We found that intraparenchymal lesions and leptomeningeal involvement were the key features of sarcoidosis involving the brain, as has reported by previous studies9,22; we also found involvement of the pituitary tract, a very specific site involved by neurosarcoidosis32, in 9% of patients, a frequency that is into the range of that previously reported (2–25%)19,21,22,23,24. Pathological studies in patients with sarcoidosis involving the brain showed microvascular changes (involving small arteries, arterioles and small veins) and granulomatous inflammation often following a perivascular distribution26, as we could see in 3 of our patients in whom brain biopsy was carried out. Brain neurosarcoidosis is characterized by a mixed cellular infiltration composed by histiocytes, macrophages, both CD4 and CD8 T lymphocytes and B lymphocytes. In our patients with brain involvement, we reported a complete/partial response in more than 90% of cases; in previous studies19,21,22,23,24, we cannot found specific data about the therapeutic response in patients with sarcoidosis involving the brain, since the main series do not separate the therapy and outcome of these patients from other patients with neurosarcoidosis.
We found cranial nerve involvement in half of our patients with neurosarcoidosis; involvement of the facial nerve represented 65% of all cranial neuropathies we identified. In the systematic review by Fritz et al.31, the facial nerve was also the most frequent cranial nerve involved (40%), followed by the optic, trigeminal and audiovestibular nerves. In some cases, the facial palsy is included into a Heerfordt’s syndrome presentation together with uveitis, parotid gland enlargement and fever. Sarcoidosis may also cause an optic neuritis, which is vision-threatening, and has a poorer GC response and outcome in comparison with other cranial nerves involved. In the 5 cases of optic neuritis that we reported, the rate of response to corticosteroids monotherapy was 40%, and most patients required the addition of immunosuppressive/biologic agents to improve the response, and a complete response was achieved in only one case. Previous studies have reported a therapeutic response to corticosteroids in 15 (52%) out of 29 patients with sarcoidosis and optic neuritis22,33,34, with a worse prognosis in those presenting with a chronic course or with bilateral involvement.
Spinal cord sarcoidosis is a rare but potentially life-threatening involvement that may present with paraplegia, paralysis, incontinence, and cauda equina syndrome, mimicking NMOSD35. Neuroimaging studies may disclose a wide spectrum of findings, including arachnoiditis, extradural or intradural lesions, and intramedullary involvement36. Among the 10 cases we have reported, most showed intramedullary involvement, as has been reported in previous studies25,36; these patients were treated with high doses of corticosteroids achieving a complete response in only 30% of cases. In previous studies, the frequency of complete response is also low, and more than 80% of cases had neurological sequelae at the end of the follow-up25,36,37,38. Meningeal involvement of sarcoidosis was the less frequent CNS involvement in our series, with only 3 cases.
PNS involvement was identified in 14% of our patients diagnosed with neurosarcoidosis according to Stern criteria10. Involvement of peripheral nerves by sarcoidosis has been little studied in comparison with CNS involvement, with some small series (10 cases or less) being reported in the last 30 years39,40,41,42. The largest study has been recently reported by Tavee et al.43, who reported small fiber neuropathy in 115 (7%) patients out of 1580 patients assessed in the sarcoidosis clinic at the Cleveland Clinic (including patients with a clinical suspicion and those with abnormal tests/skin biopsy). In contrast, the frequency of PNS involvement in our cohort was lower than 1%, probably due to a lack of systematic study searching for NNS in patients with normal electromyographic studies; in addition, the criteria proposed by Stern et al.10 do not include specifically NNS in the definition of peripheral nerve involvement of sarcoidosis.
One of the objectives of our study was to analyse how different could be the disease phenotype of the different clinical subsets of neurosarcoidosis. First, we have analysed potential differences between the two main subtypes of neurosarcoidosis (CNS and PNS involvements) and we found that, interestingly, the two subtypes do not overlap in any patient, as has been reported in other systemic autoimmune diseases such as lupus44 or Sjögren syndrome45. However, we found no significant differences in the disease phenotype between patients with CNS involvement and PNS involvement, except for a statistically-significant trend for a higher frequency of women in patients with CNS involvement and a higher frequency of renal involvement in those with PNS involvement (an association rarely reported until now)46. In contrast, the phenotypic differences were more pronounced when these two neurological subsets were compared with patients without neurosarcoidosis. Patients with CNS sarcoidosis presented with a specific phenotype defined by a lower frequency of thoracic involvement and a higher frequency of other concomitant systemic involvements (ocular and salivary gland involvement), while those with PNS involvement showed a higher frequency of liver/spleen involvement in comparison with patients without neurosarcoidosis. These results confirm that systemic involvement in sarcoidosis usually clusters the involvement of some specific organs47, with five distinct clinical phenotypes recently described, including a cluster called ocular–cardiac–cutaneous–CNS (“OCCC”; eye/heart/skin/salivary glands/CNS), in which a statistical association was found with the concomitant development of neurological, ocular and salivary gland involvements.
Since neurosarcoidosis may be associated with a significant neurological impairment, it sounds reasonable to recommend treating all patients48,49. However, a systematic review31 report no specific therapeutic intervention in 15% of patients, probably in those with milder involvements. In our series, a wait and see management was followed in 7% of patients (in patients with isolated facial palsy or mild peripheral neuropathy), of whom only one showed a spontaneous partial recovery during the follow-up. Corticosteroids are well stablished as the first line therapy of neurosarcoidosis, and were administered in 83% of cases included in the systematic review by Fritz et al.31, followed by oral immunosuppressive agents as second line therapies in 27% and TNF-alpha antagonists in 4%. In our series, the frequencies were similar for the use of biologics (4%) and somewhat higher for the use of corticosteroids (92%) and immunosuppressive agents (32%). CNS sarcoidosis is one of the extrathoracic involvements with a worse prognosis, and the ultimate target is to achieve a clinical improvement, or at least, to stabilize the neurological impairment. The complete resolution of neuroimaging involvement cannot be considered a therapeutic target, with some study50 reporting no significant changes in neuroimaging studies even when patients achieved a clinical response. In contrast, we have reported neuroimaging relapses in some asymptomatic patients who achieved a complete clinical response, suggesting that a close neurological follow-up should be carried out in patients with CNS neurosarcoidosis. The review by Fritz et al.31 showed a favourable outcome in 71% of cases after receiving first line therapies (overwhelmingly corticoids), while two additional series24,27 reported that at the last visit, only 16 (21%) were asymptomatic, with 36 (48%) having minor sequelae and 23 (31%) remaining with moderate/severe impairment.
Similarly to previous studies, which were also descriptive series of cases, our study has several potential limitations inherent to this uncontrolled design. As all retrospective analysis, it may suffer from potential selection biases. In addition, although we believe that the statistical approach that we used to compare the different neurological subsets is valid, we acknowledge that the sample sizes are small and multiple comparisons were made, and therefore, the statistically-significant findings should be interpreted with caution. In contrast, we think that a significant strength has been the use of the most recent internationally-proposed set of criteria that may have contributed to a more precise definition and classification of neurosarcoidosis in our cohort. Until now, the majority of criteria are derived from proposals from single institutions on the basis of a description of a personal series of cases51,52,53, expert opinions21,54,55 or scientific societies of single countries56. Therefore, there was a wide heterogeneity in the patients classified as neurosarcoidosis including in the series reported until now10, with some series even considering patients with muscular sarcoidosis24,29 or with ocular involvement/uveitis23,27 as neurosarcoidosis. Probably, the best classification approach was made by the WASOG consensus group13 who defined as highly/at least probable neurosarcoidosis the granulomatous inflammation of the meninges, brain, ventricular (CSF) system, cranial nerves, pituitary gland, spinal cord, cerebral vasculature or nerve roots, especially in the presence of abnormal MRI characteristic of neurosarcoidosis (abnormal enhancement following the administration of gadolinium) or a cerebrospinal fluid exam demonstrating inflammation; however, PNS involvement was excluded from the WASOG classification. Therefore, we believe that the use of the criteria recently proposed by Stern et al.10 has contributed significantly to obtain a more homogeneous picture of the involvement of the nervous system by sarcoidosis in one of the largest series of reported cases.
In a large cohort of patients from southern Europe, 5% fulfilled the criteria of neurosarcoidosis recently proposed by Stern et al.10. Among the two main subsets of neurological involvement, CNS involvement prevails significantly over PNS involvement, and both conditions do not overlap in any patient. The systemic phenotype associated to each involvement is clearly differentiated, with a lower thoracic and greater ocular and glandular expression in patients with CNS involvement, and a greater hepatosplenic expression in those with PNS involvement. This differentiated systemic expression can be helpful not only in the early identification of neurological involvement, but also in the systemic evaluation of patients diagnosed with neurosarcoidosis.
References
Baughman, R. P. et al. Sarcoidosis in America. Analysis based on health care use. Ann. Am. Thorac. Soc. 13(8), 1244–1252. https://doi.org/10.1513/AnnalsATS.201511-760OC (2016).
Brito-Zeron, P., Kostov, B., Superville, D., Baughman, R. P. & Ramos-Casals, M. Geoepidemiological big data approach to sarcoidosis: Geographical and ethnic determinants. Clin. Exp. Rheumatol. 37(6), 1052–1064 (2019).
Baughman, R. P., Lower, E. E. & du Bois, R. M. Sarcoidosis. Lancet 361(9363), 1111–1118. https://doi.org/10.1016/S0140-6736(03)12888-7 (2003).
Pereira, C. A. C., Dornfeld, M. C., Baughman, R. & Judson, M. A. Clinical phenotypes in sarcoidosis. Curr. Opin. Pulm. Med. 20(5), 496–502. https://doi.org/10.1097/MCP.0000000000000077 (2014).
Gerke, A. K. Morbidity and mortality in sarcoidosis. Curr. Opin. Pulm. Med. 20(5), 472–478. https://doi.org/10.1097/MCP.0000000000000080 (2014).
Perez-Alvarez, R. et al. Systemic phenotype of sarcoidosis associated with radiological stages. Analysis of 1230 patients. Eur. J. Intern. Med. 69, 77–85. https://doi.org/10.1016/j.ejim.2019.08.025 (2019).
Judson, M. A., Boan, A. D. & Lackland, D. T. The clinical course of sarcoidosis: Presentation, diagnosis, and treatment in a large white and black cohort in the United States. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG/World Assoc. Sarcoidosis Other Granulomatous Disord. 29(2), 119–127 (2012).
Ungprasert, P. et al. Epidemiology of sarcoidosis 1946–2013: A population-based study. Mayo Clin. Proc. 91(2), 183–188. https://doi.org/10.1016/j.mayocp.2015.10.024 (2016).
Kidd, D. P. Sarcoidosis of the central nervous system: Clinical features, imaging, and CSF results. J. Neurol. 265(8), 1906–1915. https://doi.org/10.1007/s00415-018-8928-2 (2018).
Stern, B. J. et al. Definition and consensus diagnostic criteria for neurosarcoidosis: From the Neurosarcoidosis Consortium Consensus Group. JAMA Neurol. 75(12), 1546–1553. https://doi.org/10.1001/jamaneurol.2018.2295 (2018).
Hunninghake, G. W. et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG/World Assoc. Sarcoidosis Other Granulomatous Disord. 16(2), 149–173 (1999).
Brito-Zeron, P. et al. Epidemiologic patterns of disease expression in sarcoidosis: Age, gender and ethnicity-related differences. Clin. Exp. Rheumatol. 34(3), 380–388 (2016).
Judson, M. A. et al. The WASOG sarcoidosis organ assessment instrument: An update of a previous clinical tool. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG/World Assoc. Sarcoidosis Other Granulomatous Disord. 31(1), 19–27 (2014).
Scadding, J. G. Prognosis of intrathoracic sarcoidosis in England. A review of 136 cases after five years’ observation. Br. Med. J. 2(5261), 1165–1172 (1961).
Baughman, R. P. et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am. J. Respir. Crit. Care Med. 164(10 Pt 1), 1885–1889. https://doi.org/10.1164/ajrccm.164.10.2104046 (2001).
Morimoto, T. et al. Epidemiology of sarcoidosis in Japan. Eur. Respir. J. 31(2), 372–379. https://doi.org/10.1183/09031936.00075307 (2008).
Christoforidis, G. A., Spickler, E. M., Recio, M. V. & Mehta, B. M. MR of CNS sarcoidosis: Correlation of imaging features to clinical symptoms and response to treatment. AJNR Am. J. Neuroradiol. 20(4), 655–669 (1999).
Gascón-Bayarri, J. et al. Neurosarcoidosis: Report of 30 cases and a literature survey. Eur. J. Intern. Med. 22(6), e125–e132. https://doi.org/10.1016/j.ejim.2011.08.019 (2011).
Nozaki, K., Scott, T. F., Sohn, M. & Judson, M. A. Isolated neurosarcoidosis: Case series in 2 sarcoidosis centers. Neurologist. 18(6), 373–377. https://doi.org/10.1097/NRL.0b013e3182704d04 (2012).
Kingah, P., Alam, M., Chugh, K., Kamholz, J. & Samavati, L. Role of pulmonary evaluation in diagnosis of neurosarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG. 33(3), 209–215 (2016).
Gelfand, J. M. et al. Infliximab for the treatment of CNS sarcoidosis: A multi-institutional series. Neurology 89(20), 2092–2100. https://doi.org/10.1212/WNL.0000000000004644 (2017).
Pawate, S., Moses, H. & Sriram, S. Presentations and outcomes of neurosarcoidosis: A study of 54 cases. QJM 102(7), 449–460. https://doi.org/10.1093/qjmed/hcp042 (2009).
Shah, R., Roberson, G. H. & Curé, J. K. Correlation of MR imaging findings and clinical manifestations in neurosarcoidosis. AJNR Am. J. Neuroradiol. 30(5), 953–961. https://doi.org/10.3174/ajnr.A1470 (2009).
Leonhard, S. E. et al. Neurosarcoidosis in a tertiary referral center: A cross-sectional cohort study. Medicine 95(14), e3277. https://doi.org/10.1097/MD.0000000000003277 (2016).
Sohn, M. et al. Spinal cord neurosarcoidosis. Am. J. Med. Sci. 347(3), 195–198. https://doi.org/10.1097/MAJ.0b013e3182808781 (2014).
O’Dwyer, J. P. et al. Neurosarcoidosis-related intracranial haemorrhage: Three new cases and a systematic review of the literature. Eur. J. Neurol. 20(1), 71–78. https://doi.org/10.1111/j.1468-1331.2012.03783.x (2013).
Joseph, F. G. & Scolding, N. J. Neurosarcoidosis: A study of 30 new cases. J. Neurol. Neurosurg. Psychiatry. 80(3), 297–304. https://doi.org/10.1136/jnnp.2008.151977 (2009).
Scott, T. F., Yandora, K., Kunschner, L. J. & Schramke, C. Neurosarcoidosis mimicry of multiple sclerosis: Clinical, laboratory, and imaging characteristics. Neurologist. 16(6), 386–389. https://doi.org/10.1097/NRL.0b013e3181b287df (2010).
Ferriby, D. et al. Clinical manifestations and therapeutic approach in neurosarcoidosis. Rev. Neurol. 156(11), 965–975 (2000).
Sharma, O. P. Neurosarcoidosis: A personal perspective based on the study of 37 patients. Chest 112(1), 220–228. https://doi.org/10.1378/chest.112.1.220 (1997).
Fritz, D., van de Beek, D. & Brouwer, M. C. Clinical features, treatment and outcome in neurosarcoidosis: Systematic review and meta-analysis. BMC Neurol. 16(1), 220. https://doi.org/10.1186/s12883-016-0741-x (2016).
Anthony, J., Esper, G. J. & Ioachimescu, A. Hypothalamic-pituitary sarcoidosis with vision loss and hypopituitarism: Case series and literature review. Pituitary 19(1), 19–29. https://doi.org/10.1007/s11102-015-0678-x (2016).
Beardsley, T. L., Brown, S. V., Sydnor, C. F., Grimson, B. S. & Klintworth, G. K. Eleven cases of sarcoidosis of the optic nerve. Am. J. Ophthalmol. 97(1), 62–77. https://doi.org/10.1016/0002-9394(84)90447-1 (1984).
Graham, E. M., Ellis, C. J., Sanders, M. D. & McDonald, W. I. Optic neuropathy in sarcoidosis. J. Neurol. Neurosurg. Psychiatry. 49(7), 756–763. https://doi.org/10.1136/jnnp.49.7.756 (1986).
Flanagan, E. P. et al. Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann. Neurol. 79(3), 437–447. https://doi.org/10.1002/ana.24582 (2016).
Cohen-Aubart, F. et al. Spinal cord sarcoidosis: Clinical and laboratory profile and outcome of 31 patients in a case-control study. Medicine 89(2), 133–140. https://doi.org/10.1097/MD.0b013e3181d5c6b4 (2010).
Durel, C.-A. et al. Clinical features and prognostic factors of spinal cord sarcoidosis: A multicenter observational study of 20 BIOPSY-PROVEN patients. J. Neurol. 263(5), 981–990. https://doi.org/10.1007/s00415-016-8092-5 (2016).
Bradley, D. A., Lower, E. E. & Baughman, R. P. Diagnosis and management of spinal cord sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG. 23(1), 58–65 (2006).
Said, G. et al. Nerve granulomas and vasculitis in sarcoid peripheral neuropathy: A clinicopathological study of 11 patients. Brain 125(Pt 2), 264–275 (2002).
Hoitsma, E. et al. Small fibre neuropathy in sarcoidosis. Lancet 359(9323), 2085–2086. https://doi.org/10.1016/s0140-6736(02)08912-2 (2002).
Zuniga, G., Ropper, A. H. & Frank, J. Sarcoid peripheral neuropathy. Neurology 41(10), 1558–1561. https://doi.org/10.1212/wnl.41.10.1558 (1991).
Scott, T. S., Brillman, J. & Gross, J. A. Sarcoidosis of the peripheral nervous system. Neurol. Res. 15(6), 389–390. https://doi.org/10.1080/01616412.1993.11740169 (1993).
Tavee, J. O. et al. Sarcoidosis-associated small fiber neuropathy in a large cohort: Clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir. Med. 126, 135–138. https://doi.org/10.1016/j.rmed.2017.03.011 (2017).
Hanly, J. G. et al. Peripheral nervous system disease in systemic lupus erythematosus: Results from an international inception cohort study. Arthritis Rheumatol. 72(1), 67–77. https://doi.org/10.1002/art.41070 (2020).
Brito-Zerón, P. et al. Classification and characterisation of peripheral neuropathies in 102 patients with primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 31(1), 103–110 (2013).
Dierkes-Globisch, A., Schneiderhan, W. & Mohr, H. H. Acute renal failure due to hypercalcemia and peripheral polyneuropathy–rare manifestations of sarcoidosis. Med. Klin. 95(10), 583–586. https://doi.org/10.1007/pl00002066 (2000).
Schupp, J. C. et al. Phenotypes of organ involvement in sarcoidosis. Eur. Respir. J. https://doi.org/10.1183/13993003.00991-2017 (2018).
Agnihotri, S. P., Singhal, T., Stern, B. J. & Cho, T. A. Neurosarcoidosis. Semin. Neurol. 34(4), 386–394. https://doi.org/10.1055/s-0034-1390387 (2014).
Hebel, R., Dubaniewicz-Wybieralska, M. & Dubaniewicz, A. Overview of neurosarcoidosis: Recent advances. J. Neurol. 262(2), 258–267. https://doi.org/10.1007/s00415-014-7482-9 (2015).
Agbogu, B. N., Stern, B. J., Sewell, C. & Yang, G. Therapeutic considerations in patients with refractory neurosarcoidosis. Arch. Neurol. 52(9), 875–879. https://doi.org/10.1001/archneur.1995.00540330053014 (1995).
Zajicek, J. P. et al. Central nervous system sarcoidosis–diagnosis and management. QJM 92(2), 103–117. https://doi.org/10.1093/qjmed/92.2.103 (1999).
Joseph, F. G. & Scolding, N. J. Sarcoidosis of the nervous system. Pract. Neurol. 7(4), 234–244. https://doi.org/10.1136/jnnp.2007.124263 (2007).
Marangoni, S., Argentiero, V. & Tavolato, B. Neurosarcoidosis Clinical description of 7 cases with a proposal for a new diagnostic strategy. J. Neurol. 253(4), 488–495. https://doi.org/10.1007/s00415-005-0043-5 (2006).
Krumholz, A. & Stern, B. J. Neurologic manifestations of sarcoidosis. Handb. Clin. Neurol. 119, 305–333. https://doi.org/10.1016/B978-0-7020-4086-3.00021-7 (2014).
Spiegel, D. R., Morris, K. & Rayamajhi, U. Neurosarcoidosis and the complexity in its differential diagnoses: A review. Innov. Clin. Neurosci. 9(4), 10–16 (2012).
Sakuta, M., Kumamoto, T., Iizuka, T., Nishiyama, K. & Oritsu, M. Diagnostic criteria of neurosarcoidosis. Rinsho Shinkeigaku 45(11), 837–840 (2005).
Author information
Authors and Affiliations
Consortia
Contributions
M.R-C., R.P-A. and P.B-Z. contributed to the conception and design of the work. B.K. performed the statistical analysis. M.R-C., R.P-A., P.B-Z., B.K. and Y.B. interpreted the data. All authors read, substantively revised and approved the final manuscript. All authors ensured that questions related to the accuracy or integrity of any part of the work are appropriately investigated, resolved, and the resolution documented in the literature.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ramos-Casals, M., Pérez-Alvarez, R., Kostov, B. et al. Clinical characterization and outcomes of 85 patients with neurosarcoidosis. Sci Rep 11, 13735 (2021). https://doi.org/10.1038/s41598-021-92967-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92967-6
- Springer Nature Limited
This article is cited by
-
A comprehensive diagnostic approach in suspected neurosarcoidosis
Scientific Reports (2023)
-
Neurosarcoidosis: Diagnostic Challenges and Mimics A Review
Current Allergy and Asthma Reports (2023)
-
Sarkoidose als Paradebeispiel einer granulomatösen Erkrankung
Zeitschrift für Rheumatologie (2022)