
Abstract
Background
In sudden, unexpected, non-traumatic death in young individuals, structural abnormalities of the heart are frequently identified at autopsy. However, the findings may be unspecific and cause of death may remain unclear. A significant proportion of these cases are most likely caused by inherited cardiac diseases, and the cases are categorized as sudden cardiac death (SCD). The purpose of this study was to explore the added diagnostic value of genetic testing by next-generation sequencing (NGS) of a broad gene panel, as a supplement to the traditional forensic investigation in cases with non-diagnostic structural abnormalities of the heart.
Methods and results
We screened 72 suspected SCD cases (<50 years) using the HaloPlex Target Enrichment System (Agilent) and NGS (Illumina MiSeq) for 100 genes previously associated with inherited cardiomyopathies and channelopathies. Fifty-two cases had non-diagnostic structural cardiac abnormalities and 20 cases, diagnosed with a cardiomyopathy post-mortem (ARVC = 14, HCM = 6), served as comparators. Fifteen (29 %) of the deceased individuals with non-diagnostic findings had variants with likely functional effects based on conservation, computational prediction, allele-frequency and supportive literature. The corresponding frequency in deceased individuals with cardiomyopathies was 35 % (p = 0.8).
Conclusion
The broad genetic screening revealed variants with likely functional effects at similar high rates, i.e. in 29 and 35 % of the suspected SCD cases with non-diagnostic and diagnostic cardiac abnormalities, respectively. Although the interpretation of broad NGS screening is challenging, it can support the forensic investigation and help the cardiologist’s decision to offer counselling and clinical evaluation to relatives of young SCD victims.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sudden cardiac death (SCD) is one of the most frequent causes of sudden and unexplained death in the young population (<50 years) and is mainly caused by premature ischemic heart disease, cardiomyopathies or channelopathies [1]. SCD is defined as sudden, natural and unexpected death due to cardiac or unknown causes that occurs, if witnessed, within 1 h of symptom onset and, if unwitnessed, occurring less than 24 h after the deceased was last seen alive and functioning normally [2]. In cases where an autopsy is performed, two thirds of young SCD victims have some degree of structural abnormalities of the heart [3], and in half of these individuals, the aetiology has been reported to be inherited cardiac diseases [4, 1]. However, it is not always possible to establish a definite post-mortem diagnosis as the structural findings may be unspecific and does not fulfil the diagnostic criteria for a specific disease phenotype.
In cases of sudden unexplained death, forensics is involved in most countries. The purpose of forensic investigations is to establish the mode of death and, if possible, also to identify the cause of death. However, the autopsy is also of importance for the identification—or exclusion—of the presence of an inherited cardiac disease, as this may indicate whether relatives could be at risk of SCD and if family screening is warranted [5, 6].
The introduction of molecular autopsy in forensics has proven to be valuable in sudden unexplained death (SUD) cases [7–10]. Post-mortem studies of young deceased individuals with negative autopsy (n = 15–173) have identified putative pathogenic variants in genes associated with channelopathies in 11 to 26 % of the cases [7–10]. However, only a few post-mortem studies of SCD victims with structural abnormalities of the heart have been published. Allegue et al. [11] genetically investigated 23 deceased individuals with hypertrophic cardiomyopathy (HCM) for 16 genes previously associated with HCM (MYH7, MYBPC3, TNNI3, TNNT2, TMP1, TNNC1, ACTC, MYH6, MYL2, MYL3, TCAP, GLA, PRKAG2, TTN, MYLK2 and MYO6) and found putative pathogenic variants in 2 (9 %) of the investigated cases. Zhang et al. [12] screened 25 deceased diagnosed with arrhythmogenic right ventricular cardiomyopathy (ARVC) for variants in PKP2 and identified 6 (25 %) possibly disease-causing or novel variants. Larsen et al. [13] investigated 41 deceased individuals with HCM, ARVC or dilated cardiomyopathy (DCM) for the most commonly associated genes (HCM: MYH7, MYBPC3, TNNI3, TNNT2, MYL2, MYL34; ARVC: JUP, DSP, PKP2, DSG2, DSC2, TMEM43; DCM: LMNA). This study identified four (10 %) individuals with a presumed disease-causing variant. In a recent review, we reported that the success rate of genetic testing of probands in a clinical setting is two to three times higher than that in a forensic setting [14]. This is most likely due to the challenges of defining a precise phenotype post-mortem as this is only based on structural findings, as opposed to the broad range of diagnostic tools applied in clinical cardiology, e.g. assessment of symptoms, clinical evaluations, ECG’s, imaging, pharmacological and physiological testing etc. In the SCD cases with non-diagnostic structural findings of the heart, molecular autopsy may be an important supplementary diagnostic tool, especially with next-generation sequencing (NGS), as large-scale gene sequencing can efficiently and rapidly be applied.
The aim of this study was to identify variants with functional effects in the exons of 100 genes associated with inherited cardiomyopathies and channelopathies in a cohort of young suspected SCD victims with non-diagnostic structural abnormalities of the heart in a forensic setting. For comparison, we included SCD victims fulfilling the diagnostic criteria for cardiomyopathy at autopsy. The overall purpose of this study was to explore the added diagnostic value of genetic testing by using NGS of a broad gene panel as a supplement to the conventional forensic investigation.
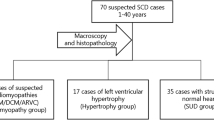
Materials
In Denmark, forensic autopsies are requested by the legal authorities and are performed in cases with suspicion of criminal involvement or in cases of unexpected death without an established cause of death after external examination. Between 2009 and 2011, a total of 829 deceased (<50 years) were autopsied at the Section of Forensic Pathology, Department of Forensic Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark. In 348 (42 %) of the cases, the death was assumed to be of natural cause and 142 (17 %) of these were categorized as suspected SCD including cases with negative autopsy. For this study, we recruited the suspected SCD cases meeting the following inclusion criteria: (a) presence of unspecific (non-diagnostic) structural findings of the heart without any other plausible cause of death or (b) a cardiomyopathy diagnosis established post-mortem without any other plausible cause of death. We excluded sudden unexpected death (SUD) cases and cases with myocarditis, primary valve disease, aortic diseases, coronary artery occlusion or myocardial infarction. The identification of eligible cases was based on autopsy reports, police reports and hospital records if available. All autopsies were performed by forensic pathologists and included general examination as well as thorough macroscopic and microscopic investigations of the heart, following a best practice protocol. Toxicology screening was performed if indicated according to the existing guidelines of the Section of Forensic Pathology. ARVC was diagnosed according to the International Task Force criteria, revised in 2010 [15], <45 % residual myocytes, fibrous replacement of the right ventricular free wall myocardium in more than one sample, with or without fatty replacement. The HCM diagnosis was fulfilled with an increased heart weight and additionally asymmetric left ventricular (LV) hypertrophy and/ or histological abnormalities of LV including disorganization of cardiac muscle cells and/or interstitial fibrosis [16]. All cardiomyopathy diagnoses were confirmed by a cardio-pathologist. Non-diagnostic findings included cases not fulfilling diagnostic criteria for any specific diagnosis and comprised isolated hypertrophy, increased heart weight, mild dilation of one or several chambers, moderate to severe fibrosis, fatty replacements or scattered inflammatory foci in the myocardium.
Methods
Genetic investigations
DNA was extracted from blood (n = 66) using the QIAamp DNA Mini Kit (Qiagen, Stockach, Germany) or from fresh frozen muscle (n = 4) and spleen (n = 2) using the Biorobot EZ1 and the EZ1 DNA Investigator Kit (Qiagen, Stockach, Germany). The extracted DNA was quantified with the Quantifiler Human DNA Quantification Kit (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol.
A total of 100 genes (Table 1) were investigated using NGS. All genes had been reported to be associated with cardiomyopathies or channelopathies and thereby predisposing to SCD. The genes were selected according to the available literature and Online Mendelian Inheritance in Man (OMIM) [17].
The 25 bp of adjacent introns and 5′- and 3′-UTR regions of the 100 investigated genes were isolated and captured using a custom design of the HaloPlex Target Enrichment system (Agilent, USA) (150 bp read lengths). The HaloPlex Target Enrichment protocol was used according to the manufacturer’s version D.5 of the protocol. In brief, the following was performed: (1) digestion of 200 ng of genomic DNA with different restriction enzymes in eight tubes and analysis of the fragments using the 2100 Bioanalyzer (Agilent Technologies, USA) and (2) hybridization of digested DNA to HaloPlex probes. The ends of the probes were complementary to the fragments of the target regions. In the hybridization, the fragments were circularized. Indices with barcodes, sequencing motifs and biotin were incorporated. (3) The target DNA was captured with HaloPlex magnetic beads. (4) The nicks in the circularized DNA fragments were repaired by ligation. (5) The captured target DNA was eluted, PCR-amplified and purified. After library build, the amount of DNA was measured using a Qubit Fluorometer 2.0 with the dsDNA HS assay (Invitrogen, USA). The size distribution of the DNA was analysed using a 2100 Bioanalyzer and the High Sensitivity DNA kit (Agilent Technologies, USA). The DNA was sequenced on a MiSeq (Illumina, USA) according to the manufacturer’s instructions with 150 bp paired-end sequencing using the MiSeq Reagent Kit V2 (300 cycles).
Data analysis and bioinformatics
SureCall (Agilent Technologies, USA) was used for post-base-calling analysis using default settings with an unknown algorithm. In brief, SureCall trimmed the reads to remove adapter sequences and low-quality reads and aligned the reads to the human genome reference (GRCh37/hg19) [18] using BWA MEM [19]. Variant calling was done by BAQ SNP Caller using SAMtools [20] for identifying single-nucleotide polymorphisms (SNPs), multiple-nucleotide polymorphisms (MNPs) and indels (insertion-deletions) (<100 bp). SureCall filtered out false-positive SNPs, MNPs and indels based on read depth and variant quality. A complete list of identified variants was created in Variant Caller Format (VCF).
The target size of the designed gene panel included 788 kbp. Overall, 99.6 % of the bases were covered >50×. Two exons were not covered sufficiently (SCN5A exon 1b NG_008934.1, JPH2 exon 3 NG_031867.1) The median coverage for all samples and regions was 528 (range: 200–710). Variants with a coverage <50 was excluded from further evaluation.
Alamut Batch v. 1.2.0 and Alamut Visual v.2.5 (Interactive Biosoftware, France) were used for the annotation and evaluation of missense, nonsense, splice site variants and small indels. Variants were selected by (1) an in house in silico analysis tool developed for determining the likelihood of variant pathogenicity, (2) frequency <1 % in the relevant reference population [21, 22] and associations to cardiac diseases reported in HGMD [23]. The in silico analysis was based on parameters of conservation (Grantham distance, AlignGVGD class, BLOSUM62, orthologues), computational prediction (MAPP, SIFT), frequency in the relevant reference population (dbSNP [22], Exome Sequencing Project (ESP) [21]), the location in the genome (distance to nearest splice site) and the coding effect. Details of the scoring of variants in the in silico analysis was described by Hertz et al. [10] The variants were evaluated and classified independently as (a) likely, (b) unknown or (c) unlikely to have functional effects by two medical doctors (CLH and MD). The available databases and literature were reviewed for each variant in order to determine the possible effect. Variants were categorized as likely to have functional effects based on the genomic alterations and the supporting literature [24]. Evidence for pathogenicity included null variants (nonsense, frameshift, near splice sites, initiation codon), known disease-causing amino acid change or residue, functional studies, prevalence of the variants in affected individuals with the associated disease, location in exon and/or functional domain with known disease-causing variants, assumed de novo, co-segregation studies and a minor-allele frequency (MAF) below the disease prevalence [24–26]. Given that rare variants are also subject to population stratification [27], we identified a local Danish reference population (n = 2000) that was whole-exome sequenced. Half of the cohort comprised metabolically healthy individuals and the other half were patients with type 2 diabetes [28]. Additionally, allele frequencies from ESP (European-American population) [21] and dbSNP [22] were used.
The software, R v. 2.11.0 [29] was used to perform the statistical analyses and for the scoring of variants in the in silico analysis using a custom script. Fisher’s exact test and Mann-Whitney-Wilcoxon test were used to compare mean and median values, respectively. P values below 0.01 were considered statistically significant. The coverage information were calculated using a custom script in python with the docopt packages and R v.3.1 [29] with dplyr, magrittr, gplots and tools packages and the Bioconductor v.2.14 [30, 31] with packages biomaRt and rtracklayer.
DNA variants were numbered according to reference sequence applied in Table 1 using HGVS nomenclature (www.HGVS.org).
Ethical standards
The study was approved by the Committees on Health Research Ethics in the Capital Region of Denmark (H-2-2012-017) and the Danish Data Protection Agency (2011-54-1262).
Results
The cohort
We identified 72 individuals who fulfilled the inclusion criteria. Of these, 50 (69 %) were males and the median age at time of death was 41 years. Overall, death occurred during sleep in 20 (28 %) cases, at rest in 14 (19 %) cases, during or after physical activity in 8 (11 %) cases and during unknown activity in 28 (39 %) cases. Twenty-two (31 %) had reported symptoms prior to death, including dizziness, syncope, chest pain, dyspnoea and/or palpitations (Table 2).
In 20 (28 %) of the cases, a cardiomyopathy diagnose (ARVC = 14, HCM = 6) was established by a forensic pathologist. The remaining 52 (72 %) individuals had unspecific and non-diagnostic post-mortem findings of the heart without any other plausible cause of death. Descriptive data of the study population is shown in Table 2.
The group of cases with diagnosed cardiomyopathies were younger than the cases with non-diagnostic findings (32 and 43 years, respectively, p = 0.004). In the group of diagnosed cardiomyopathies, 6 (30 %) victims died during physical exercise, compared to 2 (4 %) in the group of non-diagnostic structural findings (p = 0.005). Four (20 %) victims diagnosed with cardiomyopathies had reported chest pain prior to death, compared to none in the non-diagnostic group (p = 0.005). There were no statistically significant differences in the remaining symptoms prior to death, medical history, family history of SCD or toxicology results (Table 2).
Genetic findings
Cardiomyopathies
Of the 20 individuals with cardiomyopathy, 12 variants with likely functional effects were found in 7 (35 %) of the cases (Table 3). The median age of time of death was 31 years (range 21–42 years). Four (57 %) of the genotype positive deceased were males. Four (20 %) individuals had a variant in genes associated with cardiomyopathy and three (15 %) individuals had both a variant in genes associated with cardiomyopathies and in genes associated with channelopathies (Fig. 1). Of the 14 individuals diagnosed with ARVC, five (36 %) had variants with likely functional effects although only two individuals had variants in the desmosome genes. Two (40 %) of the five HCM individuals had variants with likely functional effects in genes involved in the coding for sarcomere proteins. One of the individuals had an additional variant in a gene associated with altered ion channel function.

Percentage of variants with likely functional effects in the suspected SCD cases with a cardiomyopathies and b non-diagnostic structural findings at the heart
Non-diagnostic structural findings
Of the 52 individuals with non-diagnostic structural findings of the heart, 15 (29 %) individuals had variants with likely functional effects, of which two (4 %) had more than one variant in one or more genes (Table 4). Nine (60 %) of the genotype positive individuals were males, and the median age was 45 years (range 1–50 years). Seven (14 %) individuals had variants with likely functional effects in genes associated with cardiomyopathies, and eight (15 %) individuals had a variant in genes associated with channelopathies (Fig. 1). In total, 28 (54 %) individuals had hypertrophy, either isolated or with fibrosis and/or fatty infiltrations. Eleven (39 %) of these individuals had variants with likely functional effects that were equally distributed among cardiomyopathy and channelopathy associated genes. Fourteen (27 %) individuals had isolated fibrosis or fibro-fatty replacement in the myocardium, of which three (21 %) had variants with likely functional effects in genes associated with channelopathies. In the cases with isolated fatty infiltrations (n = 3) or dilation of one or several chambers, no variants with likely functional effects were found (Table 5).
The difference between the frequency of found variants in the group of diagnosed cardiomyopathies and non-diagnostic structural findings was not statistically significant (p = 0.8).
Discussion
We investigated 72 suspected SCD victims with diagnostic or non-diagnostic cardiac findings at autopsy for variants with likely functional effects in 100 genes known to be associated with cardiomyopathies or channelopathies using NGS. Our major finding was that when expanding the genetic testing to a broad gene panel, the identified frequency of variants with likely functional effects in young suspected SCD victims with non-diagnostic and diagnostic cardiac changes, more than doubled compared to previous reports applying limited phenotype specific gene panels [11–13]. Another novel finding was that variants with likely functional effects were identified at similar rates in the two groups, hence in 29 % of the individuals with non-diagnostic structural abnormalities of the heart and in 35 % of the individuals with a cardiomyopathy diagnosis.
Post-mortem diagnosed cardiomyopathies
In the group of cases with a diagnosed cardiomyopathy, variants with likely functional effects were mainly found in genes previously associated with cardiomyopathies, i.e. a reasonable overall genotype-phenotype relationship was seen. However, in the five genotype-positive ARVC individuals, only two had gene variants coding for proteins in the desmosome complex. The remaining three genotype-positive individuals had variants in genes previously shown to be associated with DCM (LMNA and LDB3). This probably reflects the challenges in establishing specific cardiomyopathy diagnoses by post-mortem investigations. Clinically, there is also a well-known overlap between ARVC and DCM [32, 33]. Moreover, some of the assumed associations between the gene variants and specific diseases are based on studies of few cases with phenotypes that might have been misdiagnosed. On this basis it seems prudent to test a broad gene panel in suspected cardiomyopathy cases—rather than to test a narrow gene panel considered to be associated with a specific cardiomyopathy.
Non-diagnostic structural abnormalities—cardiomyopathies or channelopathies?
In the 52 cases with non-diagnostic structural findings, the frequencies of variants with likely functional effects were equally distributed in cardiomyopathy (47 %) and channelopathy (53 %) associated genes. The high frequency of identified variants in genes associated with channelopathies corresponds to previous findings made by Papadakis et al. [6] This study clinically and genetically evaluated family members of victims of either SADS (sudden arrhythmic death syndrome) (n = 163) or individuals with unspecific abnormalities of the heart (n = 41). Targeted genetic analysis was performed dependent on the suspected phenotype (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2 in long QT syndrome (LQTS); SCN5A in Brugada syndrome (BrS); RYR2 in catecholaminergic polymorphic ventricular tachycardia (CPVT)). They found a similar proportion (47 and 51 %, respectively) of channelopathic diseases in the relatives of the two groups. The predominant diagnoses were LQTS and BrS in both cohorts. This demonstrates the diversity of both the phenotypic and genotypic expression of the inherited cardiac diseases. The findings in the present study supported by previous findings by Papadakis et al. [6] suggests that SCD cases with unspecific structural findings of the heart may belong to the category of SUD, i.e. as a primary channelopathy or to the structural group of cardiomyopathies. Previous post-mortem studies of young SUD victims have genetically investigated only the genes most commonly associated with cardiac channelopathies, including LQTS, BrS and CPVT. The reported frequencies in the investigated SUD cohorts have been 11 to 26 % [7–10]. However, two recent whole-exome sequencing studies of young SUD victims reported variants in cardiomyopathy associated genes in 4 (14 %) and 6 (43 %) victims, respectively, with no or minor abnormalities of the heart [34, 35]. This could reflect that cardiomyopathies may present with malignant arrhythmia at an early stage before the overt structural phenotype has developed. On the other hand, in the cases with only variants with likely functional effects in genes associated with channelopathies, the observed unspecific structural changes in the heart may not represent significant pathological changes, but changes that may also be present in normal hearts. Thus, cardiomyopathies may present in various ways and may not be precisely defined at autopsy. Wider genetic screening, including both cardiomyopathy- and channelopathy-associated genes, is therefore a valuable supplement to forensic investigations and will increase the probability of finding the likely cause of death in these particular cases.
Multiple genetic variants
In the present study, 22 individuals (31 %) of the study cohort had variants with likely functional effects. Of these, five (23 %) had more than one variant in one or more genes. In the group of diagnosed cardiomyopathies, half of the genotype positive individuals had several variants affecting genes associated with cardiomyopathies and genes associated with channelopathies. The presence of variants affecting the function of the ion channels in the myocardium might potentially increase the risk of arrhythmias and thereby SCD in cases with structurally affected heart. The significance of several variants is not yet clear, but there might be an interaction between genes with different functions and possible modifying effects. The use of NGS in genetic screening commits to the challenge of interpreting the large amount of results that may be generated. This will most likely be increasingly common as the new genetic methods offer investigations of large numbers of genes so that the likelihood of finding rare variants with unknown significance increases.
Genetics in forensic medicine
For the prospect of implementation of genetic testing as part of forensic investigations, collaboration between all specialties involved is essential. A specific phenotype is crucial in the management of SCD cases and should be based on medical records including symptoms, predispositions and family history. Moreover, a thorough forensic autopsy comprising expert cardiac evaluation is needed. With the combined knowledge gained from adequate medical history, autopsy findings and genetic data, a comprehensive evaluation and diagnostics can be made, and counselling and clinical cardiology evaluation can be offered to the first degree relatives.
Limitations
There is a selection bias in our study, since only autopsied cases from the Department of Forensic Medicine at the University of Copenhagen were included. Furthermore, the study was retrospective and the selection was based on available records from the forensic investigation, not always including hospital records and clinical information prior to death. It was not possible to investigate the families to the deceased. Thus, segregation of the pheno- and genotypes was not explored. Several of the observed variants were novel. Therefore, the classification of variants was solely based on the coding effect, prediction of amino acid substitutions and the functional effect and previously established disease-causing variants in the nearby exon and/or protein domain. To verify the possible pathogenic impact of the observed variants, segregation studies and functional studies are needed.
We did not verify the results or cover the missing exonic regions of the design with another sequencing method. However, the genetic results presented in this study were of high quality and the vast majority of the targeted regions were highly covered, supporting the reliability of the results [36–38]. The currently used 100 gene panel was previously validated by comparing all observed variants in 47 individuals with another NGS based method (Ion AmpliSeq, Ion Personal Genome Machine, Thermo Fischer Scientific). The results were concordant [47]. We also investigated 34 arrhythmia-associated genes in 15 individuals [10] using NimbleGen capture design (Roche) and found full concordance between the results compared to the 100 gene method (unpublished data).
Conclusion
To our knowledge, this is the first large post-mortem genetic study using NGS to investigate suspected SCD victims with structural abnormalities. The study shows the potential of genetic analysis of a large number of genes associated with cardiac diseases in forensic medicine with the use of NGS, as approximately one third of the investigated cases had variants likely to have functional effects. Hence, broad genetic screening may support the medico-legal investigation, particular in cases with non-diagnostic autopsy findings and no clear cause of death. This may also be of importance for follow-up by clinical cardiac evaluation in the relatives left behind
References
Risgaard B, Winkel BG, Jabbari R, Behr ER, Ingemann-Hansen O, Thomsen JL, Ottesen GL, Gislason GH, Bundgaard H, Haunso S, Holst AG, Tfelt-Hansen J (2014) Burden of sudden cardiac death in persons aged 1 to 49 years: nationwide study in denmark. Circ Arrhythm Electrophysiol 7(2):205–211. doi:10.1161/CIRCEP.113.001421
www.who.int World Health Organization. International Classification of Disease 10 (ICD-10). http://www.who.int/classifications/icd/en/. Accessed 01 Jul 2015
Puranik R, Chow CK, Duflou JA, Kilborn MJ, McGuire MA (2005) Sudden death in the young. Heart Rhythm 2(12):1277–1282. doi:10.1016/j.hrthm.2005.09.008
Ferrero‐Miliani L, Holst AG, Pehrson S, Morling N, Bundgaard H (2010) Strategy for clinical evaluation and screening of sudden cardiac death relatives. Fundamental Clin Pharmacol 24(5):619–635. doi:10.1111/j.1472-8206.2010.00864.x
Behr ER, Dalageorgou C, Christiansen M, Syrris P, Hughes S, Esteban MTT, Rowland E, Jeffery S, McKenna WJ (2008) Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J 29(13):1670–1680. doi:10.1093/eurheartj/ehn219
Papadakis M, Raju H, Behr ER, Noronha SVD, Spath N, Kouloubinis A, Sheppard MN, Sharma S (2013) Sudden cardiac death with autopsy findings of uncertain significance potential for erroneous interpretation. Circ Arrhythm Electrophysiol 6(3):588–596. doi:10.1161/CIRCEP.113.000111
Skinner JR, Crawford J, Smith W, Aitken A, Heaven D, Evans C-A, Hayes I, Neas KR, Stables S, Koelmeyer T, Denmark L, Vuletic J, Maxwell F, White K, Yang T, Roden DM, Leren TP, Shelling A, Love DR (2011) Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm 8(3):412–419. doi:10.1016/j.hrthm.2010.11.016
Winkel BG, Larsen MK, Berge KE, Leren TP, Nissen PH, Olesen MS, Hollegaard MV, Jespersen T, Yuan L, Nielsen N, Haunsø S, Svendsen JH, Wang Y, Kristensen IB, Jensen HK, Tfelt-Hansen J, Banner J (2012) The prevalence of mutations in KCNQ1, KCNH2, and SCN5A in an unselected national cohort of young sudden unexplained death cases. J Cardiovasc Electrophysiol 23(10):1092–1098. doi:10.1111/j.1540-8167.2012.02371.x
Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ (2012) Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc 87(6):524–539. doi:10.1016/j.mayocp.2012.02.017
Hertz CL, Christiansen SL, Ferrero-Miliani L, Fordyce SL, Dahl M, Holst AG, Ottesen GL, Frank-Hansen R, Bundgaard H, Morling N (2014) Next-generation sequencing of 34 genes in sudden unexplained death victims in forensics and in patients with channelopathic cardiac diseases. Int J Legal Med. doi:10.1007/s00414-014-1105-y
Allegue C, Gil R, Blanco-Verea A, Santori M, Rodríguez-Calvo M, Concheiro L, Carracedo Á, Brion M (2011) Prevalence of HCM and long QT syndrome mutations in young sudden cardiac death-related cases. Int J Legal Med 125(4):565–572. doi:10.1007/s00414-011-0572-7
Zhang M, Tavora F, Oliveira JB, Li L, Franco M, Fowler D, Zhao Z, Burke A (2012) PKP2 mutations in sudden death from arrhythmogenic right ventricular cardiomyopathy (ARVC) and sudden unexpected death with negative autopsy (SUDNA). Circ J 76(1):189–194. doi:10.1253/circj.CJ-11-0747
Larsen MK, Nissen PH, Berge KE, Leren TP, Kristensen IB, Jensen HK, Banner J (2012) Molecular autopsy in young sudden cardiac death victims with suspected cardiomyopathy. Forensic Sci Int 219(1–3):33–38. doi:10.1016/j.forsciint.2011.11.020
Hertz CL, Ferrero-Miliani L, Frank-Hansen R, Morling N, Bundgaard H (2014) A comparison of genetic findings in sudden cardiac death victims and cardiac patients: the importance of phenotypic classification. Europace. doi:10.1093/europace/euu210
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DMY, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia proposed modification of the task force criteria. Eur Heart J 31(7):806–814. doi:10.1093/eurheartj/ehq025
Maron BJ, Carney KP, Lever HM, Lewis JF, Barac I, Casey SA, Sherrid MV (2003) Relationship of race to sudden cardiac death in competitive athletes with hypertrophic cardiomyopathy. J Am Coll Cardiol 41(6):974–980. doi:10.1016/S0735-1097(02)02976-5
www.omim.org Online Mendelian Inheritance in Man. http://omim.org/. Accessed 01 Mar 2014
Kent W, Sugnet C, Furey T, Roskin K, Pringle T, Zahler A, Haussler D (2002) The human genome browser at USCS. Genome Res 12(6):996–1006. doi:10.1101/gr.229102
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25:1754–1760. doi:10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. doi:10.1093/bioinformatics/btp352
www.evs.gs.washington.edu/EVS Exome Variant Server. http://evs.gs.washington.edu/EVS/. Accessed 02 Jan 2014
www.ncbi.nlm.nih.gov National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/. Accessed 10 Feb 2014
Stenson P, Ball E, Mort M, Phillips A, Shiel J, Thomas N, Abeysinghe S, Krawczak M, Copper D (2003) The Human Gene Mutation Database (HGMD). Hum Mutat 21:577–581. doi:10.1002/humu.10212
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med : Off J Am Coll Med Genet 17(5):405–423. doi:10.1038/gim.2015.30
Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, Lewis KL, Cooper DN, Stenson PD, Mullikin JC, Biesecker LG, Program NIHISCCS (2013) Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet 6(4):337–346. doi:10.1161/CIRCGENETICS.113.000039
Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, Bennett RL, Jones KL, Tokita MJ, Bennett JT, Kim JH, Rosenthal EA, Kim DS, National Heart L, Blood Institute Grand Opportunity Exome Sequencing P, Tabor HK, Bamshad MJ, Motulsky AG, Scott CR, Pritchard CC, Walsh T, Burke W, Raskind WH, Byers P, Hisama FM, Nickerson DA, Jarvik GP (2013) Actionable, pathogenic incidental findings in 1,000 participants' exomes. Am J Hum Genet 93 (4):631–640. doi:10.1016/j.ajhg.2013.08.006
Mathieson I, McVean G (2012) Differential confounding of rare and common variants in spatially structured populations. Nat Genet 44(3):243–246. doi:10.1038/ng.1074
Lohmueller KE, Sparso T, Li Q, Andersson E, Korneliussen T, Albrechtsen A, Banasik K, Grarup N, Hallgrimsdottir I, Kiil K, Kilpelainen TO, Krarup NT, Pers TH, Sanchez G, Hu Y, Degiorgio M, Jorgensen T, Sandbaek A, Lauritzen T, Brunak S, Kristiansen K, Li Y, Hansen T, Wang J, Nielsen R, Pedersen O (2013) Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet 93(6):1072–1086. doi:10.1016/j.ajhg.2013.11.005
Team RC (2014) R: a language and environment for statistical computing. Available via R Foundation for Statistical Computing. http://www.R-project.org
Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, Hahne F, Hansen KD, Irizarry RA, Lawrence M, Love MI, MacDonald J, Obenchain V, Oles AK, Pages H, Reyes A, Shannon P, Smyth GK, Tenenbaum D, Waldron L, Morgan M (2015) Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12(2):115–121. doi:10.1038/nmeth.3252
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5(10):R80. doi:10.1186/gb-2004-5-10-r80
Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M (1996) Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 94(5):983–991
Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ (2008) Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 52(25):2175–2187. doi:10.1016/j.jacc.2008.09.019
Bagnall RD, Das KJ, Duflou J, Semsarian C (2014) Exome analysis-based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm 11(4):655–662. doi:10.1016/j.hrthm.2014.01.017
Narula N, Tester DJ, Paulmichl A, Maleszewski JJ, Ackerman MJ (2014) Post-mortem whole exome sequencing with gene-specific analysis for autopsy-negative sudden unexplained death in the young: a case series. Pediatr Cardiol. doi:10.1007/s00246-014-1082-4
Millat G, Chanavat V, Rousson R (2014) Evaluation of a new NGS method based on a custom AmpliSeq library and Ion Torrent PGM sequencing for the fast detection of genetic variations in cardiomyopathies. Clin Chim Acta; Int J Clin Chem 433:266–271. doi:10.1016/j.cca.2014.03.032
Sikkema-Raddatz B, Johansson LF, de Boer EN, Almomani R, Boven LG, van den Berg MP, van Spaendonck-Zwarts KY, van Tintelen JP, Sijmons RH, Jongbloed JD, Sinke RJ (2013) Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat 34(7):1035–1042. doi:10.1002/humu.22332
Mook ORF, Haagmans MA, Soucy J-F, van de Meerakker JBA, Baas F, Jakobs ME, Hofman N, Christiaans I, Lekanne Deprez RH, Mannens MMAM (2013) Targeted sequence capture and GS-FLX Titanium sequencing of 23 hypertrophic and dilated cardiomyopathy genes: implementation into diagnostics. J Med Genet 50(9):614–626. doi:10.1136/jmedgenet-2012-101231
Zimmerman RS, Cox S, Lakdawala NK, Cirino A, Mancini-DiNardo D, Clark E, Leon A, Duffy E, White E, Baxter S, Alaamery M, Farwell L, Weiss S, Seidman CE, Seidman JG, Ho CY, Rehm HL, Funke BH (2010) A novel custom resequencing array for dilated cardiomyopathy. Genet Med : Off J Am Coll Med Genet 12(5):268–278. doi:10.1097/GIM.0b013e3181d6f7c0
Roux-Buisson N, Gandjbakhch E, Donal E, Probst V, Deharo J, Chevalier P, Klug D, Masencal N, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller D, Hidden-Lucet F, Trapani J, Fouret P, Frank R, Fressart V, Fauré J (2014) Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: results of a systematic screening. Heart Rhythm 11(11):1999–2009. doi:10.1016/j.hrthm.2014.07.020
Abraham RL, Yang T, Blair M, Roden DM, Darbar D (2010) Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J Mol Cell Cardiol 48(1):181–190. doi:10.1016/j.yjmcc.2009.07.020
Liu H, El Zein L, Kruse M, Guinamard R, Beckmann A, Bozio A, Kurtbay G, Megarbane A, Ohmert I, Blaysat G, Villain E, Pongs O, Bouvagnet P (2010) Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ Cardiovasc Genet 3(4):374–385. doi:10.1161/CIRCGENETICS.109.930867
Stallmeyer B, Zumhagen S, Denjoy I, Duthoit G, Hebert JL, Ferrer X, Maugenre S, Schmitz W, Kirchhefer U, Schulze-Bahr E, Guicheney P, Schulze-Bahr E (2012) Mutational spectrum in the Ca(2+)—activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum Mutat 33(1):109–117. doi:10.1002/humu.21599
Nielsen NH, Winkel BG, Kanters JK, Schmitt N, Hofman-Bang J, Jensen HS, Bentzen BH, Sigurd B, Larsen LA, Andersen PS, Haunso S, Kjeldsen K, Grunnet M, Christiansen M, Olesen SP (2007) Mutations in the Kv1.5 channel gene KCNA5 in cardiac arrest patients. Biochem Biophys Res Commun 354(3):776–782. doi:10.1016/j.bbrc.2007.01.048
Gonzalez-Garay ML, McGuire AL, Pereira S, Caskey CT (2013) Personalized genomic disease risk of volunteers. Proc Natl Acad Sci U S A 110(42):16957–16962. doi:10.1073/pnas.1315934110
Hassel D, Dahme T, Erdmann J, Meder B, Huge A, Stoll M, Just S, Hess A, Ehlermann P, Weichenhan D, Grimmler M, Liptau H, Hetzer R, Regitz-Zagrosek V, Fischer C, Nurnberg P, Schunkert H, Katus HA, Rottbauer W (2009) Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat Med 15(11):1281–1288. doi:10.1038/nm.2037
Hertz CL, Christiansen SL, Larsen MK, Dahl M, Ferrero-Miliani L, Weeke PE, Pedersen O, Hansen T, Grarup N, Ottesen GL, Frank-Hansen R, Banner J, Morling N (2015) Genetic investigations of sudden unexpected deaths in infancy using next-generation sequencing of 100 genes associated with cardiac diseases. Eur J Human Genet Accept. In press
Acknowledgments
The authors thank Francisc-Raul Kantor for bioinformatics support. For the screening of variants among Danish controls, we thank LuCamp, The Lundbeck Foundation Centre for Applied Medical Genomics in Personalized Disease Prediction, Prevention, and Care (www.lucamp.org), and the Novo Nordisk Foundation Center for Basic Metabolic Research, which is an independent Research Center at the University of Copenhagen partially supported by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk).
Funding sources
This work was supported by Ellen and Aage Andersen’s foundation and The A.P. Møller Foundation.
Conflic of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
C. L. Hertz and S. L. Christiansen have shared first authorship and contributed equally to the work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 479 kb)
Rights and permissions
About this article

Cite this article
Hertz, C.L., Christiansen, S.L., Ferrero-Miliani, L. et al. Next-generation sequencing of 100 candidate genes in young victims of suspected sudden cardiac death with structural abnormalities of the heart. Int J Legal Med 130, 91–102 (2016). https://doi.org/10.1007/s00414-015-1261-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-015-1261-8