
Abstract
Sudden cardiac death (SCD) is responsible for a large proportion of sudden deaths in young individuals. In forensic medicine, many cases remain unexplained after routine postmortem autopsy and conventional investigations. These cases are called sudden unexplained deaths (SUD). Genetic testing has been suggested useful in forensic medicine, although in general with a significantly lower success rate compared to the clinical setting. The purpose of the study was to estimate the frequency of pathogenic variants in the genes most frequently associated with SCD in SUD cases and compare the frequency to that in patients with inherited cardiac channelopathies. Fifteen forensic SUD cases and 29 patients with channelopathies were investigated. DNA from 34 of the genes most frequently associated with SCD were captured using NimbleGen SeqCap EZ library build and were sequenced with next-generation sequencing (NGS) on an Illumina MiSeq. Likely pathogenic variants were identified in three out of 15 (20 %) forensic SUD cases compared to 12 out of 29 (41 %) patients with channelopathies. The difference was not statistically significant (p = 0.1). Additionally, two larger deletions of entire exons were identified in two of the patients (7 %). The frequency of likely pathogenic variants was >2-fold higher in the clinical setting as compared to SUD cases. However, the demonstration of likely pathogenic variants in three out of 15 forensic SUD cases indicates that NGS investigations will contribute to the clinical investigations. Hence, this has the potential to increase the diagnostic rate significantly in the forensic as well as in the clinical setting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sudden cardiac death (SCD) is a major cause of unexpected, non-traumatic deaths in young and apparently healthy individuals. The major aetiologies of SCD include cardiomyopathies, channelopathies and ischaemic heart diseases. In about two thirds of SCD cases, structural abnormalities are evident at autopsy [1–3], whereas no structural pathogenic variation of the heart is found in the remaining cases. These cases are called sudden unexplained deaths (SUD). Many of the SUD cases are suspected to be caused by inherited cardiac channelopathies [4] among which the most common ones are long QT syndrome (LQTS), Brugada syndrome (BS), catecholaminergic polymorphic ventricular tachycardia (CPVT) and short QT syndrome (SQTS). More than 100 genes have been shown to be associated with inherited SCD [5–8]. With modern DNA technology like next-generation sequencing (NGS), it is possible to investigate large numbers of genes and thus support the clinical investigation and increase the possibility to establish a likely cause of death. This may be of importance for legal reasons but may also be valuable information when considering screening of family members for the same disease.
In the forensic setting, SUD is a major challenge. Investigation of pathogenic variants in genes associated with cardiac diseases is possible, but the diagnostic yield is not well known. The aim of this study was to estimate the frequency of pathogenic variants in 34 of the genes most frequently associated with channelopathic diseases, comparing SUD cases and patients with clinically diagnosed inherited cardiac channelopathies.
Material and methods
Study population
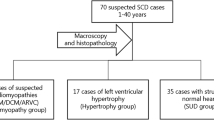
A total of 15 SUD cases from the Section of Forensic Pathology, Department of Forensic Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark, were investigated. The individuals were <50 years of age and randomly selected if (i) deceased within a time period of 1 year (2009–2010), (ii) they had died suddenly, unexpectedly and with no diagnosis after thorough macro- and microscopic investigations by the forensic pathologists and (iii) the toxicology screenings were negative. The SUD cases included 11 males with a median age of 38 years (range 3–49) and four females with a median age of 19 years (range 1–42). The majority, 12 (80 %), was last seen alive and healthy less than 24 h prior to the death and none had a known family history of SCD.
A total of 29 clinically affected patients from the Department of Cardiology, National University Hospital, Rigshospitalet, Copenhagen, Denmark, were included. Twelve (41 %) patients had LQTS, nine (31 %) Brugada syndrome, one (3 %) SQTS and seven (24 %) non-diagnostic phenotypes with symptoms and findings suggestive of channelopathic diseases. The median age at time of diagnosis was 36 years (8–64) with 22 (79 %) under the age of 50 years. A total of 17 (59 %) were males. Family history of SCD was reported in 10 (34 %) patients (Table 1).
Genetic investigation of single nucleotide variants and large insertion-deletions
DNA was extracted from anti-coagulated whole blood using the QIAamp DNA Mini Kit (Qiagen, Germany) and quantified with the Quantifiler Human DNA Quantification Kit (Thermo Fisher, USA) following the manufacturer’s protocol. The 15 SUD samples were investigated with the AmpFlSTR Identifiler PCR Amplification Kit (Thermo Fisher, USA) to measure the degree of DNA degradation. Full STR profiles were found, thus no SUD sample was degraded. Two patient samples were whole genome amplified due to low DNA concentrations, using the REPLI-g Midi kit (Qiagen, Germany) following the manufacturer’s protocol.
A total of 34 genes (ABCC9, AKAP9, ANK2, CACNA1C, CACNB2, CASQ2, CAV3, DPP6, GJA5, GPD1L, HCN4, KCNA5, KCNE1, KCNE1L, KCNE2,KCNE3, KCNE4, KCNH2, KCNJ2, KCNJ5, KCNJ8, KCNQ1, MYH6, MYH7, NPPA, PRKAG2, RYR2, SCN1B, SCN2B, SCN3B, SCN4B, SCN5A, SNTA1 and TRPM4) were investigated using NGS. All genes have been reported to be associated with inherited cardiac diseases often presented with arrhythmia. The genes were selected from the literature and Online Mendelian Inheritance in Man (OMIM) [9]. The gene MYH7, which is primarily associated with structural cardiac diseases, was included as a result of the similarity to MYH6 and the capture design. The total length of the target regions was 387,395 bp and involved 639 target regions. Target regions included exons, 250 bp of adjacent introns and 5′- and 3′-UTR regions. Targeted sequencing capture probes were custom designed by Roche NimbleGen (USA). Library preparation was performed by using the TruSeq LT DNA Sample Preparation Guide (Illumina, USA). This protocol involved the steps “Fragment Genomic DNA” to “Ligate Indexed Paired-End Adapters”. Following this, the preparation of the libraries were continued using the NimbleGen SeqCap EZ Library SR User’s Guide v4.0 (Roche, Germany) according to the manufacturer’s recommendations. After library build, the samples were analysed on Agilent’s 2100 Bioanalyzer and the DNA concentrations were measured using the Qubit 2.0 Fluorometer (Thermo Fisher, Germany). Libraries were sequenced on an Illumina MiSeq using the MiSeq Reagent kit v2 (500 cycles) with 250-bp paired-end sequencing.
Genetic investigations of large insertion-deletions (indels) in KCNQ1, KCNH2, KCNE1, KCNE2 and SCN5A were performed with multiplex ligation-dependent probe amplification (MLPA) [10] using pre-designed kits (SALSA MLPA P114 Long-QT probe mix, SALSA MLPA P108 SCN5A probe mix (MRC-Holland, Holland). The analyses were performed according to the protocol provided by the supplier. In brief, the procedure involved denaturation of the DNA, hybridisation, ligase reaction, PCR and fragment separation by means of electrophoresis. Each sample was investigated in duplicate. If a rearrangement was observed, a third investigation was performed. Only concordant results were reported.
Data analysis and annotation
MiSeq Reporter (Illumina, USA) was used for the analysis of fastq-files generated by the MiSeq with default settings for PCR amplicon analysis. The human genome sequence UCSC/hg19 [11] was used as reference sequence. The following steps were performed using the MiSeq Reporter: Reads were aligned to the reference genome using BWA [12]. Variant calling was carried out using The Genome Analysis Toolkit (GATK) [13]. Aligned reads were provided in BAM-files (Binary compressed SAM format) and lists of variants in variant caller format (VCF). A total of 98.4 % of the target regions were covered more than 10 times. The median coverage for all regions and samples was 1216 (range 619–3191). In total, all exons in the investigated 34 genes were sequenced except for exon 1 and the 5′-UTR region in SCN1B. Moreover, the first exons and 5′-UTR regions of GPD1L, KCNH2, SCN5A and SNTA1 were not sufficiently covered (maximum coverage <50). Variants were included according to the following criteria: (1) passing the MiSeq Reporter quality filter on default settings based on coverage depth, variant score, mapping quality and quality score and (2) the coverage was above 50.
Alamut-HT v. 1.1.8 (Interactive Biosoftware, France) and Alamut v.2.2 (Interactive Biosoftware, France) was used for the annotation and evaluation of variants. In brief (Fig. 1), variants were selected by (1) frequency in reference populations [14, 15] and associations to cardiac diseases in HGMD [16], (2) in silico analysis of the likelihood of pathogenicity (Tables 2 and 3) and (3) evaluation and classification of each variant by two medical doctors. Variants were classified as likely pathogenic or of unknown significance based on the allele frequency of the variant, segregation evidence, in vitro functional studies, number of affected individuals found with the variant, computational prediction of the functional effect of amino acid substitutions and status as a de novo mutation. The algorithm used for in silico analysis was inspired by van Spendonck-Zwarts et al. [17].

Selection and evaluation of variants. After annotation (step 1), variants were selected manually (step 2) or through the in silico analysis (step 3). Using Alamut (Version 2.2, Interactive Biosoftware, LLC, France), variants were evaluated in terms of clinical relevance (step 4) and categorised as “likely pathogenic”, “unknown pathogenicity” or “unlikely pathogenic”. ESP (EA) minor-allele frequency in European American population in NHLBI GO Exome Sequencing Project (ESP) [14]; dbSNP variation global minor allele in the single nucleotide polymorphism database [15], HGMD Human Gene Mutation Database [16], VUS variant of unknown significance
The fragment lengths of the MLPA investigations were analysed with GeneMapper v. 4.0 (Thermo Fisher, USA) and visual inspection of the electropherograms. Probe ratios below 0.7 or above 1.3 were regarded as heterozygous deletions or duplications, respectively.
The software, R v. 2.11.0 [18] was used to perform the statistical analysis of medians, means and standard deviations. The genetic results of the two investigated groups were compared with Fisher’s exact test.
Ethical standards
The study was approved by the Committees on Health Research Ethics in the Capital Region of Denmark (H-2-2012-017), local ethics committee and the Danish Data Protection Agency (2011-54-1262).
Results
Possibly pathogenic variants in the SUD victims and cardiac patients
Among the 44 investigated samples, the mean number of variants per sample that passed the MiSeq Reporter quality filter was 528 (s.d. 124). After selection of candidate variants by the process described in Fig. 1 and Tables 2 and 3, a mean of 40 (s.d. 8) possibly pathogenic variants per sample were further evaluated and classified.
Among the 15 SUD victims, three likely pathogenic variants including one novel, were identified in three (20 %) cases (Table 4). No larger rearrangement (indels) was found. In 12 (41 %) of the 29 patients, 18 likely pathogenic variants including three novel, were identified. Large deletions were found in a Brugada patient (SCN5A deletion in exon 23) and a LQTS patient (KCNQ1 deletion in exon 7, 8 and 9). The coverage in the deleted areas was approximately 50 % of the average coverage. Both deletions were verified with MLPA. No other insertion or deletion of large gene segments was observed. Details of all possibly pathogenic variants are described in Table 4. The difference in the number of observed likely pathogenic variants in the SUD cases and the patients was not statistically significant (p = 0.1).
Variants of unknown significance
In all of the SUD victims and in 26 (90 %) of the patients, variants previously associated with cardiac diseases but with an unknown significance, were identified (Supplementary material online, Table S1). Two previously identified variants (K1459N and A1777T in MYH7) in patients diagnosed with hypertrophic cardiomyopathy (HCM) were found in two of our BS patients. Both variants were categorised as being of unknown significance because of the phenotypic variation between the patients.
Discussion
With NGS, we identified likely pathogenic variants in three of the 15 SUD victims (20 %). In the patients, the corresponding frequency was 41 %. In addition, two larger deletions of whole exons were identified in the patient cohort, increasing the diagnostic rate of the genetic investigation to 48 %. The frequency of identified likely pathogenic variants in the clinically diagnosed patients was >2-fold compared with that of the SUD victims, although not statistically significant (p = 0.1). The difference is most likely due to the reduced possibility of phenotypic characterisation of the forensic victims [19].
Previous genetic studies of SUD cases showed pathogenic variants in 11–17 % of the deceased [20–22] when investigating up to five genes associated with channelopathies (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2). However, Tester et al. [4] found putative pathogenic variants in 26 % of the individuals in the same five genes when investigating a large cohort comprising 173 SUD victims. In the 26 % with a pathogenic variant, the median age was 16 years. The corresponding number in our study was 37 years. Moreover, Tester et al. [4] found a higher frequency of pathogenic variants in the female SUD cases compared to that of the male SUDs (39 and 18 %, respectively). In our study, the males constituted 11 of the 15 SUD cases. Disease-causing variants in patients diagnosed with LQTS, BS and CPVT were previously identified in 19–50 % of the investigated cases [23–27]. None of the studies were performed with NGS and the number of investigated genes was smaller.
Compared to previous studies with similar cohorts, we found higher frequencies of likely pathogenic variants in both deceased and living patients when investigating 34 genes associated with channelopathies. This suggests that the gene panel used in this study is relevant in both clinical and forensic investigations. Due to the wide screening, we found that five of the cardiac patients carried several likely pathogenic variants in two or more genes. This may result in a compound effect, worsening the phenotypic trait of the disease. We also observed an overlap in the established phenotypes and the identified genotypes, specifically two BS diagnosed patients were identified with two variants previously found in patients diagnosed with hypertrophic cardiomyopathy [28, 29]. This could suggest that the diseases are expressed differently among individuals or not related to the described phenotypes and low frequent variants. In both the clinical and forensic settings, many cases have unspecific symptoms or presentation that makes it impossible to establish a clear phenotypic diagnosis. Hence, a wider genetic screening would be preferable in these cases and could contribute to the clinical investigation. Papadakis et al. [30] evaluated family members of victims of either sudden arrhythmic death syndrome (SADS) (n = 163) or with unspecific abnormalities of the heart (n = 41). They found a similar proportion (47 and 51 %, respectively) of channelopathic diseases in the relatives of the two groups. The predominant diagnoses were LQTS and BS in both cohorts. This demonstrates that there is diversity in both phenotypic and genotypic expression and that we should also include genes associated with structural cardiac diseases when investigating arrhythmogenic diseases.
We found a high frequency of variants associated with a cardiac disease with unknown significance. The variants were present in ESP and dbSNP in higher frequencies than the expected phenotype prevalence or present in several individuals in our study cohort. Similarly, Andreasen et al. [31] recently investigated the frequency of previously cardiomyopathy-associated variants in population-based exome data. They found a significant overrepresentation of variants previously thought to be pathogenic in the control population which indicated that the genetic variants may not be disease-causing but may have a modifying effect or may even be benign.
To establish the real pathogenicity of the described variants in this study, family co-segregation studies and functional testing should be performed. Non-coding and regulatory regions were not analysed unless they had previously been found to be associated with a cardiac disease or symptom. A large amount of genetic information that may be of importance has not yet been published. For future work, focus ought to be extended to the intronic and UTR regions in order to fully understand the genetics behind the diseases causing SCD. Also, epigenetic factors may have a possible influence on the pathogenesis and the medical relevance of epigenetic regulation remains largely unexplored in cardiac pathology [32].
This study shows that NGS is a valuable tool in a postmortem forensic setting with enhanced results to previous first-generation sequencing studies. Additionally, it is possible to use NGS to detect large deletions of entire exons. As described above, we found inconsistencies between phenotypes and genotypes, suggesting that there is an overlap among the channelopathic and cardiomyopathic diseases. For future studies, wide genetic screening of both channelopathic and cardiomyopatic associated genes should be performed in suspected SCD victims to fully understand the phenotype-genotype correlation.
References
Winkel BG, Holst AG, Theilade J, Kristensen IB, Thomsen JL, Ottesen GL, Bundgaard H, Svendsen JH, Haunsø S, Tfelt-Hansen J (2011) Nationwide study of sudden cardiac death in persons aged 1–35 years. Eur Heart J 32(8):983–990. doi:10.1093/eurheartj/ehq428
Wisten A, Forsberg H, Krantz P, Messner T (2002) Sudden cardiac death in 15–35-year olds in Sweden during 1992–99. J Intern Med 252(6):529–536
Puranik R, Chow CK, Duflou JA, Kilborn MJ, McGuire MA (2005) Sudden death in the young. Heart Rhythm 2(12):1277–1282. doi:10.1016/j.hrthm.2005.09.008
Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ (2012) Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc 87(6):524–539. doi:10.1016/j.mayocp.2012.02.017
Tester DJ, Ackerman MJ (2009) Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Annu Rev Med 60:69–84. doi:10.1146/annurev.med.60.052907.103838
Rodríguez-Calvo MS, Ma B, Allegue C, Concheiro L, Carracedo A (2008) Molecular genetics of sudden cardiac death. Forensic Sci Int 182(1–3):1–12. doi:10.1016/j.forsciint.2008.09.013
Hedley PL, Jørgensen P, Schlamowitz S, Moolman‐Smook J, Kanters JK, Corfield VA, Christiansen M (2009) The genetic basis of Brugada syndrome: a mutation update. Hum Mutat 30(9):1256–1266. doi:10.1002/humu.21066
Hedley PL, Jorgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, Kanters JK, Corfield VA, Christiansen M (2009) The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat 30(11):1486–1511. doi:10.1002/humu.21106
Online Mendelian Inheritance in Man, OMIM. McKusick-Nathans Institute of Genetic Medicine, John Hopkins University (Baltimore, MD)
MRC-Holland. http://mlpa.com/WebForms/WebFormMain.aspx?Tag=_fNPBLedDVp38p-CxU2h0mQ
Kent WJSC, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at USCS. Genome Res 12(6):996–1006
Li HDR (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2002) From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit Best Practices Pipeline. In: Current Protocols in Bioinformatics. Wiley
Exome Variant Server. http://evs.gs.washington.edu/EVS/.
National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/.
Stenson PDBE, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Copper DN (2003) The Human Gene Mutation Database (HGMD). Hum Mutat 21:577–581
Spaendonck-Zwarts KY, Rijsingen IAW, Berg MP, Deprez RHL, Post JG, Mil AM, Asselbergs FW, Christiaans I, Langen IM, Wilde AAM, Boer RA, Jongbloed JDH, Pinto YM, Tintelen JP (2013) Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience. Eur J Heart Fail 15(6):628–636. doi:10.1093/eurjhf/hft013
Team RC (2014) R: A language and environment for statistical computing. Available via R Foundation for Statistical Computing. http://www.R-project.org
Hertz CL F-ML, Frank-Hansen R, Morling N, Bundgaard H (2014) A comparison of genetic findings in sudden cardiac death victims and cardiac patients: the importance of phenotypic classification. Europace In press. doi:10.1093/europace/euu210
Chugh SS, Senashova O, Watts A, Tran PT, Zhou Z, Gong Q, Titus JL, Hayflick SJ (2004) Postmortem molecular screening in unexplained sudden death. J Am Coll Cardiol 43(9):1625–1629. doi:10.1016/j.jacc.2003.11.052
Skinner JR, Crawford J, Smith W, Aitken A, Heaven D, Evans C-A, Hayes I, Neas KR, Stables S, Koelmeyer T, Denmark L, Vuletic J, Maxwell F, White K, Yang T, Roden DM, Leren TP, Shelling A, Love DR (2011) Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm 8(3):412–419. doi:10.1016/j.hrthm.2010.11.016
Winkel BG, Larsen MK, Berge KE, Leren TP, Nissen PH, Olesen MS, Hollegaard MV, Jespersen T, Yuan L, Nielsen N, Haunsø S, Svendsen JH, Wang Y, Kristensen IB, Jensen HK, Tfelt-Hansen J, Banner J (2012) The prevalence of mutations in KCNQ1, KCNH2, and SCN5A in an unselected national cohort of young sudden unexplained death cases. J Cardiovasc Electrophysiol 23(10):1092–1098. doi:10.1111/j.1540-8167.2012.02371.x
Tester DJ, Will ML, Haglund CM, Ackerman MJ (2005) Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2(5):507–517. doi:10.1016/j.hrthm.2005.01.020
Berge KE, Haugaa KH, Früh A, Anfinsen OG, Gjesdal K, Siem G, Øyen N, Greve G, Carlsson A, Rognum TO, Hallerud M, Kongsgård E, Amlie JP, Leren TP (2008) Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand J Clin Lab Investig 68(5):362–368. doi:10.1080/00365510701765643
Nakajima T, Kaneko Y, Saito A, Irie T, Tange S, Iso T, Kurabayashi M (2011) Identification of six novel SCN5A mutations in Japanese patients with Brugada syndrome. Int Heart J 52(1):27–31
Liang P, Liu W, Li C, Tao W, Li L, Hu D (2010) Genetic analysis of Brugada syndrome and congenital long-QT syndrome type 3 in the Chinese. J Cardiovasc Dis Res 1(2):69–74. doi:10.4103/0975-3583.64437
Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FES, Vignati G, Benatar A, DeLogu A (2002) Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106(1):69–74. doi:10.1161/01.CIR.0000020013.73106.D8
Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ (2004) Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 44(3):602–610. doi:10.1016/j.jacc.2004.04.039
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet J-P, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M, for the EHFP (2003) Hypertrophic cardiomyopathy. Circulation 107(17):2227–2232. doi:10.1161/01.CIR.0000066323.15244.54
Papadakis M, Raju H, Behr ER, Noronha SVD, Spath N, Kouloubinis A, Sheppard MN, Sharma S (2013) Sudden cardiac death with autopsy findings of uncertain significance potential for erroneous interpretation. Circ Arrhythm Electrophysiol 6(3):588–596. doi:10.1161/CIRCEP.113.000111
Andreasen C, Refsgaard L, Nielsen JB, Sajadieh A, Winkel BG, Tfelt-Hansen J, Haunso S, Holst AG, Svendsen JH, Olesen MS (2013) Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can J Cardiol 29(9):1104–1109. doi:10.1016/j.cjca.2012.12.002
Ordovas JM, Smith CE (2010) Epigenetics and cardiovascular disease. Nat Rev Cardiol 7(9):510–519. doi:10.1038/nrcardio.2010.104
Acknowledgments
This work was supported by Ellen and Aage Andersen’s foundation. The authors thank Francisc-Raul Kantor for the bioinformatics support.
Conflict of interest
The authors declare that they have no conflict of interest. AGH is an employee of Novo Nordisk A/S, Denmark.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
(DOCX 19 kb)
Rights and permissions
About this article

Cite this article
Hertz, C.L., Christiansen, S.L., Ferrero-Miliani, L. et al. Next-generation sequencing of 34 genes in sudden unexplained death victims in forensics and in patients with channelopathic cardiac diseases. Int J Legal Med 129, 793–800 (2015). https://doi.org/10.1007/s00414-014-1105-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-014-1105-y