
Abstract
Annually, thousands of sudden deaths in individuals under 35 years remain unexplained following comprehensive medico-legal autopsy. Previously, post-mortem genetic analysis by Sanger sequencing of four major cardiac channelopathy genes revealed that approximately one-fourth of these autopsy-negative sudden unexplained death in the young (SUDY) cases harbored an underlying mutation. However, there are now over 100 sudden death-predisposing cardiac channelopathy-, cardiomyopathy-, and metabolic disorder-susceptibility genes. Here, we set out to determine whether post-mortem whole exome sequencing (WES) is an efficient strategy to detect ultra-rare, potentially pathogenic variants. We performed post-mortem WES and gene-specific analysis of 117 sudden death-susceptibility genes for 14 consecutively referred Caucasian SUDY victims (average age at death 17.4 ± 8.6 years) to identify putative SUDY-associated mutations. On average, each SUDY case had 12,758 ± 2,016 non-synonymous variants, of which 79 ± 15 localized to these 117 genes. Overall, eight ultra-rare variants (seven missense, one in-frame insertion) absent in three publically available exome databases were identified in six genes (three in TTN, and one each in CACNA1C, JPH2, MYH7, VCL, RYR2) in seven of 14 cases (50 %). Of the seven missense alterations, two (T171M-CACNA1C, I22160T-TTN) were predicted damaging by three independent in silico tools. Although WES and gene-specific surveillance is an efficient means to detect rare genetic variants that might underlie the pathogenic cause of death, accurate interpretation of each variant is challenging. Great restraint and caution must be exercised otherwise families may be informed prematurely and incorrectly that the root cause has been found.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Annually, 300,000–400,000 individuals die suddenly in the United States, with the majority involving the elderly and coronary artery disease [34]. Sudden death in the young is relatively uncommon, with an incidence of 1.3–8.5 per 100,000 patient-years [13, 22]. Yet, tragically each year, 1,000–5,000 otherwise healthy individuals aged 1–35 years die suddenly. The cause is often identifiable during comprehensive medico-legal investigation, including autopsy, and attributed to structural cardiovascular abnormalities [7, 26]. However, up to half of these cases remain unexplained [24, 30, 33] and are termed autopsy-negative sudden unexplained death in the young (SUDY).
Long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS) are potentially lethal, heritable cardiac channelopathies associated with syncope, seizures, and sudden cardiac arrest in the setting of a structurally normal heart and may account for a significant number of SUDY. Additionally, heritable cardiomyopathies, including hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and arrhythmogenic cardiomyopathy (ACM) can display minimal structural abnormalities deemed inconclusive, or completely missed, and may underlie a significant portion of SUDY.
Post-mortem genetic investigation may elucidate the pathogenic basis for SUDY. Post-mortem genetic analysis of the four most common channelopathy-associated genes (KCNQ1 [LQT1], KCNH2 [LQT2], SCN5A [LQT3, BrS1], and RYR2 [CPVT]) have implicated LQTS, CPVT, and BrS as a pathogenic basis for approximately 25–30 % of SUDY [32].
Because accurate diagnosis from molecular analysis of an SUDY victim may be crucial to surviving family members who may also be genetically susceptible to life-threatening arrhythmia syndromes, recent guidelines for autopsy investigations of SUDY suggest that post-mortem genetic testing should become the new standard of care in evaluation of SUDY cases [2, 3, 8, 29]. However, with over 100 sudden death-susceptibility genes, the traditional “one gene, one exon at a time” Sanger sequencing approach to post-mortem genetic testing is often too time-consuming and cost-prohibitive for the medical examiner/coroner/forensic pathologist community to provide this level of care given the financial landscape and unwillingness of major insurance companies to provide coverage/reimbursement for post-mortem genetic testing.
Next-generation whole exome sequencing (WES), allowing for the simultaneous genetic analysis of an individual’s entire library of ~20,000 genes, is an attractive, cost-effective ($1,000–$2,000 per sample), and time-conducive (few weeks) alternative technique for a comprehensive post-mortem genomic study [19]. In fact, we recently provided the first ever proof-of-principle case report of a WES-based comprehensive molecular autopsy of a previously healthy 16-year-old SUDY victim [19]. Subsequently, Bagnall and colleagues completed a WES-based post-mortem genetic analysis in their cohort of sudden death cases [6].
Herein, using a cohort of 14 consecutively referred, unrelated autopsy-negative SUDY victims, we provide a replication study that illustrates the potential benefits as well as inherent complexity and daunting task of variant interpretation when performing a WES-based molecular autopsy in SUDY.
Materials and Methods
Medical Examiner-Referred Autopsy-Negative SUDY Cases
From May 2011 to February 2013, 14 consecutive, unrelated autopsy-negative SUDY cases (eight males, mean age 17.4 ± 8.6 years, range 1.3–29 years) were referred to Mayo Clinic’s Windland Smith Rice Sudden Death Genomics Laboratory for research-based genetic testing. To be included, (a) the death had to have occurred between the ages of 1–35 years, (b) the autopsy had to be absent of any findings deemed causative of death, and (c) there was no ante-mortem diagnosis of any cardiac channelopathy (BrS, CPVT, LQTS) or cardiomyopathy (HCM, DCM, ACM) in the victim or any relative. Mayo Clinic Institutional Review Board-approved protocol for molecular autopsy was performed following informed written consent from the decedent’s next-of-kin.
Whole Exome Next-Generation DNA Sequencing
Three micrograms (µg) of genomic DNA isolated from 10 mL of autopsy blood using the Gentra Puregene Blood Kit (Qiagen, Germantown, MD) following the manufacturer’s protocol, was submitted to Mayo Clinic’s Medical Genome Facility (Rochester, MN), supported by the Mayo Center for Individualized Medicine for WES of all 14 SUDY victims. Following exome capture with the SureSelect XT Human All Exon V4 plus UTR Target Enrichment System (Agilent, Santa Clara, CA), 71-MB paired-end sequencing at 96 % coverage with a read depth of 35× was carried out on the Illumina HiSeq 2000 platform using V3 reagents. Variant alignment to the latest available human genome (hg19), mapping and assembly with quality (Maq) single nucleotide variant (SNV) detection [21], Burrows–Wheeler Alignment insertion/deletion (INDEL) detection [20], Maq and Genome Analysis Toolkit-based SNV/INDEL calling, SeattleSeq/Sorting Intolerant from Tolerant (SIFT) annotation, and allele frequencies for variants in the Single Nucleotide Polymorphism database (dbSNP) and 1,000 genomes was carried out using the automated Targeted RE-sequencing Annotation Tool (TREAT) analytical pipeline developed at Mayo Clinic (Rochester, MN) [5].
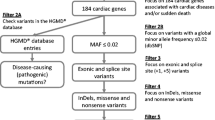
An annotated list of all SNVs/INDELs that met quality control standards was provided in an Excel (Microsoft, Redmond, WA) spreadsheet with links for variant visualization, tissue expression, and biologic pathway/process. Following WES and variant annotation, variant filtration involving the exclusion of all non-coding regions and synonymous variants (i.e., DNA nucleotide alteration amino acid sequence of the protein) and gene-specific analysis of the 117 channelopathy-(LQTS, CPVT, and BrS), cardiomyopathy-(HCM, DCM, and ACM), and metabolic disorder-susceptibility genes was performed to identify possible pathogenic mutation(s).
To be considered a possible pathogenic mutation responsible for sudden death, any variant discovered had to be absent in three publicly available exome databases including the 1,000 Genome Project (n = 1,094 subjects; 381 Caucasian, 246 African-American, 286 Asians, and 181 Hispanics) [11], the National Heart, Lung and Blood Institute Grand Opportunity (NHLBI GO) Exome Sequencing Project (n = 6,503 subjects; 4,300 Caucasians and 2,203 African-Americans) [35], and the Exome Chip Design (n = 12,000 subjects) [1].
All possible pathogenic mutations were confirmed in the SUDY case’s genomic DNA using standard polymerase chain reaction (PCR) and Sanger DNA sequencing methods. PCR primers, conditions, and sequencing methods are available upon request.
Results
Cohort Description
Demographic characteristics for our cohort are shown in Table 1. The cohort contained 14 consecutively referred, unrelated autopsy-negative SUDY individuals (100 % Caucasian, mean age 17.4 ± 8.6 years, range 1.3–29 years). There were eight males (average age 18.2 ± 8.5 years, range 1.5–29 years) and six females (average age 16.4 ± 9.4 years, range 1.3–27 years). Event at time of death was sleep in 9 of 14 (64.3 %), non-specific in 3 (21.4 %), and unknown in two. Exact time of death is known for 6 (42.9 %), with the majority of these deaths occurring in the morning (4/6, 66.7 %). There was no contributory past medical history in 11 of 13 (84.6 %), and unknown in one. In the remaining two, past medical history was notable for unexplained pulmonary embolism 7 months prior to SUD, previous cardiac arrest, and prior syncopal episode in one individual (case 3), and an episode of diaphoresis and hypotension 17 months prior to SUD in another (case 10). At that time, troponin was elevated, and the individual underwent cardiac catheterization, which demonstrated questionable apical hypokinesis. Follow-up echocardiogram revealed normal ejection fraction (EF) of 65 %, some concentric left ventricular hypertrophy, and normal estimated right ventricular systolic pressure (RSVP) of 25–30 mmHg. Eight months later, follow-up echocardiogram showed EF 50–55 % and elevated RSVP of 35–40 mmHg. The individual was noted subsequently to have hypertension and sleep apnea.
Three individuals (21.4 %) had a known family history of cardiac abnormalities: SUD occurred in the mother of one (case 1) 5 years preceding that of the victim, with cause of death including a large mural thrombus involving the right ventricle, resulting in a fatal cardiac dysrhythmia; cardiomegaly, dilated cardiomyopathy, and myocarditis were additional diagnoses noted on autopsy in this relative, thought to be secondary to systemic lupus erythematosus. Family history of one (case 2) was significant for cardiac arrhythmias. In another (case 6), family history was significant for myocardial infarction at young age in the father and grandfather, and several cases of sudden infant death syndrome (SIDS) on the maternal side.
Prevalence of Ultra-Rare Non-Synonymous Possibly Pathogenic Mutations
Following WES, an average of 77,836,271 total reads was produced with an average of 49,155,829 (63 %) reads mapped to the exome-targeted region per sample. The overall average gene level coverage at ≥10 reads (×10) was 94.3 ± 1.8 %, and for the 117 targeted genes, the average coverage at 10× was 93.4 ± 8.6 %. For the most common channelopathy and cardiomyopathy genes, the average coverage at 10× was 88.6 ± 3.0 % for KCNQ1, 93.9 ± 1.8 % for KCNH2, 97.4 ± 2.1 % for SCN5A, 97.2 ± 1.7 % for RYR2, 90.6 ± 1.9 % for MYH7, and 94.6 ± 1.9 % for MYBPC3.
On average, each SUDY case had 12,758 ± 2,016 non-synonymous single nucleotide variants (12,048 ± 1,914 missense mutations, 58 ± 10 splice site mutations, 95 ± 27 nonsense mutations) and coding region insertions/deletions (219 ± 30 frame shift mutations, 290 ± 39 in-frame mutations, 47 ± 4 splice site mutations). Of these variants, 79 ± 15 localized to the 117 surveyed genes (Table 2).
Eight ultra-rare possibly pathogenic mutations (seven missense, one in-frame insertion) absent in three publically available exome databases were detected in six genes (D4301 N-TTN, I22160T-TTN, 9928_9929insE-TTN, T171M-CACNA1C, A1744S-MYH7, A189T-JPH2, S434Y-VCL, H4552R-RYR2) in 7 of 14 victims (50 %, Table 3). Only two of seven missense mutations (I22160T-TTN and T171M-CACNA1C) were predicted to be damaging by at least three of four in silico prediction tools (Polyphen2 [25], SIFT [15], Provean [23], Mutation Assessor [9]), and four missense mutations (D4301 N-TTN, A1744S-MYH7, A189T-JPH2, and H4552R-RYR2) were predicted to be either benign, tolerated, low or neutral by all four in silico prediction tools (Table 4).
Despite the absence of gross or microscopic findings at time of autopsy, 6 of 14 cases (42.9 %) hosted rare variants in cardiomyopathy-associated genes (three with TTN, one with JPH2, one with MHY7, one with VCL mutation). Only 1 of 14 (7.1 %) had a mutation (H4552R-RYR2) in one of the four most common cardiac channelopathy genes. One individual (case 10) had two possibly pathogenic mutations (T171M-CACNA1C, A1744S-MYH7).
Discussion
Sudden cardiac death can be the sentinel event in young, otherwise healthy individuals and may represent the initial means of uncovering a familial sudden death-predisposing disorder. In the case of a negative autopsy, post-mortem genetic testing may reveal an underlying responsible genetic substrate, such as non-synonymous mutations within cardiac channelopathy and cardiomyopathy genes [32]. This information may be vitally important in identification and prophylactic treatment of surviving relatives genetically susceptible to this tragic fate [12]. Additionally, it provides for important bio-epidemiological information enabling an accurate determination of cause and manner of death.
Comprehensive post-mortem genetic testing (or “molecular autopsy”) is becoming part of the standard of care in these cases and has been addressed extensively [2, 3, 8, 27], with WES being especially relevant, as over 100 cardiac channelopathy-, cardiomyopathy-, and metabolic disorder-susceptibility genes associated with SUDY have been discovered. In fact, we recently provided the first ever proof-of-principle case report of a WES-based comprehensive molecular autopsy of an otherwise healthy 16-year-old SUDY victim, where we identified a pathogenic MYH7 mutation, previously described with familial HCM, sudden death, and impaired MHC-β actin-translocating and actin-activated ATPase activity [19]. This case illustrated the potential efficiency and cost-effectiveness of WES in the comprehensive genetic evaluation of a SUDY victim with mutation identification and subsequent genetic interrogation of surviving family members also at risk for HCM and possible sudden death.
WES allows for rapid genetic analysis of an individual’s complete complement genes at a relatively low cost, using a small amount of DNA. This makes it an appealing approach for post-mortem genetic analysis of SUDY, where funding and source DNA is often limited, but timely identification of the responsible pathogenic substrate can bring much needed closure to the family, and perhaps more importantly, identify others at risk. Unfortunately, as illustrated in our current study and that recently by Bagnall et al. [6], the WES-based approach may not be ready for prime time in the post-mortem setting.
While the comprehensive nature of WES may be beneficial, it creates the daunting task of scrutinizing thousands of non-synonymous genetic variants for each exome, many of which may be rare, predicted in silico to be deleterious, and reside within biologically plausible genes. In our SUDY cohort, over 12,000 non-synonymous variants were, on average, detected in each individual, of which approximately 80 on average localized to 117 surveyed sudden death-susceptibility genes. Additionally, tens to hundreds of other rare, non-synonymous variants may be identified in genes whose encoded protein products have not yet been established as disease causing but have biological plausibility for contribution to a sudden death-associated phenotype. Variant interpretation must therefore be performed carefully, given the tremendous psychological consequences of potential misdiagnosis. In fact, the recent American College of Medical Genetics (ACMG) policy warns that “it is critical that the standards for what is reportable be high to avoid burdening the health-care system and consumers with what could be very large numbers of false-positive results” [4]. This is especially important in regards to incidental findings that may not be consistent with the individual’s disease phenotype and is particularly difficult in autopsy-negative SUDY, where there are no evidentiary clues to guide genetic testing.
Half of our SUDY cases contained at least one ultra-rare variant among 117 sudden death-associated genes, with nearly 43 % of cases having mutations in cardiomyopathy-associated genes, despite having an autopsy without overt structural pathology. Importantly, however, an ultra-rare variant does not always equal a pathogenic one. This concept of a rare variant being just that “just there, just rare, just because” must be considered critically, and distinguishing a rare, innocuous variant from a truly pathogenic mutation that may be responsible for the overall phenotype is vitally important [18, 31]. Whether or not the JPH2, MYH7, TTN, and VCL, rare variants discovered in this cohort, are pathogenic or simply exceedingly rare but nevertheless non-disease causing requires extensive functional studies. Given the high degree of background genetic noise in TTN-encoded titin and the lack of structural pathology identified at autopsy, we suspect that the ultra-rare non-synonymous TTN missense variants identified are non-contributory to the SUD.
Among the four most common cardiac channelopathy-associated genes previously identified as the pathogenic basis for approximately 15–35 % of SUDY [16, 28, 32], only a single case (7 % of our cohort) had a mutation (H4552R-RYR2) in one of these genes. Similarly, Bagnall et al. [6] in their recent post-mortem, WES-based post-mortem genetic analysis, identified a rare genetic variant in the four major channelopathy genes in 7 % of their cases. In our previous, denaturing high-performance liquid chromatography and Sanger sequencing-based post-mortem genetic analysis of 173 unrelated autopsy-negative SUDY cases, we demonstrated that approximately one-fourth (13 % in either KCNQ1, KCNH2, or SCN5A and 12 % in RYR2) harbored mutations in the major cardiac channelopathy genes [32]. This raises the possibility that our sentinel molecular autopsy studies may have been influenced unwittingly by a referral bias whereby medical examiner’s had elected to send their unsolved SUDY cases that struck them as “channelopathic” because of the circumstances, triggers, setting, and so forth.
Two cases had subtle histopathologic alterations, including fibrosis and hypertrophy (case 8), and intramural coronary artery changes (case 9). While these changes were not significant enough per the referring medical examiner to be diagnostic of cardiomyopathy, they are clearly abnormal. Gross and histologic changes in nascent cardiomyopathies are not well described, which is particularly problematic in a young cohort. Large-scale studies involving comprehensive genotype–phenotype correlation will be imperative in ascribing more definitive significance to these often subtle findings [10].
One SUDY victim (case 10) was unresponsive behind the steering wheel of a car and experienced cardiac arrest in the Emergency Department. This individual had the mutation in one of the channelopathy-associated genes (T171M-CACNA1C), predicted to be deleterious by three independent in silico tools. Given the absence of family history, this may represent a de novo mutation responsible for SUD. However, without parental DNA, this suspicion cannot be confirmed. Conversely, no putatively pathogenic mutations were discovered in three cases (cases 2, 3, 6) that contained suggestive personal or family history. This could suggest the involvement of a novel disease gene/mechanism responsible for the SUD or a potential mutation detection failure by WES in these three cases as WES is not as sensitive as Sanger sequencing.
While demographic differences between cohorts can, in part, explain discrepancies in mutation detection yield, there is a possibility with WES, that exon coverage may not be optimal for each gene analyzed, leading to false-negative results. Bagnall and colleagues [6] have highlighted the potential short-comings of the current generation of WES by performing gene-targeted coverage analysis indicating deficiencies in both KCNQ1 and KCNH2 exome coverages, where nearly 25 % of KCNH2 had inadequate sequencing coverage. This suggests the potential for mutation detection failure in these two genes. In fact, the coverage of the exome capture technology, the sequencing quality, and read mapping all contribute to the sensitivity of detecting mutations [36]. Whether potential WES coverage issues have resulted in mutation detection misses in our cohort is unknown.
In contrast, false-positive variants (i.e., sequencing artifact) as a result of library construction biases, errant polymerase reactions, difficulty in short sequence read mapping, and misalignment with a genomic reference sequence can be produced during WES [14]. As such, it is extremely important to validate any putative mutation identified by WES using standard Sanger sequencing protocols, and this should be done regardless of the WES variant quality score and/or read depth. In our study, all variants reported have been Sanger sequence validated.
WES is a promising time- and cost-effective technique for discovering the genetic basis of SUDY. However, limitations of WES for mutation discovery and the heavy burden of genetic variant interpretation must be recognized. Given the complexities of inheritance patterns, expressivity, penetrance, and variability of phenotypes in channelopathies and cardiomyopathies, strong collaboration between multiple experts, including cardiovascular specialists, geneticists, and genetic counselors is paramount [17, 31]. Perhaps, the only thing worse than being unable to tell a grieving family what caused their loved one’s sudden death is to prematurely and incorrectly tell them that the genetic root cause has been found.
References
Abecasis G, Neale B (2011) Exome Chip Design. Accessed August 2012
Ackerman M (2009) State of postmortem genetic testing known as the cardiac channel molecular autopsy in the forensic evaluation of unexplained sudden cardiac death in the young. Pacing Clin Electrophysiol 32(Suppl 2):S86–S89
Ackerman M, Priori S, Willems S, Berul C, Brugada R, Calkins H, Camm A, Ellinor P, Gollob M, Hamilton R, Hershberger R, Judge D, Le Marec H, McKenna W, Schulze-Bahr E, Semsarian C, Towbin J, Watkins H, Wilde A, Wolpert C, Zipes D (2011) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 8(8):1308–1339
ACMG Board of Directors (2012) Points to consider in the clinical application of genomic sequencing. Genet Med 14 (8):759–761
Asmann YW, Middha S, Hossain A, Baheti S, Li Y, Chai HS, Sun Z, Duffy PH, Hadad AA, Nair A, Liu X, Zhang Y, Klee EW, Kalari KR, Kocher JP (2011) TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics. doi:10.1093/bioinformatics/btr612
Bagnall RD, Das KJ, Duflou J, Semsarian C (2014) Exome analysis-based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm 11(4):655–662. doi:10.1016/j.hrthm.2014.01.017
Basso C, Calabrese F, Corrado D, Thiene G (2001) Postmortem diagnosis in sudden cardiac death victims: macroscopic, microscopic and molecular findings. Cardiovasc Res 50(2):290–300
Basso C, Burke M, Fornes P, Gallagher PJ, de Gouveia RH, Sheppard M, Thiene G, van der Wal A, Association for European Cardiovascular P (2008) Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch 452(1):11–18
Boczek NJ, Best JM, Tester DJ, Giudicessi JR, Middha S, Evans JM, Kamp TJ, Ackerman MJ (2013) Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ Cardiovasc Genet 6(3):279–289
Brugada R, Campuzano O, Brugada P, Brugada J, Hong K (2005 [Updated 2012]) Brugada Syndrome. GeneReviews [Internet]. University of Washington, Seattle
Clarke L, Zheng-Bradley X, Smith R, Kulesha E, Xiao C, Toneva I, Vaughan B, Preuss D, Leinonen R, Shumway M, Sherry S, Flicek P (2012) The 1000 Genomes Project: data management and community access. Nat Methods 9(5):459–462. doi:10.1038/nmeth.1974
Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, Papagiannis J, Feldkamp MD, Rathi SG, Kunic JD, Pedrazzini M, Wieland T, Lichtner P, Beckmann B-M, Clark T, Shaffer C, Benson DW, Kaab S, Meitinger T, Strom TM, Chazin WJ, Schwartz PJ, George AL, Jr (2013) Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127:1009–1017. doi:10.1161/CIRCULATIONAHA.112.001216
Driscoll DJ, Edwards WD (1985) Sudden unexpected death in children and adolescents. J Am Coll Cardiol 5(6 Suppl):118B–121B
Fuentes Fajardo KV, Adams D, Program NCS, Mason CE, Sincan M, Tifft C, Toro C, Boerkoel CF, Gahl W, Markello T (2012) Detecting false-positive signals in exome sequencing. Hum Mutat 33(4):609–613. doi:10.1002/humu.22033
Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ (2011) Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 8(7):1024–1032. doi:10.1016/j.hrthm.2011.02.021
Gladding P, Evans C, Crawford J, Chung S, Vaughan A, Webster D, Neas K, Love D, Rees M, Shelling A, Skinner J (2010) Posthumous diagnosis of long QT syndrome from neonatal screening cards. Heart Rhythm 7(4):481–486
Hershberger RE, Cowan J, Morales A, Siegfried JD (2009) Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail 2(3):253–261
Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ (2009) Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 120(18):1752–1760
Kupershmidt S, Yang IC, Sutherland M, Wells KS, Yang T, Yang P, Balser JR, Roden DM (2002) Cardiac-enriched LIM domain protein fhl2 is required to generate I(Ks) in a heterologous system. Cardiovasc Res 56(1):93–103
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. doi:10.1093/bioinformatics/btp324
Li H, Ruan J, Durbin R (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18(11):1851–1858. doi:10.1101/gr.078212.108
Liberthson RR (1996) Sudden death from cardiac causes in children and young adults. N Engl J Med 334(16):1039–1044
MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, Conrad DF, Lunter G, Zheng H, Ayub Q, DePristo MA, Banks E, Hu M, Handsaker RE, Rosenfeld JA, Fromer M, Jin M, Mu XJ, Khurana E, Ye K, Kay M, Saunders GI, Suner MM, Hunt T, Barnes IH, Amid C, Carvalho-Silva DR, Bignell AH, Snow C, Yngvadottir B, Bumpstead S, Cooper DN, Xue Y, Romero IG, Wang J, Li Y, Gibbs RA, McCarroll SA, Dermitzakis ET, Pritchard JK, Barrett JC, Harrow J, Hurles ME, Gerstein MB, Tyler-Smith C (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science 335(6070):823–828. doi:10.1126/science.1215040
Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO (1996) Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA 276(3):199–204
Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, Lewis KL, Cooper DN, Stenson PD, Mullikin JC, Biesecker LG (2013) Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet. doi:10.1161/CIRCGENETICS.113.000039
Puranik R, Chow C, Duflou J, Kilborn M, McGuire M (2005) Sudden death in the young. Heart Rhythm 2(12):1277–1282
Skinner JR, Duflou JA, Semsarian C (2008) Reducing sudden death in young people in Australia and New Zealand: the TRAGADY initiative. Med J Aust 189(10):539–540
Skinner J, Crawford J, Smith W, Aitken A, Heaven D, Evans C, Hayes I, Neas K, Stables S, Koelmeyer T, Denmark L, Vuletic J, Maxwell F, White K, Yang T, Roden D, Leren T, Shelling A, Love D (2010) Prospective, population-based long QT molecular autopsy study of post-mortem negative sudden death in 1–40 year olds. Heart Rhythm. doi:10.1016/j.hrthm.2010.11.016
Tester DJ, Ackerman MJ (2006) The role of molecular autopsy in unexplained sudden cardiac death. Curr Opin Cardiol 21(3):166–172
Tester DJ, Ackerman MJ (2009) Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Annu Rev Med 60:69–84
Tester DJ, Ackerman MJ (2011) Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation 123(9):1021–1037
Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ (2012) Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc 87(6):524–539
Tsuji K, Akao M, Ishii TM, Ohno S, Makiyama T, Takenaka K, Doi T, Haruna Y, Yoshida H, Nakashima T, Kita T, Horie M (2007) Mechanistic basis for the pathogenesis of long QT syndrome associated with a common splicing mutation in KCNQ1 gene. J Mol Cell Cardiol 42(3):662–669. doi:10.1016/j.yjmcc.2006.12.015
Virmani R, Burke A, Farb A (2001) Sudden cardiac death. Cardiovasc Pathol 10(6):275–282
Exome Variant Server NESPE, Seattle, WA. http://evs.gs.washington.edu/EVS/
Zhi D, Chen R (2012) Statistical guidance for experimental design and data analysis of mutation detection in rare monogenic mendelian diseases by exome sequencing. PLoS One 7(2):e31358. doi:10.1371/journal.pone.0031358
Acknowledgments
This work was supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program, the Sheikh Zayed Saif Mohammed Al Nahyan Fund in Pediatric Cardiology Research, the Dr. Scholl Fund, and the Hannah M. Wernke Memorial Fund. This Project was also supported in part by funding from Mayo Clinic’s Center for Individualized Medicine (CIM).
Conflict of interest
DJT, MJA, and Mayo Clinic receive royalties from Transgenomic for their FAMILION-LQTS and FAMILION-CPVT genetic tests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nupoor Narula and David J. Tester are co-equal first authors.
Rights and permissions
About this article

Cite this article
Narula, N., Tester, D.J., Paulmichl, A. et al. Post-mortem Whole Exome Sequencing with Gene-Specific Analysis for Autopsy-Negative Sudden Unexplained Death in the Young: A Case Series. Pediatr Cardiol 36, 768–778 (2015). https://doi.org/10.1007/s00246-014-1082-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-014-1082-4