
Abstract
The degradation and recycling of waste epoxy resins is an urgent environmental problem, encouraging the use of degradable thermosetting epoxies. In this study, a high-performance thermosetting epoxy resin material that can be easily degraded and recycled was prepared using a low-viscosity and high-activity epoxy monomer, tetrahydrophthalic acid diglycidyl ester. Owing to the breakable ester bond in this epoxy monomer, the thermosetting three-dimensional epoxy cross-linked structure can be rapidly degraded using ethylene glycol at atmospheric pressure. After further depolymerization of the epoxy resin/glycol solution with NaOH, sodium cyclohexene-2-carboxylate was obtained. The sodium salt was acidified, epoxidized, and then re-prepared to obtain the epoxy monomer diglycidyl tetrahydrophthalate. The recycled epoxy monomer possesses the same thermal and mechanical properties as the original epoxy monomer, thus realizing the economic and environmentally friendly degradation and recycling of the thermosetting epoxy resin under mild conditions, and this recycling method is applicable to epoxy systems with ester bonding in the cured material.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Thermosetting epoxy resins are widely used in various fields owing to their high strength, corrosion resistance, and good adhesion [1,2,3,4,5,6,7]. However, owing to their dense three-dimensional cross-linked structure, they are difficult to recycle and reuse [8,9,10,11,12,13], and their waste causes serious environmental and resource problems [14,15,16,17].
Recycling thermosetting epoxy resins is a common problem worldwide, making it a hot topic in current research. There are two main strategies for recycling epoxy resin: one is to synthesize a special thermosetting polymer through vitrification, where the resin is introduced into the dynamic covalent bonds, such as disulfides [18], esters [19], acetal [20], Schiff bases [21,22,23], Diels–Alder addition structures [24, 25], and hemiacetal/hemiketal ester bonds [26]. Under external stimuli (e.g., heat, light, and pH), the dynamic network structure in epoxy can be cleaved or attacked by other compounds, which can change the bond arrangement and topology, resulting in the degradation of thermosetting epoxy resins. Si et al. [27] proposed a high-performance epoxy vitrification using aromatic disulfide bond cross-linking, which could be degraded by environmentally harmful thiols. Pan et al. [28] established a dynamic acetal–epoxy resin system degraded by acid hydrolysis. Li et al. [29] investigated a kinetic system based on imine bonding that decomposed into small molecules in strongly acidic solvents. However, this recycling route cannot cope with the current situation of recycling waste commercial thermosetting resins.
Another route is to utilize the degradable bonds in monomers to achieve degradation recycling. Glycidyl ester epoxy resins can be degraded and recycled owing to their unique ester bond structure. Moreover, glycidyl ester epoxy resin is high performing owing to its excellent mechanical properties, heat resistance, and electrical insulation; it possesses low viscosity, high activity, low temperature resistance, and is usually used as a coating, diluent, and large-scale vacuum-infusion device [30]. Current research on the recycling of thermosetting epoxy resins includes mechanical, physical, and chemical methods, where chemical recycling is relatively effective [31, 32]. For example, Ahrens et al. [33] studied the recycling of thermosetting epoxy used for wind turbine blades and recovered bisphenol A and fiber from it. Knappich et al. [34] found that supercritical water can degrade epoxy resins, but temperatures above 400 °C and pressures above 600 bar are required, resulting in high energy consumption. Liu et al. [35] studied the methanol degradation of an anhydride-cured epoxy under subcritical conditions in the presence of potassium hydroxide. Xing et al. [36] proposed a mild and effective method for the decomposition of brominated Epoxy using subcritical acetic acid. Lu et al. [37] degraded anhydride-cured epoxy in a hot solution of ethylene glycol for up to 72 h. Liu et al. [38] used an aqueous solution of environmentally friendly phosphotungstic acid as a catalyst to selectively cleave ester bonds in the cross-linked structure.
However, epoxy polymers obtained by the chemical degradation and recycling of thermosetting epoxy resins are often of low molecular weight; obtaining epoxy monomers is considerably difficult (Zhao et al. [39]). Dattilo et al. [40] found that the thermosetting resin EP can be completely degraded in the H2O2/phosphotungstic acid system at 80 ℃ over 4 h, and the macromolecular product can be used for the synthesis of supramolecular adhesive raw materials. Zhao et al. [41] degraded thermosetting epoxy in aqueous acetic acid solution at 80 ℃ and used the product to synthesize polyurethane. They added the degradates to the original epoxy resin as fillers, and the low additive amount of degradates had little effect on the thermal and mechanical properties of the resin; however, the method did not have a high degradation rate.
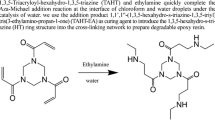
Owing to the difficulties in the utilization of epoxy oligomers with a low polymerization degree, in this study, we chose epoxy resin curing compositions containing ester-bonded epoxy resin monomers and amine curing agents. First, we degraded them with ethylene glycol at atmospheric pressure and then separated and recovered the epoxy degradation solution (DEP) to obtain epoxy resin monomers with the same structure. The effects of reaction temperature, reaction time, ratio of DEP to NaOH, and NaOH concentration on the purity of the product in the alkaline decomposition process of the epoxy degradation solution were explored using an orthogonal experimental design. The optimal process conditions for the preparation of sodium cyclohexene-2-carboxylate (R-THA-Na) were obtained, and the sodium salt was prepared through acidification and epoxidation treatment to obtain epoxy monomers having the same chemical structure as the original epoxy monomers, as shown in Scheme 1. The performance of the recycled epoxy resins was comprehensively assessed, and a new method was proposed for the separation and reuse of thermosetting epoxy resins with the presence of ester-bonded structures.

Flow chart of preparation and recycling of Epoxy curing compounds
Materials and methods
Reagents
The following reagents were used: industrial-grade tetrahydrophthalic acid diglycidyl ester (TADE; Sichuan Dongshu New Material Co., Ltd.); industrial-grade isophorone diamine (IPDA; Sichuan Dongshu New Material Co. Ltd.); ethylene glycol (EG; analytical reagent (AR), Chengdu Cologne Chemical Co., Ltd.); sodium hydroxide (NaOH; AR, Shanghai McLean Biochemical Technology Co., Ltd.); concentrated sulfuric acid (AR, Shanghai McLean Biochemical Technology Co., Ltd.); anhydrous ethanol (AR, Chengdu Cologne Chemical Co., Ltd.); sodium cyclohexene-2-carboxylate (THA-Na; AR, Chengdu Cologne Chemical Co., Ltd.); epichlorohydrin (ECH; AR, Chengdu Cologne Chemical Co., Ltd.); and benzyltrimethylammonium chloride (AR, Chengdu Cologne Chemical Co., Ltd.).
Procedure
Preparation of epoxy curing compounds
The epoxy monomer TADE and curing agent IPDA were mixed uniformly and poured into the mold coated with silicone release agent. The mold was placed in a vacuum drying oven, degassed, and then cured for 6 h in a blower oven set to 80 ℃. Afterwards, it was removed and cooled to room temperature (25℃). The epoxy resin-cured material sample was labeled EP and cut into a uniform block with dimensions of 10 mm × 10 mm × 2 mm using a cutting machine.
Two-step degradation of epoxy cures
EG and the uniform sample block with dimensions of 10 mm × 10 mm × 2 mm were added to a three-necked flask in a mass ratio of 1:9 (sample:EG). After purging the device with nitrogen, the temperature was raised to 180 °C for degradation, and then the degraded mixture was distilled under reduced pressure to remove EG. The viscous epoxy degradation product (DEP) remained. The solid epoxy samples were removed from the solution at regular intervals, cleaned, and weighed to monitor the change in residual mass over time. The catalytic degradation rate of the epoxy resin was evaluated by measuring the ratio (Mt /M0) of the residual mass (Mt) to the original solid mass (M0) at time t.
DEP was further degraded with NaOH to obtain a sodium salt. The liquid obtained from the alkaline degradation was passed through a rotary evaporator to remove water, and ethanol was added to precipitate the sodium salt (R-THA-Na). To investigate the effect of NaOH on the separation of DEP, four factors were considered (Table 1): reaction temperature (A), reaction time (B), ratio of DEP to sodium hydroxide (C), and sodium hydroxide concentration (D). Each factor included three levels: A: 60, 80, and 100 °C; B: 2.5, 5, and 10 h; C: 10:2, 10:2.5, and 10:3; D: 5%, 10%, and 20%.
DEP recycling
Acidification of recycled sodium salt
The dried sodium salt was dissolved in water, and a 5 mol/L sulfuric acid solution was added dropwise to adjust the pH of the solution to 2. A colorless precipitate was obtained, which was filtered and dried to obtain the acid (R-THA).
Synthesis of the recycled epoxy
To a three-necked flask equipped with a thermometer, stirring device, and reflux condenser tube, R-THA, ECH, and benzyltrimethylammonium chloride were added in a molar ratio of 1:13:0.015. The temperature was raised to 100 ℃ reaction for 1 h, and then the reaction was cooled to 55 ℃. Then, 20 wt% aqueous NaOH (pH = 7) was added dropwise through the dispensing funnel to separate the upper aqueous layer, and rotary evaporation was used to remove ECH, yielding the recycled epoxy monomer (R-EP).
The R-EP epoxy monomer and IPDA curing agent were mixed evenly, poured into a mold coated with a silicone release agent, and placed in a vacuum drying oven until the bubbles were completely discharged. After degassing, the mold was placed in a blower oven at 80 ℃ for 6 h and then cooled to room temperature and removed. The recycled epoxy-cured sample was obtained and labeled R-EP + IPDA.
Testing and characterization
Proton nuclear magnetic resonance spectroscopy (1H-NMR)
The sodium salt (R-THA-Na), acid (R-THA), and epoxy (R-EP) were characterized using 1H-NMR spectroscopy using a Bruker spectrometer (Switzerland), where R-THA-Na was dissolved in deuterated H2O, R-THA and R-EP were dissolved in deuterated dimethyl sulfoxide (DMSO). Tetramethylsilane was used as an internal standard.
Fourier-transform infrared (FT-IR) spectroscopy
The IR spectra of R-THA-Na and R-THA were obtained using a Nicolet-5700 FT-IR spectrometer (Nicolet, USA). The samples were prepared using the KBr pellet method.
Differential scanning calorimetry (DSC)
A STA 449 C differential scanning calorimeter (NETZSCH) was used to determine the glass transition temperatures of the synthesized products (R-EP + IPDA). The temperature range of the test was 20–250 °C, the ramp rate was 10 °C/min, and the atmosphere was N2.
Gel permeation chromatography (GPC)
The molecular weight and distribution of DEP were characterized using a gel chromatograph (PL-GPC220, PL, UK) calibrated with a polystyrene standard using tetrahydrofuran as the eluent.
Thermal weight loss
The thermal stability of R-EP + IPDA was determined using a STA449C (NETZSCH, Germany) integrated thermal analyzer under a nitrogen atmosphere with a gas flow rate of 30 mL/min over a temperature range of 30–800 °C and temperature increase rate of 10 °C/min.
Mechanical performance test
The mechanical properties of the cured samples (R-EP + IPDA) were tested using an electronic universal testing machine (ETM-104C, Shenzhen Wanjie Testing Company, China). The experiment was conducted according to the GB/T 1447–2005 test standard at a tensile rate of 2 mm/min.
Epoxy value test
The epoxy value of the product was determined using the acetone–hydrochloride method. Epoxy resin m (g) was added to an experimental bottle containing the same volume of HCl–acetone solution (configured according to the 1:40 volume ratio of HCl:acetone) and recorded as the experimental group, with a total of five groups. Two or three drops of phenolphthalein indicator were added, and the solution was left to stand for ≈30 min to completely dissolve the epoxy. A blank control group for the three groups was established. Configure NaOH ethanol solution with c = 0.1 mol/L for titration, titrate the volume V1 (mL), and titrate the blank control group V2. Calculate the mean value of V2 as V.
Calculated according to Eq. 1–1, and the average of 5 sets of values was taken.
Viscosity test
The viscosity of the epoxy was determined using a digital viscometer (NDJ-8S) from Shanghai Fangrui Instrument Co.
Acid value test
A total of 0.8 ~ 1.5 of the acid sample was dissolved in 20 mL of DMSO. Phenolphthalein was used as the indicator. The sample was titrated with NaOH standard titration solution as a titrant until the color of the sample turned light red for 30 s. The volume of NaOH added (mL) was recorded, and the acid value was calculated using the formula \(\frac{c*V*56.1}{1000m}\) (g KOH/g).
Liquid chromatography mass spectrometry (LC–MS)
The composition of DEP was analyzed by liquid chromatography-mass spectrometry (Varian 1200 LC/MS) from Varian, USA.
Results and discussion
Degradation of epoxy cures into oligomers in glycol
The low-viscosity glycidyl ester/amine curing compound selected in this study can be cured at low temperatures (< 90 °C) and used for large-scale potting devices such as wind turbine blades. It degrades outwardly via surface erosion through the combination of ester exchange and alcohol diffusion [42], satisfying the shrinking core model. The alcoholysis reaction between the small glycol molecule and high-molecular-weight glycidyl ester/amine curing agents tends to produce diol esters, polyols, and some oligomers in the equilibrium state, as shown in Fig. 1. The epoxy resin can be completely degraded under atmospheric pressure at 180 ℃; its degradation efficiency is shown in Fig. 1(a). From the figure, it can be seen that the degradation is completed within 123 min, and the degradation process can be divided into two phases. The dissolution rate of the first phase is greater than the dissolution rate of the second phase, and when the maximal rate is reached, it is greater than the dissolution rate. According to the GPC analysis shown in Fig. 1(b) and Table 2, the molecular weight of DEP was 1014, the molecular weight distribution was 1.08. The liquid chromatography-mass spectrometry of the degradation product is shown in Fig. 2(a), and the mass spectrometry is shown in Fig. 2(b) and (c). It can be seen from the liquid chromatograms that there are different substances flowing out under different elution times, and the mass spectrometry of the elution times of 0.845 min and 1.171 min are selected, and the graphs show the peak of molecular ions with a mass-to-charge ratio of 258.75, and the molecular ion peak with a mass-to-charge ratio of 258.75. It can be seen that the molecular weight of the effluent material single of 0.845 min was 258.75, which was consistent with the molecular weight of the diol ester S-1. Figure 2(c) shows a molecular ion peak with a mass-to-charge ratio of 468.40, which is consistent with the molecular weight of polyol S-2, as well as fragment ion peaks with one hydroxyl group and two hydroxyl groups missing at m/z 451.50 and 432.60, and fragment ion peak with propylene glycol missing at mass-to-charge ratio 394.35. Therefore, it can be determined by liquid-mass spectrometry that the three-position network structure of the epoxy resin block was destroyed by the solvent and degraded into small molecule structures, and the alcoholic degradation solution contained mainly diol structures containing ester bonds and polyol structures. the cured polymeric material transformed into an oligomer, and the final product consisted of a mixture of various oligomers, as shown in Fig. 3.

a Degradation efficiency of epoxy block glycol system at varying times. b Gel permeation chromatogram of the degradates

Compositional analysis of degradation product DEP a Liquid chromatogram of DEP. b Mass chromatogram with an elution time of 0.845 min. c Mass spectra with an elution time of 0.845 min

Alcoholysis mechanism of epoxy resin in alcohol solution
Further degradation of oligomers
DEP was further degraded by NaOH via the reaction equation shown in Fig. 4. First, glycol alcoholysis was used to degrade the epoxy-cured material into an oligomer and small-molecule system as opposed to directly mixing the alcohol and alkali in equal proportions using the joint depolymerization method [43]; this avoids sodium cyclohexene-2-carboxylate deposition on the surface of the epoxy-cured material, lowering the reaction rate and reducing side reactions.

NaOH-induced degradation mechanism of DEP
The IR and 1H-NMR spectra of the sodium salt isolated from DEP resembled those of the standard material, confirming the generation of the sodium salt during alkaline depolymerization; however, the separated sodium salt still retained the structure of the amine curing agent. We used the 1H-NMR spectrum of the sodium salt as a reference for assessing the purity of the product, comparing the NMR spectra of the products obtained from each test with those of the standard product (Fig. 5). The 1H-NMR spectrum, including the integrations, of the isolated sodium salt contains peaks at chemical shifts of 5.71 and 2.79 ppm, corresponding to the hydrogen atoms on the double bond of cyclohexene; the peaks at 2.43 and 2.25 ppm are characteristic of the methylene hydrogen atoms of cyclohexene; and the peak at 2.79 ppm is attributed to the hydrogen atoms attached to the ester bond. The integrated area ratio of the corresponding hydrogens was 1:1:1:1, confirming the successful separation of the sodium salt. Compared with the standard material, the sodium salt obtained from each experiment also showed peaks at 5.76, 2.59, 2.44, and 2.07 ppm, with integrated area ratios of 1:1:1:1, corresponding to the trans configuration of the separated sodium salt. In addition, the isolated substance also showed peaks at chemical shifts of 1.19, 1.06, and 0.96 ppm, which are characteristic of the amine functional group and attributed to impurities in the product. In this study, the purity of the product was determined by comparing the ratio of the integral area of the hydrogen atoms on the double bond of cyclohexene to the integral of the impurity peak at 1.06 ppm (Table 4). The polar analysis results are listed in Table 3. From the polar analysis, it can be seen that the order of influencing factors is NaOH concentration, reaction time, ratio between DEP and NaOH, reaction temperature, and NaOH concentration, which are the most critical factors in alkaline decomposition. The optimal process conditions were as follows: reaction temperature of 60 °C, reaction time of 2.5 h, DEP:NaOH ratio of 10:2, and NaOH concentration of 10%.

Orthogonal 1H-NMR spectra of the sodium salt
The sodium salt obtained under the optimal process conditions for alkaline decomposition was characterized using FT-IR and NMR spectroscopies, as well as elemental analysis, as shown in Fig. 6 and 6(a). The C = O of the carboxylate group undergoes symmetric and antisymmetric stretching vibrations, and the IR spectrum of R-THA-Na reveals characteristic peaks at 1582 cm–1 and 1378 cm–1 corresponding with the antisymmetric and symmetric stretching of the sodium salt, respectively; both peaks are more intense than those of THA-Na but appear at basically the same positions. The one-dimensional 1H-NMR spectra of THA-Na and R-THA-Na are shown in Fig. 6(b). Both contain peaks at 5.71, 2.79, 2.43, and 2.25 ppm, where the peak at 5.71 ppm is characteristic of the hydrogen atoms on the double bond of cyclohexene, while those at 2.43 ppm and 2.25 ppm correspond to the methylidene hydrogen atoms. The quartet centered at 2.79 ppm is characteristic of the hydrogen atom bonded to the ester group, with an integrated area ratio of 1:1:1:1. The peaks at 5.76, 2.59, 2.44, 2.07 ppm (1:1:1:1) indicate that the isolated sodium salts adopt a trans configuration. Tables 5 show the results of the elemental analysis of THA-Na and R-THA-Na, indicating that both contain C, H, and a negligible amount of N. These results reveal that the isolated sodium salt was almost as pure as the standard THA-Na monomer.

Spectroscopic analysis of R-THA-Na and THA-Na: a IR b 1H-NMR spectra
Recycling of epoxy resin degradates
Sodium salt acidification
In this study, the isolated R-THA-Na was further acidified to prepare the acid (R-THA), whose FT-IR spectrum is shown in Fig. 7(a). The most obvious change is the disappearance of the two strong absorption peaks (corresponding to the C = O and C–O stretching vibrations of the ester group in the sodium salt) at 1540 cm–1 and 1398 cm–1, respectively, and the characteristic peaks of the carboxylic acid at 1740 cm–1 and 920 cm–1. This indicated that ACOOK was protonated to form ACOOH. The 1H-NMR spectra supported this result (Fig. 7(b)). The sodium salt and acid gave rise to chemical shifts of 5.71 ppm and 5.58 ppm, respectively, corresponding to the cyclohexene structure. Owing to the hydrolysis of the ester group to a carboxylic acid group, a carboxylic acid proton with a chemical shift of 12.13 ppm appeared in the spectrum of the acid.

Structural characterization of R-THA: a IR and b 1H-NMR spectra
Synthesis of recycled epoxides
The acid was reacted with epichlorohydrin to prepare the epoxy resin monomer product R–EP, and its corresponding IR and 1H-NMR spectra closely resembled those of the standard material (Fig. 8). The 1H-NMR spectrum of the product contains a peak at 5.67 ppm, which is characteristic of the hydrogen atoms on the cyclohexene ring. The signals at 4.38 ppm and 3.85 ppm (quartet) correspond to the hydrogen atoms on –O–CH2– and CH2, respectively, while the hydrogen atom on the CH of the epoxy group gives rise to two sets of quartets at 2.62 ppm and 2.37 ppm. The signal at 3.17 ppm is characteristic of the CH proton on the epoxy group, and that at 3.3 ppm is typical of the hydroxyl protons in water. The integral area ratio of 1:2:1:2:2:1:2, corresponding to positions a, b, c, d, e, and f, prove the successful synthesis of diglycidyl tetrahydrodipecanoate. The IR spectra is similar, where the telescopic vibration peak at 1640 cm–1 is due to C = C in the product cyclohexene. The peaks in the range of 650 ~ 730 cm–1 were identified as telescopic vibrations of the double-bond C–H, while the peak at 3500 cm–1 signals the telescopic vibration of –OH. The acid gives rise to a characteristic C = O peak of the carboxyl group at 1690 cm–1, the C = O stretching vibration of the ester bond in the product appears at 1728 cm–1, and the strong absorption at 1126 cm–1 is the antisymmetric stretching vibration of C–O–C. The product gives rise to an epoxy absorption peak at 910 cm–1, which indicates that the acid has been epoxidized.

Structural analysis of R-EP: a IR and b 1H-NMR spectra
Comprehensive R-EP performance
The experimentally synthesized diglycidyl tetrahydrodiacetate is shown in Fig. 9(a). The product was a light yellow, transparent liquid at room temperature (25 °C), and its viscosity at room temperature was 797 MPa s, as determined using a viscometer; its epoxide value was 0.63 mol/100 g, as determined by the hydrochloric acid–acetone method. The comprehensive properties of R-EP + IPDA and S-EP + IPDA (in this paper, the resin synthesized from the original monomers was named S-EP + after curing) were tested using DSC, DTG, and a universal tensile machine, as shown in Fig. 9(b), (c), and (d). The thermal and mechanical properties of the recycled epoxy did not change significantly compared with those of the original epoxy, and the Tg of the recycled epoxy was 104 °C. From the heat-loss data (Table 6), the T5%, T10%, and Tmax were found to stabilize near 282, 295, and 342 °C, respectively, which, taken together, showed that the recycled epoxy possessed the same excellent thermal properties as those of the original epoxy. From the stress–strain curves, it can be seen that the mechanical strength of the recycled epoxy was 65 MPa, and the elongation at break was 8.8%, demonstrating its superior mechanical properties compared with the original epoxy.

Comprehensive performance of R-EP + IPDA: a epoxy resin R-EP, b DSC analysis of R-EP + IPDA, c DTG analysis of R-EP + IPDA, and d stress–strain diagram of R-EP + IPDA
Conclusion
In this paper, an ester epoxy/amine curing system was studied. The curing compound was degraded, and the monomers that could be used for the preparation of feedstock epoxy were separated from the degraded products, and this recycling method is applicable to epoxy systems with ester bonding in the cured material.. The influencing factors of the separated alkaline decomposition process were analyzed using an orthogonal experimental design, and the availability of the recycled epoxy was explored. The following conclusions were drawn:
-
1.
EG and NaOH-induced double-decomposition of the polyester epoxy/amine curing material can realize rapid and complete depolymerization separation of the curing material at atmospheric pressure, demonstrating the efficacy of this method for the recycling ester epoxy/amine curing materials.
-
2.
The optimal process conditions for alkaline decomposition were as follows: reaction temperature of 60 °C, reaction time of 2 h, DEP:NaOH ratio of 10:2, and 10% addition of NaOH. Under these conditions, the purity of R-THA-Na is optimal.
-
3.
The acidity value obtained after acidification of the sodium salt was 467.3 mg KOH/g, with a purity of 99%. After the acid was epoxidized, the structure of the original epoxy resin monomer was characterized, and its comprehensive performance was determined to be comparable to that of the original epoxy resin.
Data availability
No datasets were generated or analysed during the current study.
References
Yaroslavov AA, Arzhakov MS, Khokhlov AR (2022) The life cycle of polymer materials: problems and prospects. Herald Russ Acad Sci 92:18–24. https://doi.org/10.1134/S1019331622010087
Da Silva DJ, Wiebeck H (2020) Current options for characterizing, sorting, and recycling polymeric waste. Prog Rubber Plast Recy Technol 36:284–303. https://doi.org/10.1177/1477760620918603
Duan H, Chen Y, Ji S, Hu R, Ma H (2019) A novel phosphorus/nitrogen-containing polycarboxylic acid endowing epoxy resin with excellent flame retardance and mechanical properties. Chem Eng J 375:121916. https://doi.org/10.1016/j.cej.2019.121916
Kumar S, Samal SK, Mohanty S, Nayak SK (2019) Curing kinetics of bio-based epoxy resin toughened DGEBA epoxy resin blend: synthesis and characterization. J Therm Anal Calorim 137:1567–1578. https://doi.org/10.1007/s10973-019-08080-4
Marotta A, Faggio N, Ambrogi V, Cerruti P, Gentile G, Mija A (2019) Curing behavior and properties of sustainable furan-based epoxy/anhydride resins. Biomacromol 20:3831–3841. https://doi.org/10.1021/acs.biomac.9b00919
Mora A-S, Tayouo R, Boutevin B, David G, Caillol S (2019) Synthesis of biobased reactive hydroxyl amines by amination reaction of cardanol-based epoxy monomers. Eur Polym J 118:429–436. https://doi.org/10.1016/j.eurpolymj.2019.06.020
Xu Y-J, Chen L, Rao W-H, Qi M, Guo D, Liao W, Wang Y (2018) Latent curing epoxy system with excellent thermal stability, flame retardance and dielectric property. Chem Eng J 347:223–232. https://doi.org/10.1016/j.cej.2018.04.097
Miao J-T, Yuan L, Guan Q, Liang G, Gu A (2017) Biobased heat resistant epoxy resin with extremely high biomass content from 2,5-furandicarboxylic acid and eugenol. ACS Sustainable Chem Eng 5:7003–7011. https://doi.org/10.1021/acssuschemeng.7b01222
Pourchet S, Sonnier R, Ben-Abdelkader M, Gaillard Y, Ruiz Q, Placet V, Plasseraud L, Boni G (2019) New reactive isoeugenol based phosphate flame retardant: toward green epoxy resins. ACS Sustainable Chem Eng 7:14074–14088. https://doi.org/10.1021/acssuschemeng.9b02629
Wan JT, Zhao JQ, Gan B, Li C, Molina-Aldareguia J, Zhao Y, Pan Y, Wang D (2016) Ultrastiff biobased epoxy resin with high Tg and low permittivity: from synthesis to properties. ACS Sustainable Chem Eng 4:2869–2880. https://doi.org/10.1021/acssuschemeng.6b00479
Xu X, Ma S, Wu J, Yang J, Wang B, Wang S, Li Q, Feng J, You S, Zhu J (2019) High-performance, command-degradable, antibacterial Schiff base epoxy thermosets: synthesis and properties. J Mater Chem A 7:15420–15431. https://doi.org/10.1039/C9TA05293C
Yi J, Li S, Xia J, Li M, Ding H, Xu L, Yang X (2019) The design, preparation, and properties of dual crosslinking copolymerized systems based on hemp oil. New J Chem 43:14928–14937. https://doi.org/10.1039/C9NJ03385H
Zheng S, Bellido-Aguilar DA, Wu X, Zhan X, Huang Y, Zeng X, Zhang Q, Chen Z (2019) Durable waterborne hydrophobic bioepoxy coating with improved anti-icing and self-cleaning performance. ACS Sustainable Chem Eng 7:641–649. https://doi.org/10.1021/acssuschemeng.8b04203
Jambeck J, Hardesty BD, Brooks AL, Friend T, Teleki K, Fabres J, Beaudoin Y, Bamba A, Francis J, Ribbink AJ, Baleta T, Bouwman H, Knox J, Wilcox C (2018) Challenges and emerging solutions to the land-based plastic waste issue in Africa. Mar Policy 96:256–263. https://doi.org/10.1016/j.marpol.2017.10.041
Vollmer I, Jenks MJF, Mayorga González R, Meirer F, Weckhuysen BM (2021) Plastic waste conversion over a refinery waste catalyst. Angew Chem Int Ed Engl 60:16101–16108. https://doi.org/10.1002/anie.202104110
Pruksawan S, Lambard G, Samitsu S, Sodeyama K, Naito M (2019) Prediction and optimization of epoxy adhesive strength from a small dataset through active learning. Sci Technol Adv Mater 20:1010–1021. https://doi.org/10.1080/14686996.2019.1673670
Fortman DJ, Brutman JP, De Hoe GX, Snyder RL, Dichtel WR, Hillmyer MA (2018) Approaches to sustainable and continually recyclable cross-linked polymers. ACS Sustainable Chem Eng 6:11145–11159. https://doi.org/10.1021/acssuschemeng.8b02355
Amamoto Y, Otsuka H, Takahara A, Matyjaszewski K (2012) Self-healing of covalently cross-linked polymers by reshuffling thiuram disulfide moieties in air under visible light. Adv Mater 24:3975–3980. https://doi.org/10.1002/adma.201201928
Hayashi M, Yano R, Takasu A (2019) Synthesis of amorphous low T g polyesters with multiple COOH side groups and their utilization for elastomeric vitrimers based on post-polymerization cross-linking. Polym Chem 10:2047–2056. https://doi.org/10.1039/C9PY00293F
Ma S, Wei J, Jia Z, Yu T, Yuan W, Li Q, Wang S, You S, Liu R, Zhu J (2019) Readily recyclable, high-performance thermosetting materials based on a lignin-derived spiro diacetal trigger. J Mater Chem A 7:1233–1243. https://doi.org/10.1039/C8TA07140C
Wang S, Ma S, Li Q, Xu X, Wang B, Yuan W, Zhou S, You S, Zhu J (2019) Facile in situ preparation of high-performance epoxy vitrimer from renewable resources and its application in nondestructive recyclable carbon fiber composite. Green Chem 21:1484–1497. https://doi.org/10.1039/C8GC03477J
Memon H, Liu H, Rashid MA, Chen L, Jiang Q, Zhang L, Wei Y, Liu W, Qiu Y (2020) Vanillin-Based epoxy vitrimer with high performance and closed-loop recyclability. Macromolecules 2:621–630. https://doi.org/10.1021/acs.macromol.9b02006
Rashid MA, Zhu S, Zhang L, Jin K, Liu W (2023) High-performance and fully recyclable epoxy resins cured by imine-containing hardeners derived from vanillin and syringaldehyde. Eur Polym J 187:111878. https://doi.org/10.1016/j.eurpolymj.2023.111878
Trovatti E, Lacerda T, Carvalho A, Gandini A (2015) Recycling tires? Reversible crosslinking of poly (butadiene). Adv Mater 27:2242–2245. https://doi.org/10.1002/adma.201405801
Luo K, Xie T, Rzayev J (2013) Synthesis of thermally degradable epoxy adhesives. J Polym Sci Part A: Polym Chem 51:4992–4997. https://doi.org/10.1002/pola.26926
González L, Ferrando F, Ramis X, Salla JM, Mantecón A, Serra A (2009) Characterization of new reworkable thermosetting coatings obtained by cationic and anionic curing of DGEBA and some Meldrum acid derivatives. Prog Org Coat 65:175–181. https://doi.org/10.1016/j.porgcoat.2008.10.007
Si H, Zhou L, Wu Y, Song L, Kang M, Zhao X, Chen M (2020) Rapidly reprocessable, degradable epoxy vitrimer and recyclable carbon fiber reinforced thermoset composites relied on high contents of exchangeable aromatic disulfide crosslinks. Compos B Eng 199:108278. https://doi.org/10.1016/j.compositesb.2020.108278
Pan C, Lu J, Wu B, Wu L, Li B (2017) Effect of monomer structure on crystallization and glass transition of flexible copolyesters. J Polym Environ 25:1051–1061. https://doi.org/10.1007/s10924-016-0881-5
Li H, Bai J, Shi Z (2016) Environmentally friendly polymers based on Schiff-base reaction with self-healing, remolding and degradable ability. Polymer 85:106–113
Luo HY, Yin YQ, Wang Y, Li Q, Tang A, Liu Y (2022) Enhanced properties of a soybean adhesive by modification with a cycloaliphatic epoxy resin. Int J Adhes Adhes 114:103026. https://doi.org/10.1016/j.ijadhadh.2021.103026
Huang Z, Deng Z, Dong C, Fan J, Ren Y (2022) A closed-loop recycling process for carbon fiber reinforced vinyl ester resin composite. Chem Eng J 446:137254. https://doi.org/10.1016/j.cej.2022.137254
Chan CH, Wakisaka M, Nishida H (2019) Specific oligomer recovery behavior from cured unsaturated polyester by superheated steam degradation. Polym Degrad Stab 161:1–6. https://doi.org/10.1016/j.polymdegradstab.2018.12.025
Ahrens A, Bonde A, Sun H, Wittig NK, Hammershøj HCD, Batista GMF, Sommerfeldt A, Frølich S, Birkedal H, Skrydstrup T (2023) Catalytic disconnection of C-O bonds in epoxy resins and composites. Nature 617:730–737. https://doi.org/10.1038/s41586-023-05944-6
Knappich F, Klotz M, Schlummer M, Wölling J, Mäurer A (2019) Recycling process for carbon fiber reinforced plastics with polyamide 6, polyurethane and epoxy matrix by gentle solvent treatment. Waste Manag 85:73–81. https://doi.org/10.1016/j.wasman.2018.12.016
Liu Y, Kang H, Gong X, Jiang L, Liu Y, Wu S (2014) Chemical decomposition of epoxy resin in near-critical water by an acid-base catalytic method. RSC Adv 4:22367–22373. https://doi.org/10.1039/C4RA02023E
Xing M, Li Y, Zhao L, Song X, Fu Z, Du Y, Huang X (2020) Swelling-enhanced catalytic degradation of brominated epoxy resin in waste printed circuit boards by subcritical acetic acid under mild conditions. Waste Manag 102:464–473. https://doi.org/10.1016/j.wasman.2019.11.011
Lu Y, Liu T, Wang SJ, Sun Y, Zhang Y, Kang J, Li B, Gao Y, Gao X, Fan W (2022) A mild, environmental, and low-destructive recycling strategy for the 3D multiaxial braided composites cured by the vacuum-assisted resin transformed molding process. Compos Commun 35:101354. https://doi.org/10.1016/j.coco.2022.101354
Liu T, Guo X, Liu W, Hao C, Wang L, Hiscox WC, Liu C, Jin C, Xin J, Zhang J (2017) Selective cleavage of ester linkages of anhydride-cured epoxy using a benign method and reuse of the decomposed polymer in new epoxy preparation. Green Chem 19:4364–4372. https://doi.org/10.1039/C7GC01737E
Zhao X, Liu X, Feng K, An WL, Tian F, Du R, Xu S, Chen L, Wu G, Wang YZ (2022) Multicycling of epoxy thermoset through a two-step strategy of alcoholysis and hydrolysis using a self -Separating catalysis system. Chemsuschem 15:e202101607. https://doi.org/10.1002/cssc.202101607
Dattilo S, Cicala G, Riccobene PM, Puglisi C, Saitta L (2022) Full recycling and re-use of bio-based epoxy thermosets: chemical and thermomechanical characterization of the recycled matrices. Polymers 14:4828. https://doi.org/10.3390/polym14224828
Zhao X, Wang XL, Tian F, An W, Xu S, Wang Y (2019) A fast and mild closed-loop recycling of anhydride-cured epoxy through microwave-assisted catalytic degradation by trifunctional amine and subsequent reuse without separation. Green Chem 21:2487–2493. https://doi.org/10.1039/C9GC00685K
Kuang X, Shi Q, Zhou Y, Zhao Z, Wang T, Qi HJ (2018) Dissolution of epoxy thermosets via mild alcoholysis: the mechanism and kinetics study. RSC Adv 8:1493–1502. https://doi.org/10.1039/c7ra12787a
Liu X, Zhao X, An W, Du R, Wu G, Xu S, Zhang F, Wang YZ (2022) Exploiting valuable supramolecular materials from waste plastics. Mater Horiz 9:2993–3001. https://doi.org/10.1039/d2mh00781a
Acknowledgements
This work was supported by the Science and Technology Department of Sichuan Province (2022ZHCG0079 and 2023YFG0235).
Funding
I declare that this work was supported by the Science and Technology Department of Sichuan Province (2022ZHCG0079 and 2023YFG0235).
Author information
Authors and Affiliations
Contributions
H's contributions included designing the experiments, performing the manipulations, analyzing the data, organizing the data, and writing the manuscript. M's contribution included identifying the selected topic, data analysis, and manuscript review. Z's contribution included manuscript review and supervision. D's contribution included manuscript review and supervision. L's contribution due to this revision, especially in the data processing and integration of LC-MS.
Corresponding author
Ethics declarations
Ethical approval
I declare that this study does not involve human and/or animal research.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, X., Ma, H., Zhou, B. et al. Rapid degradation of thermosetting ester epoxies and monomer recovery methods. Colloid Polym Sci 302, 1467–1478 (2024). https://doi.org/10.1007/s00396-024-05287-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-024-05287-2