
Abstract
Reparative inflammation is an important protective response that eliminates foreign organisms, damaged cells, and physical irritants. However, inappropriately triggered or sustained inflammation can respectively initiate, propagate, or prolong disease. Post-hemorrhagic (PHH) and post-infectious hydrocephalus (PIH) are the most common forms of hydrocephalus worldwide. They are treated using neurosurgical cerebrospinal fluid (CSF) diversion techniques with high complication and failure rates. Despite their distinct etiologies, clinical studies in human patients have shown PHH and PIH share similar CSF cytokine and immune cell profiles. Here, in light of recent work in model systems, we discuss the concept of “inflammatory hydrocephalus” to emphasize potential shared mechanisms and potential therapeutic vulnerabilities of these disorders. We propose that this change of emphasis could shift our thinking of PHH and PIH from a framework of life-long neurosurgical disorders to that of preventable conditions amenable to immunomodulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydrocephalus is generally defined as the progressive distension of the brain’s ventricular system due to inadequate movement of CSF from its site of production at the choroid plexus epithelium (ChP) to its site(s) of reabsorption. As our understanding of hydrocephalus advances, however, the complex nature of CSF homeostasis has become evident, with increased appreciation of other important physiological factors such as cardiac pulsatility [1, 2], and the proposal of alternative anatomic sites of CSF secretion and absorption. Furthermore, recent human genomic analysis is highlighting important differences between congenital and acquired hydrocephalus, with data showing up to 25% of sporadic congenital hydrocephalus arising from de novo mutation and impaired neural stem cell fate [3]. In those forms of hydrocephalus associated with active CSF accumulation, intracranial pressure increases as the ventricles enlarge, often resulting in irreversible brain damage. If untreated, hydrocephalus can lead to progressive neurological decline, coma and, ultimately, death [4].
Although hydrocephalus is a frequent complication of many central nervous system (CNS) insults [4,5,6,7], post-hemorrhagic and post-infectious are the two most common forms of acquired hydrocephalus worldwide [4, 7]. Pathological CSF dynamics observed in these conditions have historically been attributed to intraventricular obstruction or inadequate CSF reabsorption [8, 9]. However, many patients with post-hemorrhagic hydrocephalus (PHH) [10] and post-infectious hydrocephalus (PIH) [11] demonstrate no discernible physical impediment to CSF flow through either the ventricular system or subarachnoid spaces. Further, recent work is demonstrating pathological changes in CSF hypersecretion and flow dynamics, ependymal integrity, and damage/scarring of the intraventricular and parenchymal CSF pathways underlie the pathogenesis of acquired hydrocephalus [4, 6, 10, 12]. Even with these advancements in our understanding, however, the mainstay of treatment for PIH and PHH remains surgical CSF diversion, commonly through placement of a ventricular-peritoneal shunt. Newer treatments based on a more complete molecular understanding of disease pathophysiology are required.
Here, we review the current epidemiology, etiology, and clinical management of PIH and PHH, and highlight recent findings suggesting that these most common forms of acquired hydrocephalus share a common inflammatory-dependent pathophysiological mechanism. In so doing, we emphasize the role of the choroid plexus epithelium (ChP), which serves as both the major part of the blood-CSF barrier (much like the blood–brain barrier in the brain parenchyma) and the main producer of CSF [13]. An improved understanding of the shared pathophysiology of these forms of acquired hydrocephalus may catalyze discovery of effective pharmaceutical and/or biological treatments, thus reducing the need for invasive neurosurgical procedures.
Hydrocephalus: classification, etiology, and epidemiology
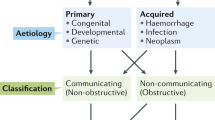
Hydrocephalus has been generally classified as non-communicating (obstructive) or communicating (non-obstructive), defined by the presence or absence of a physical blockage preventing CSF flow through the ventricular system. These broad definitions are further defined by whether hydrocephalus is congenital (i.e., primary) or acquired (i.e., secondary). Acquired hydrocephalus often occurs as a result of intracranial hemorrhage, infection, tumor, or head trauma. As numerous insults can result in the development of hydrocephalus, the high burden of disease affects individuals of all age groups, imposing a heavy physical, emotional, and socioeconomic impact worldwide [14].
A paucity of reliable data for disease incidence rates and prevalence has long prevented accurate estimates of the global burden of hydrocephalus [7]. Two recent meta-analyses, however, have provided much needed insight into the burden of childhood and adult hydrocephalus. Dewan et al. estimate nearly 400,000 new cases of childhood hydrocephalus worldwide each year [7]. Furthermore, Isaacs et al. indicate a global prevalence of hydrocephalus of 88 cases per 100,000 individuals under 18 years of age, 11 cases per 100,000 individuals between ages 19 and 64, and 175 cases per 100,000 individuals over the age of 64 [14]. Interestingly, disparities in the burden of hydrocephalus between lower and higher income countries were also noted, with both analyses reporting significantly higher incidence rates among lower- and middle-income countries [7, 14]. Thus, Isaacs et al. found low- and middle-income countries had an annual incidence per 100,000 of nearly 30 cases more than that of high-income countries [14]. Dewan et al. similarly reported annual rates of 123 cases of pediatric hydrocephalus per 100,000 in low-to-middle income countries vs. 79 cases per 100,000 in high income countries [7]. Thus, the burden of hydrocephalus is driven to a large degree by socio-economic status. As discussed below, a major reason for this disparity is the predominance of PIH among children of poorer countries in which neonatal and infant infections are more common [7].
Worldwide, PIH is the most common cause of hydrocephalus in the pediatric population [4], with the highest prevalence in Africa, Latin America, and Southeast Asia [7]. Lower- and middle-income countries report ~123,000 cases of PIH each year [7]. By contrast, PIH is seldom seen in higher income regions. This discrepancy likely reflects higher rates of perinatal infections secondary to unsanitary practices during childbirth and with newborns, and lack of advanced obstetric care [15, 16]. Furthermore, many agents cause PIH, and predominant pathogens vary with geographic location [17], likely reflecting regional differences in bacterial flora, living conditions (e.g., proximity to farm animals, housing standards) [17], the standard of perinatal care [7, 17], and seasonal changes in rainfall [18]. For example, and perhaps unsurprisingly, rates of PIH within the African meningitis belt increase in parallel with seasonal peaks in meningitis infections [19]. Furthermore, congenital Zika virus is a cause of life-threatening hydrocephalus in Brazil [20], while in endemic areas such as South Africa [21], India [22, 23], China [24], and Philippines [25], post-tuberculosis hydrocephalus constitutes a considerable disease burden. In higher income countries, PIH is most commonly due to prenatal infections of the mother transmitted to the fetus in utero. Such cases are commonly due to Toxoplasma gondii and cytomegalovirus (CMV) [4, 17, 26]. Postnatal and pediatric etiologies are typically bacterial meningitides caused by Escherichia coli, Streptococcus agalactiae, and Listeria monocytogenes [4, 17, 26]. The most common causes of bacterial PIH in adults include Neisseria meningitidis and Streptococcus pneumoniae [27]. Viral, fungal, and protozoan infections also contribute a significant hydrocephalus burden in immunocompromised patients [28, 29].
In contrast, PHH is the most common cause of acquired hydrocephalus in higher-income countries, occurring in ~38 neonates per 100,000 live births [7, 30]. PHH predominates in very low birth weight (VLBW) preterm neonates (< 1500 g), due to rupture of the highly vascularized germinal matrix [31, 32]. Without adequate prenatal, neonatal intensive, and neurosurgical care, neonatal PHH is often fatal [33]. Accordingly, infantile PHH is underrepresented in countries that lack the perinatal resources to save and maintain the lives of preterm infants [7]. For example, East Africa has ~1 neurosurgeon per 10,000,000 people [15] and few neonatal resources; thus, most VLBW neonates fail to survive [31]. PHH is also common in adults [31, 34, 35], and most frequently caused by blood leaking into the ventricular system following hypertensive hemorrhage, aneurysm rupture, or traumatic brain injury [36].
Management of acquired hydrocephalus
Regardless of the etiology of acquired hydrocephalus, treatment typically involves CSF diversion through the placement of a ventricle-peritoneal shunt or endoscopic third ventriculostomy (ETV). One caveat is the presence of an intracranial mass causing ventricular obstruction, for which surgical resection of the mass lesion would first be attempted to restore normal CSF flow. However, as PIH and PHH are the most common etiologies of acquired hydrocephalus, shunting is perhaps the most common treatment for hydrocephalus among all age groups [4, 26, 37,38,39,40].
Ventriculo-peritoneal shunting involves tunneling a silicone elastomer tube under the skin running from the ventricles in the head into the abdominal cavity, with a valve in place that regulates CSF drainage [4]. Shunts provide a lower rate of failure in the early months following surgery [40, 41], faster improvement in ventricular size [4, 40], and moderate level of required technical expertise [42]. However, complications arise frequently, typically in the form of mechanical obstruction/malfunction, tubing complications, and infection. This high likelihood of shunt failure requires long-term, consistent, and emergent access to neurosurgical care [41]. Indeed, shunt failure is the most common medical device failure in the USA, with 2- and 10-year failure rates of > 50% and 70%, respectively, after primary shunt placement [4, 43]. Furthermore, nearly 26% of revision operations fail within 30 days of placement [44]. Thus, as many patients develop shunt dependence after PIH/PHH, a lifelong need for accessible neurosurgical care is common. In one retrospective chart review, pediatric patients with shunts required on average 2.66 revisions, with ~85% of patients requiring at least one revision and ~5% of patients requiring 10 or more [45]. These frequent and potentially life-threatening complications and the interventions required to correct them explain many patients’ reports of significantly decreased quality of life after shunt placement [4, 43].
Alternatively, in some cases, an endoscopic third ventriculostomy (ETV) with or without choroid plexus cauterization (CPC) can be performed. In this procedure, an endoscope is used to perform fenestration of the third ventricular floor, providing an alternate pathway for CSF flow and reabsorption. Choroid plexus cauterization employs electro-thermal destruction of the ChP in attempt to decrease CSF production. ETV/CPC has been increasingly utilized for the treatment of infants with both PHH and PIH worldwide [46, 47]. In comparison to shunt placement, ETV/CPC shows decreased failure rates long-term [41], and eliminates hardware complications [48] (specifically, infection and failure). In addition, although the change in ventricular size on imaging may be less robust, cognitive development and brain growth in infants younger than 6 months of age are comparable after ETV/CPC and after shunting [26, 49]. The most significant limitations to ETV/CPC are the requirement for advanced technical expertise to perform the procedure [50], a higher rate of short-term failure, and an unclear impact on other critical functions of the ChP, including immune function, nutrient reabsorption, and neurodevelopment [51]. However, recent data suggests that ETV/CPC is becoming the preferred treatment for hydrocephalus, especially in developing countries with limited access to urgent neurosurgical care [26].
Lastly, neuroendoscopic lavage (NEL), although a less commonly used technique, has been reported as a potential early intervention for patients with hemorrhagic and infectious hydrocephalus. This procedure is performed similarly to an ETV; however, instead of fenestrating the third ventricular floor, the endoscope is used to irrigate the ventricular system under direct visualization. A septostomy can also be performed during the procedure to reach the contralateral lateral ventricle for further washout of blood/infectious material. Unlike VPS and ETV, NEL does not provide CSF diversion; rather, the goal of the procedure is to remove as much of the irritant (blood or infection) as possible to restore a physiological ventricular environment and CSF flow. A recent retrospective analysis in preterm infants with IVH demonstrated lower permanent shunt rate and improved neurological outcome in patients undergoing early NEL [52], and an earlier study showed improved shunt survival for those who did progress to require shunt placement [53]. Another recent case report and literature review determined positive outcomes in adult patients with ventriculitis that underwent NEL in addition to EVD and septostomy for further washout [54]. These reported outcomes, although modest, strongly suggests that the presence of blood and infectious debris stimulate an acute response, that, if attenuated early, may provide restoration of CSF homeostasis and prevent permanent damage that leads to hydrocephalus. Furthermore, given this technique allows direct access and irrigation of the ventricular system, it could provide an additional method of drug delivery for new and promising therapeutic targets to treat or prevent acquired hydrocephalus.
Pathogenesis of hydrocephalus
Hydrocephalus is the clinical manifestation of an alteration in fluid homeostasis in the central nervous system, leading to accumulation of CSF in the ventricular system of the brain. Classical models of CSF dynamics describe CSF production by the choroid plexus and absorption by extra-ventricular arachnoid granulations. Current theories of both post-hemorrhagic and post-infectious hydrocephalus center around the disruption of homeostatic CSF dynamics due to obstruction of intraventricular CSF flow and/or dysfunction of the arachnoid granulations decreasing reabsorption capacity [9, 33]. Older studies report findings of fourth ventricular outflow tract obstruction due to fibrous thickening of the leptomeninges creating a tetra-ventricular hydrocephalus in post-hemorrhagic brains [55, 56], while some have suggested that blood and its breakdown products acutely obstruct the narrow CSF passages in the brain (i.e., cerebral aqueduct) [57, 58]. For cases of non-obstructive/communicating post-hemorrhagic hydrocephalus, arachnoid granulation dysfunction has been implicated, proposed to result from microthrombi and IVH-related debris plugging arachnoid villi and impairing CSF reabsorption. Indeed, obliterative arachnoiditis in the posterior fossa may also lead to impaired CSF flow [59].
Although some cases of hydrocephalus occurring after hemorrhage or infection do result from frank obstruction due to an intraventricular blood clot or infectious scarring of the aqueduct, it may fail to fully explain the clinical presentation. The “plugged drain” paradigm overlooks the role of increased CSF secretion [5, 6] and ChP inflammation observed in human samples and animal models [6, 60,61,62,63] and is supported by limited experimental evidence [6, 31]. The absence of identifiable arachnoid granulations in human infants and most hydrocephalus animal models [64, 65], the presence of additional sites of CSF absorption (ventricular ependyma, perineural space, leptomeninges, glymphatics, and nasal mucosa nasal) [59, 64, 66,67,68], and evidence showing ChP villous hyperplasia and tumors cause CSF hypersecretion potentially sufficient to cause hydrocephalus [5] together argue for an underlying mechanism of acquired hydrocephalus more complex (and perhaps more elegant) than straightforward ventricular outlet obstruction.
Choroid plexus as secretory and immuno-modulatory epithelium
Secretory function of the ChP
The structure and function of the ChP is that of a polarized, secretory epithelium, consisting of a monolayer of cuboidal epithelial cells surrounding a core of fenestrated capillaries and forming the blood-CSF barrier. Although the ChP’s location deep within the ventricles of the brain has made it difficult to study, improved knowledge of its activity and regulation is critical to advancing our understanding of brain development, physiology, and pathophysiology. ChP cells in animal models produce the majority of CSF (~80%) [69] through active secretion of sodium (Na+), potassium (K+), and chloride (Cl−) ions, with osmotically driven movement of water into the ventricular space. As the most actively secreting epithelium, the ChP produces CSF at a rate of ~400–500 mL per day (~25 mL/hour) [13], receives the most blood flow per gram of tissue in the body, and expends the most energy through ATP utilization than any other epithelium [13, 70].
A novel organoid model system recently developed by Pellegrini et al. allows in vitro generation of a ChP-like epithelium from human pluripotent stem cells. These ChP organoids demonstrate secretion of a CSF-like fluid, expresses ChP markers (transthyretin (TTR), aquaporin 1 (AQP1), and other ChP-related proteins), and many of the transcriptomics and proteomic features of in vivo ChP. Single-cell RNA sequencing (scRNAseq) of the organoid cells has suggested heterogenous population of “dark” and “light” cells in the ChP and uncovered a new myoepithelial cell type in the ChP, with initial findings suggesting these different cell types exhibit distinct secretory roles in CSF production and secretion [71].
Given the historical difficulty of studying the ChP, the proteins and mechanisms essential to CSF production and secretion remain debated and actively investigated. Multiple models have been proposed, many centering on the unique polarity of transporters on the apical membrane of the ChP, especially Na+/K+-ATPase, and changes in osmotic and ion concentrations across the epithelium [13]. Currently, ChP-mediated CSF secretion is thought to be accomplished by a combination of basolateral and apical membrane transporters. The basolateral membrane components such as anion exchanger 2 (AE2) and Ncbe, function to take up Na+, Cl−, and HCO3− from the blood, and at the apical membrane, Na+, Cl−, and HCO3− secretion into the ventricles, is mediated by Na+/K+-ATPase, Na+-HCO3− cotransporters (NBCs), K+-Cl− cotransporters (KCCs), and Na+/K+/Cl− cotransporter 1 (NKCC1), with water uptake and secretion mediated by aquaporin expression, cotransporters (glucose transporter 1, GLUT1), and possibly paracellular transport through tight junctions [1, 13, 72].
Experimental evidence clearly supports the view that apical NKCC1 participates in the secretion of cerebrospinal fluid in the mature brain: Na+ transport from blood to CSF is inhibited by ventricular application of furosemide [73]. Interestingly, loss-of-function NKCC1 mutations in humans cause encephalopathy and impaired epithelial secretion throughout the body that is accompanied by increased CSF damage biomarkers and “slit ventricles” suggestive of decreased CSF production [74]. Outstanding questions remain as to the mechanism by which NKCC1 affects the rate of CSF secretion: by transporting outwardly as suggested by Steffensen et al. [75] or by inward transport to maintain cell volume and/or Cl− levels [76]. The controversy over the direction of net NKCC1-mediated transport might result from the transporter operating quite close to its equilibrium, and therefore operating in different directions based on CSF ionic gradients and other factors, as in certain disease states. More in vivo work is clearly needed to settle the issue.
ChP functions as an immune barrier
The CNS has been described as an “immune-privileged” tissue; however, this concept is being redefined as our understanding of the neuro-immune landscape advances [77]. While the CNS parenchyma is characterized by limited adaptive and innate immune response, endothelial and epithelial brain barriers, located at the superficial leptomeningeal vessels, parenchymal vessels, and ChP, provide immune surveillance and regulate immune cell entry [78]. The ChP forms an important interface between the CNS and periphery, allowing communication between the blood and CSF spaces. Floating in the brain’s ventricles, epithelial cells surround a connective stroma, through which penetrate blood vessels containing fenestrated capillaries. In contrast to the open capillary/stromal interface, the epithelial cells of the ChP are joined by tight junctions that compose the blood-CSF barrier (BCSFB) [79, 80].
Microglia, the resident immune cells found in the parenchyma of the brain, play an important role in brain function and homeostasis, guard the CNS from insults, and maintain their region-specific densities through self-renewal [80, 81]. The vast majority of the immunological diversity of the CNS, however, is now thought to be restricted to its border regions. This diverse population of immune cells, called border-associated macrophages (BAMs), are found in the perivascular space, meninges, and ChP, and are an area of active investigation. As work advances, new insights into the neuro-immune landscape of the CNS are being revealed [80, 82].
BAMs found in the ChP (ChP BAMs) have historically been elusive and challenging to study; however, advance techniques using live in vivo imaging and analysis using single-cell RNA sequencing (scRNAseq) are allowing a more detailed look into this population of cells. Recent studies describe a heterogenous population of ChP immune cells, including stromal macrophages, blood-borne macrophages, and epiplexus (Kolmer) cells located on the CNS side of the BCSFB [80, 83]. These studies are also helping clarify the origin and function of these cells. Epiplexus cells, long described as “macrophages” appear to share properties of microglia, including ontogeny, transcriptome, and self-renewal capacity, suggesting they may, in fact, be a subset of microglia inhabiting the ChP [81]. In contrast, stromal ChP macrophages are replaced via CCR2-dependent recruitment of circulating Ly6C+ blood monocytes [84, 85]. Epiplexus cells also demonstrate similarities to disease associated macrophages (DAMs), a phenotype of microglia presenting in response to brain pathology. Gene ontology network analysis shows common ontologies of DAMs and epiplexus cells, related to lipid metabolism, leukocyte differentiation/migration, and stimulus detection [80], suggesting an important role of these cells in disease states.
Dendritic and T cells are also found in the ChP stroma and play important roles in normal physiology and disease. As a response to lipopolysaccharide (LPS)-induced inflammation, T cells in the ChP undergo proliferation, and their stimulation shifts the monocyte/dendritic cells toward a leukocyte recruitment and antigen presentation phenotype [77]. Recent scRNAseq analysis identified a dendritic cell cluster in the ChP reminiscent of migratory dendritic cells, with expression of Ccr7 and Nudt17, suggesting they may also migrate to draining lymph nodes [81]. In humans, dendritic cells are densely present in the normal ChP [86], and in rodents, they have been described nestled between ChP cells with extension of their processes to the apical ChP surface [79, 86, 87], serving a key role in immunosurveillance. Even in healthy states, T cells are found in the CSF and ChP, and ~80% of cells in normal human CSF are CD4+ T cells, further supporting the role of the ChP in lymphocyte trafficking and regulation [88].
The ChP demonstrates a unique immunological plasticity, functioning as a highly regulated entrance for circulating peripheral immune cells into the CSF, in response to disease processes [89]. Surprisingly, however, compared to our understanding of immune cell migration and translocation across the blood brain barrier (BBB) [90], our understanding of the mechanisms controlling cellular movement across the choroid plexus remains in its infancy. Several mechanism of immune transmigration have been reported, including paracellular and transcellular in the ChP, and influx of stromal macrophages through choroid plexus epithelial cells during development has been termed “emperipolesis” [88]. During inflammation, the paracellular route, through the spaces between ChP epithelial cells appears to predominate [91]; however, disrupted cell–cell interactions is also a likely route of immune cell entry into the CSF [88]. This aspect of the ChP immune regulation remains an active area of research, and it will be important to elucidate these mechanisms of immune cell trafficking across the BCSFB [92].
Interestingly, recently studies have demonstrated an early development role of the ChP, in response to maternal immune activation. Cui et al. demonstrate a proinflammatory maternal environment increases the pro-inflammatory cytokines of embryonic mice, and enhances activation and migration of ChP macrophages across the embryonic BCSFB [93, 94]. These findings suggest a variety of neurodevelopmental processes and diseases may be driven by the ChP immune environment very early on in development. In adults, ChP immune function has been described to play a role in multiple human pathologies, including lyme disease [95], multiple sclerosis [86], stroke [96, 97], and neuropsychiatric disorders [98].
Multiple models and advanced tools now exist to precisely study the activation and response of the immune cells in the ChP [92]. Currently, one of the most common is intraperitoneal injection of LPS. Peripheral exposure to LPS causes systemic inflammation, which induces an inflammatory response in the ChP, including elevation of pro-inflammatory cytokines, IL-1β, CC-motif ligand 2 (CCL2), CXC-motif ligand 1 (CXCL1), CXCL2, and Il-6 [99], as well as extracellular vesicles and pro-inflammatory miRNAs [100]. Further, the specific response of the epithelia and stromal cells is important to distinguish, as these components of the BCSFB likely affect the regulation of apical and basal membrane expression differently, leading to differential changes in CSF composition and stromal expression [101]. This is demonstrated by studies showing proinflammatory cytokines altering the apical cation channel transient receptor potential vanilloid-4 (TRPV4) resulting in changes in ion flux and potentially CSF production [102], as well as reciprocal interactions between epithelial and stromal cells, demonstrated by similar cytokine receptors being found on cells in both locations [99].
Toll-like receptors (TLRs) are the main players in the activation of inflammatory pathways through the innate immune system. TLRs are found on both the apical and basolateral membranes of the ChP [6, 55, 97], but most highly expressed in macrophages and other immune cell types, including epiplexus cells. Along with other pattern recognition receptors (e.g., NOD-like receptors (NOD)), they bind pathogen-associated molecular patterns (PAMPs) to activate non-specific innate immune responses [103, 104] and exhibit ligand-specific regulation in response to pro-inflammatory stimuli [6, 60, 61]. PAMPs that trigger cytokine production through TLRs including LPS on gram negative bacteria, peptidoglycans in the cell wall of bacteria, and dsRNA/DNA of viral pathogens. LPS, a ligand of TLR4, causes upregulation of cytokines in the ChP, and interestingly, causes upregulation of cellular adhesion molecules, and down-regulation of tight junction components [92, 105]. A recent study in mice demonstrated TLR2-mediated transmigration of neutrophils and monocytes in postnatal day 8 mice after exposure to LPS, as well as cytoskeleton remodeling [106].
Furthermore, recent work has focused on endogenous, non-canonical ligands also bound by TLRs, and how they may contribute to inflammatory responses. For example, TLRs have been shown to bind damage-associated molecular patterns (DAMPs) released from dying or injured tissues [107], including matrix degradation products, heat shock proteins, S100A8/S100A9 [108], LPA [109], and blood breakdown products (e.g., methemoglobin [metHgb], heme, and iron) present in the setting of intraventricular hemorrhage (IVH) [110,111,112,113]. In a preterm rabbit pup model of IVH, TLR4 upregulation of mRNA for TLR4 and nuclear factor κB (NF-κB), as well as proinflammatory molecules, was seen in the ChP, and found to be reversed with injection of a hemoglobin scavenger [60]. In a recent study using CSF samples from premature infants with IVH, an upregulation of proinflammatory miRNAs was detected in the acute setting, and found to be decreased at later timepoints once oxidized hemoglobin products were no longer detected [114]. In a novel rat model of IVH where autologous blood is directly injected into the brain ventricles, immune cell activation and NF-κB expression were found to be upregulated through a TLR4 dependent-mechanism [6]. These findings suggest that the ChP immune response is similarly activated in the presence of pathogens like LPS, as well as other irritants like intraventricular blood products.
CSF hypersecretion as a response to neuroinflammation in PIH
Outside the CNS, secretory epithelial tissues commonly respond to a pro-inflammatory environment with increased fluid secretion [115], which functions to clear pathogens and/or debris from the epithelial surface [116, 117]. However, sustained or dysregulated inflammation and fluid hypersecretion in many organs can exacerbate disease states, as in chemical, autoimmune, or infectious pleuritis, colitis, pancreatitis, and other conditions [4, 13]. Increased and/or unregulated activity of the ChP secretory epithelium leads to pathological CSF accumulation in the ventricular system of the brain, often producing tissue ischemia and death without urgent surgical intervention.
Pathological CSF accumulation in the ventricles is a well-known complication of neuroinflammatory pathologies, including intraventricular infection. Study of the effect of inflammation on the ChP secretory response has been challenging historically due to the lack of techniques to measure and manipulate CSF secretion in vivo. However, our recent development of a novel microsurgical technique has provided the ability to directly measure real-time CSF secretion rate in living rats [118] and mice [119], uncovering novel observations of CSF dynamics [6]. Additional recent, exciting work is beginning to demonstrate important mechanistic links between the secretory and immune functions of the ChP, detecting similarities among different etiologies of hydrocephalus, including infectious and hemorrhagic.
TLR4-dependent ChP inflammation
Inflammation resulting from any underlying etiology causes tissue damage, which often propagates and amplifies the response to the initial insult. In addition to the presence of PAMPs endogenous to the infectious agent, secondary tissue injury initiates the release of host-derived DAMPs from damaged epithelial barriers and other tissues. Both PAMPs and DAMPs can bind and activate TLRs located on microglia and ChP cells [60]. In the setting of infection, LPS binds TLR4 receptors and triggers a signaling cascade through NF-κB dependent pathways, and peripheral injection of LPS is a stimulus strong enough to activate brain microglia [120]. However, and perhaps more surprisingly, a similar TLR4-mediated pathway is triggered with intracerebral injections of autologous blood [6, 62].
In patients with intraventricular hemorrhage (IVH), the ChP is one of the first tissues in the brain exposed to these mis-localized blood products [60, 121, 122]. Intraventricular injection of autologous blood into the lateral ventricle of rodents causes ventriculomegaly, NF-κB activation, and cytokine production in the ChP. Furthermore, in human infants with IVH, methemoglobin (metHgb), a blood breakdown product found in the CSF after intracranial hemorrhage, correlates with TLR4-dependent production of the cytokine TNFα. Experimental in vivo IVH models using rabbit pups show effects similar to those observed clinically. In this model, direct delivery of cell-free metHgb is sufficient to cause ventriculomegaly associated with increases in TLR4-NF-κB, TNF-α, and IL-1β in the ChP [60, 61]; furthermore, physiological levels of metHgb typical of clinical IVH are sufficient to activate TLR4 homodimers and/or TLR4/2 heterodimers [82, 83]. In the IVH rodent model, ChP inflammation was characterized by robust phosphorylation of NF-κB, the upregulation of TNF-α and IL-1β, and infiltration of activated ED-1+ microglia and macrophages [6]. These findings suggest that blood products, and metHgb more specifically, function as DAMPs to promote TNFα secretion through a TLR4-dependent mechanism of neuroinflammation [110, 123, 124].
TLR4-dependent, SPAK-mediated ChP hypersecretion of CSF
In addition to a robust TLR4-NF-κB-dependent ChP inflammatory response, rats injected intraventricularly with autologous blood also demonstrate a threefold increase in CSF production. In this model, CSF hypersecretion is seen as early as 24 h after IVH, is sustained for approximately 7 days, and is sufficient to cause ventriculomegaly or PHH [6]. Interestingly, the increase in CSF secretion was found to be sensitive to bumetanide, an inhibitor of the NKCC1 cotransporter. Further investigation demonstrated a TLR4-dependent activation of NKCC1 through phosphorylation by the NF-κB-regulated STE20/SPS1-related, proline-alanine-rich kinase (SPAK). SPAK binds and phosphorylates NKCC1 in the apical membrane of the ChP, upregulating its activity and resulting in CSF hypersecretion [6, 125, 126].
Looking to well-studied secretory epithelial tissues outside the nervous system gives further mechanistic insights into this pathway of inflammatory-mediated, SPAK-dependent hypersecretion. SPAK integrates and transduces environmental signals intracellularly. In animal models of inflammatory bowel disease, both TNF-α and IFN-γ [127, 128] activate SPAK in an NF-κB-dependent pathway, which results in increased epithelial transport and permeability [128,129,130]. Conversely in a mouse model, SPAK knockout animals showed milder colitis, as well as decreased proinflammatory cytokine secretion, and reduced epithelial permeability [131]. In IgA nephropathy, SPAK knockout in mice prevents production of inflammatory mediators and T cell activation, and macrophages show decreased production of proinflammatory cytokines, as well as reduced NF-κB/MAPK activation [132]. Furthermore, SPAK binds the TNF receptor, RELT (receptor expressed in lymphoid tissues) to directly activate both p38 and JNK1/2 signaling pathways [133]. Similar findings are demonstrated in a rodent model of acute lung injury (ALI), in which increased NKCC1 expression aggravates ALI, and hyperglycemic induction of NKCC1 causes pulmonary edema, lung inflammation, increased expression of pro-inflammatory cytokines, and infiltration of immune cells. Bumetanide administration inhibited activation of the WNK4 (with no lysine kinase 4)-SPAK-NKCC1 pathway and reduced the inflammatory and secretory responses in the animals [134]. And in a separate model, activation of the WNK4-SPAK-NKCC1 cascade modulated macrophage activation in LPS-induced lung inflammation and injury [135].
Interestingly, SPAK is more abundant in the ChP than in most other tissues and demonstrates robust expression in the apical membrane of the ChP [136]. Upstream regulation of SPAK is mediated through TLR4-NF-κB signaling, and SPAK in turn binds and activates multiple downstream ion transporters, including NKCC1. Therefore, SPAK may serve as a critical mechanistic link between TLR4-dependent inflammation and CSF hypersecretion. In further support of this hypothesis, genetic inhibition of either TLR4 or SPAK, or pharmacological inhibition of TLR4-NF-κB or SPAK-NKCC1, suffice to normalize CSF secretion in PHH through reduction in IVH-induced NKCC1 phosphorylation and activation [6]. Taken together, these findings argue that intraventricular hemorrhage induces CSF hypersecretion through an inflammatory mechanism dependent on TLR4 and SPAK.
Notably, there remains some debate regarding the role of NKCC1 in CSF production and secretion at the ChP. Recently, Xu et al. found that in early postnatal development, the expression of NKCC1 is increased, and in their mouse model, engineered elevation of NKCC1 levels was associated with decreased CSF and reduced ventriculomegaly [137]. Gregoriades et al. present findings that suggest NKCC1 functions in net influx to maintaining intracellular chloride and water content at a concentration to allow CSF secretion [76]. However, findings by Steffenson et al. demonstrate bidirectional NKCC1 flow, with a new outward secretory function for direct CSF production [75]. Furthermore, the findings of SPAK-mediated PHH CSF hypersecretion and sensitivity to NKCC1 inhibition with bumetanide [6] argue that NKCC1 directly contributes to CSF production and hypersecretion in pathological disease states. As our understanding of choroid plexus physiology continues to grow, it will likely reveal nuanced regulation of vectorial fluid transport dependent on specific factors such as ion concentrations, basal activity versus pathological states, and developmental stages of the organism.
ChP inflammatory response: potential pathologic feed-forward mechanism
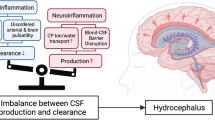
In the setting of intraventricular infection or hemorrhage, the inflammatory response appears to cause acute CSF hypersecretion which results in ventriculomegaly and clinical hydrocephalus. This timeline tracks well with the development of acute post-infectious and post-hemorrhagic hydrocephalus seen in patients. However, some patients who initially do not require permanent CSF diversion may develop delayed hydrocephalus requiring later intervention [138, 139] and many patients remain dependent on CSF diversion lifelong. This then raises the question of a chronic process that prevents return of normal, physiological CSF fluid dynamics, with important implications for development of pharmacological treatments of acquired hydrocephalus.
One explanation for the continued ventriculomegaly in these patients is the possibility that additional TLR-dependent or other innate immune mechanisms sustain a basal neuroinflammatory state, leading to inflammation-induced scarring, cilia dysfunction, or damage to other CSF homeostatic pathways, promoting aberrant, even reversed CSF dynamics after resolution of the initial insult. The glymphatic system may also play an important role in both acute and chronic inflammation, as a site of entry for immune cells [140] and a component of CSF drainage, immune cell trafficking, and neuroinflammation [141, 142]. After resolution of an initial insult, microglial and epithelial derived cytokines are released to promote epithelial proliferation and repair [143]; however, in the setting of chronic inflammation or tissue damage, a feedforward mechanism may prevent longer term resolution and exacerbate tissue damage and inflammation, as in many peripheral organs such as intestinal and respiratory epithelial tissues [144, 145]. This theory is further supported by neuro-endoscopic observations of damaged and “burnt out” ChP, intraventricular fibrosis, and friable ependyma in patients with previous intraventricular hemorrhage or infection. Ependymal denudation and ventricular zone disruption in experimental chronic PHH have also been reported [146].
Opportunities for pharmacological treatment of “inflammatory” hydrocephalus
As our understanding of acquired hydrocephalus evolves, similarities between the mechanistic underpinnings of infectious and hemorrhagic hydrocephalus are becoming clear. Infectious pathogens and blood products, through PAMPs and DAMPs, respectively, trigger innate immune responses in the ChP and likely also in brain parenchyma to drive a signaling cascade leading to aberrant CSF fluid dynamics and hydrocephalus. These similarities raise the compelling possibility that pharmacotherapeutics targeting this common inflammatory process could modulate the ventricular response to these insults—preventing significant morbidity and mortality from both PIH and PHH.
In the acute setting, many patients with acquired hydrocephalus, especially hemorrhagic-mediated injury, require urgent CSF diversion. This is typically achieved through the placement of an external ventricular drain (EVD), Ommaya reservoir, or lumbar drain. These devices conveniently allow access to CSF spaces in the CNS, which could be used for intraventricular or intrathecal administration of medications. This targeted administration in fact is used clinically to administer highly potent antibiotics in the setting of severe ventriculitis [147]. New agents, such as those targeting TLR4 or SPAK, could be infused through these devices to short-circuit the hydrocephalus-promoting inflammatory response and potentially prevent long-term CSF diversion in the form of a VP shunt or ETV/CPC. One such agent, Tak242, a small-molecule inhibitor of TLR4-mediated signaling, has demonstrated therapeutic effects in acute experimental PHH [6] and is being used in clinical trials for treatment of patients with sepsis [148]. Additionally, other anti-inflammatory agents targeting the TLR4-NF-κB pathway are being investigated, including pyrrolidine dithiocarbamate [149] and melatonin [150, 151]. Systemic administration of anti-inflammatory agents in the acute setting of the infectious or hemorrhagic insult may also be beneficial, as suggested by experimental PHH models showing response to intraperitoneal injection of Tak242 [6]. Intraperitoneal minocycline, a second-generation tetracycline with anti-inflammatory properties, reduces ventriculomegaly in spontaneously hypertensive SHR rats through attenuation of macrophage/microglial activation [152]. Other agents, including neutralizing antibodies or decoy receptors targeting DAMP/PAMP or cytokine receptors may also be beneficial, with potentially fewer off-target side effects than those of less specific pharmacotherapeutics.
Modulation of the SPAK/NKCC1 pathway is also a promising avenue for development of drugs targeting pathological inflammatory-mediated CSF hypersecretion. Unfortunately, current clinically available NKCC1-inhibitors, such as bumetanide and its derivatives, are systemically administered and have poor CNS penetration [6, 153]. Although bumetanide given in combination with phenobarbital for neonatal epilepsy was associated with hearing loss [154], it has been used successfully in clinical trials for treatment of autism in children [155,156,157]. Intraventricular or intrathecal administration may provide improved benefit of these drugs; however, more work is needed to understand the efficacy and safety of this route of delivery.
Targeting of SPAK may be a particularly advantageous route, given its role in both CSF hypersecretion and activation of the TLR4-dependent inflammatory response [6]. And although a master regulator of many other important targets [125], SPAK expression is highest in the ChP, and offers potential to be a powerful modulator of both PIH and PHH. The recently synthesized, novel, potent, and selective SPAK inhibitor, 5-chloro-N-(5-chloro-4-((4-chlorophenyl)(cyano)methyl)-2-methylphenyl)-2-hydroxybenzamide (ZT-1a), has indeed shown promise in reducing CSF secretion in experimental PHH with intracerebroventricular administration, and protects against brain damage in a stroke model when administered systemically [158]. Mounting data thus suggests that the immune-secretory plasticity of the choroid plexus is central to development of acquired, inflammatory hydrocephalus, and that pharmacological targeting of TLR4 and/or SPAK offers the promise of non-invasive therapies for patients.
Conclusions
The development of novel non-surgical, mechanism-based therapies for PIH/PHH could help avoid the adverse effects and complications of current invasive treatments and make treatments more accessible to areas of the world lacking neurosurgeons and related resources. Emerging data are shedding light into hydrocephalus pathophysiology, beginning to suggest that prevention of PIH/PHH is a feasible goal. We have proposed that the concept of “inflammatory hydrocephalus” conveys more accurately the pathogenic mechanisms and therapeutic vulnerabilities shared between PHH and PIH than does the term “secondary hydrocephalus.” We believe this change of nomenclature could catalyze a shift in thinking about these types of hydrocephalus from the category of life-long neurosurgical brain plumbing disorders to that of preventable neuro-inflammatory conditions. Much future work nonetheless remains before any treatment strategies can be considered, including: (i) continued identification of specific inflammatory mechanisms and targets contributing to pathogenesis of PHH and PIH, (ii) development of pharmacologic agents modulating these targets, and (iii) pre-clinical trials of these drugs in relevant experimental models. An approach addressing neuro-inflammation may not only prevent shunt dependence, but also ameliorate associated neurodevelopmental sequelae, including cerebral palsy and secondary inflammation-induced tissue damage, unaddressed by surgical CSF diversion. Such an approach would reduce the lifelong economic burden and morbidity associated with shunt placement and offer life-saving assistance in regions with limited neurosurgical access.
Abbreviations
- PHH:
-
Post-hemorrhagic hydrocephalus
- PIH:
-
Post-infectious hydrocephalus
- TLR:
-
Toll-like receptor
- CSF:
-
Cerebrospinal fluid
- ChP:
-
Choroid plexus epithelium
- CPC:
-
Choroid plexus cauterization
- NKCC1:
-
Na-K-Cl cotransporter 1
- SPAK:
-
STE20/SPS1-related, proline-alanine-rich kinase
- IVH:
-
Intraventricular hemorrhage
References
Brinker T, Stopa E, Morrison J, Klinge P (2014) A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 11:10
Benveniste H, Lee H, Volkow ND (2017) The glymphatic pathway: waste removal from the CNS via cerebrospinal fluid transport. Neuroscientist 23(5):454–465
Furey CG, Choi J, Jin SC et al (2018) De novo mutation in genes regulating neural stem cell fate in human congenital hydrocephalus. Neuron 99(2):302–14.e4
Kahle KT, Kulkarni AV, Limbrick DD Jr, Warf BC (2016) Hydrocephalus in children. Lancet 387(10020):788–799
Karimy JK, Duran D, Hu JK et al (2016) Cerebrospinal fluid hypersecretion in pediatric hydrocephalus. Neurosurg Focus 41(5):E10
Karimy JK, Zhang J, Kurland DB et al (2017) Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med
Dewan MC, Rattani A, Mekary R et al (2018) Global hydrocephalus epidemiology and incidence: systematic review and meta-analysis. J Neurosurg 1–15
Cherian S, Whitelaw A, Thoresen M, Love S (2004) The pathogenesis of neonatal post-hemorrhagic hydrocephalus. Brain Pathol 14(3):305–311
Strahle J, Garton HJ, Maher CO, Muraszko KM, Keep RF, Xi G (2012) Mechanisms of hydrocephalus after neonatal and adult intraventricular hemorrhage. Transl Stroke Res 3(Suppl 1):25–38
Tully HM, Wenger TL, Kukull WA, Doherty D, Dobyns WB (2016) Anatomical configurations associated with posthemorrhagic hydrocephalus among premature infants with intraventricular hemorrhage. Neurosurg Focus 41(5):E5
Visagan R, Livermore LJ, Kelly D, Magdum S (2017) Subclinical meningoventriculitis as a cause of obstructive hydrocephalus. BMJ Case Rep 2017
Klebe D, McBride D, Krafft PR, Flores JJ, Tang J, Zhang JH (2020) Posthemorrhagic hydrocephalus development after germinal matrix hemorrhage: established mechanisms and proposed pathways. J Neurosci Res 98(1):105–120
Damkier HH, Brown PD, Praetorius J (2013) Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev 93(4):1847–1892
Isaacs AM, Riva-Cambrin J, Yavin D et al (2018) Age-specific global epidemiology of hydrocephalus: Systematic review, metanalysis and global birth surveillance. PLoS One 13(10):e0204926
Warf BC (2010) East African Neurosurgical Research C. Pediatric hydrocephalus in East Africa: prevalence, causes, treatments, and strategies for the future. World Neurosurg 73(4):296–300
Muir RT, Wang S, Warf BC (2016) Global surgery for pediatric hydrocephalus in the developing world: a review of the history, challenges, and future directions. Neurosurg Focus 41(5):E11
Li L, Padhi A, Ranjeva SL et al (2011) Association of bacteria with hydrocephalus in Ugandan infants. J Neurosurg Pediatr 7(1):73–87
Schiff SJ, Ranjeva SL, Sauer TD, Warf BC (2012) Rainfall drives hydrocephalus in East Africa. J Neurosurg Pediatr 10(3):161–167
Aziz IA (1976) Hydrocephalus in the sudan. J R Coll Surg Edinb 21(4):222–224
van der Linden V, de Lima Petribu NC, Pessoa A et al (2018) Association of severe hydrocephalus with congenital Zika syndrome. JAMA Neurol
Kamat AS, Gretschel A, Vlok AJ, Solomons R (2018) CSF protein concentration associated with ventriculoperitoneal shunt obstruction in tuberculous meningitis. Int J Tuberc Lung Dis 22(7):788–792
Aranha A, Choudhary A, Bhaskar S, Gupta LN (2018) A randomized study comparing endoscopic third ventriculostomy versus ventriculoperitoneal shunt in the management of hydrocephalus due to tuberculous meningitis. Asian J Neurosurg 13(4):1140–1147
Rajshekhar V (2009) Management of hydrocephalus in patients with tuberculous meningitis. Neurol India 57(4):368–374
Li K, Tang H, Yang Y et al (2017) Clinical features, long-term clinical outcomes, and prognostic factors of tuberculous meningitis in West China: a multivariate analysis of 154 adults. Expert Rev Anti Infect Ther 15(6):629–635
Lee LV (2000) Neurotuberculosis among Filipino children: an 11 years experience at the Philippine Children’s Medical Center. Brain Dev 22(8):469–474
Kulkarni AV, Schiff SJ, Mbabazi-Kabachelor E et al (2017) Endoscopic treatment versus shunting for infant hydrocephalus in Uganda. N Engl J Med 377(25):2456–2464
Thigpen MC, Whitney CG, Messonnier NE et al (2011) Bacterial meningitis in the United States, 1998–2007. N Engl J Med 364(21):2016–2025
Pyrgos V, Seitz AE, Steiner CA, Prevots DR, Williamson PR (2013) Epidemiology of cryptococcal meningitis in the US: 1997–2009. PLoS One 8(2):e56269
Liu J, Chen ZL, Li M et al (2018) Ventriculoperitoneal shunts in non-HIV cryptococcal meningitis. BMC Neurol 18(1):58
Warf BC, Dagi AR, Kaaya BN, Schiff SJ (2011) Five-year survival and outcome of treatment for postinfectious hydrocephalus in Ugandan infants. J Neurosurg Pediatr 8(5):502–508
Chen Q, Feng Z, Tan Q et al (2017) Post-hemorrhagic hydrocephalus: Recent advances and new therapeutic insights. J Neurol Sci 375:220–230
Tsitouras V, Sgouros S (2011) Infantile posthemorrhagic hydrocephalus. Childs Nerv Syst 27(10):1595–1608
Murphy BP, Inder TE, Rooks V et al (2002) Posthaemorrhagic ventricular dilatation in the premature infant: natural history and predictors of outcome. Arch Dis Child Fetal Neonatal Ed 87(1):F37-41
Bir SC, Patra DP, Maiti TK et al (2016) Epidemiology of adult-onset hydrocephalus: institutional experience with 2001 patients. Neurosurg Focus 41(3):E5
Chahlavi A, El-Babaa SK, Luciano MG (2001) Adult-onset hydrocephalus. Neurosurg Clin N Am 12(4):753–760, ix
Cioca A, Gheban D, Perju-Dumbrava D, Chiroban O, Mera M (2014) Sudden death from ruptured choroid plexus arteriovenous malformation. Am J Forensic Med Pathol 35(2):100–102
Warf BC (2005) Comparison of endoscopic third ventriculostomy alone and combined with choroid plexus cauterization in infants younger than 1 year of age: a prospective study in 550 African children. J Neurosurg 103(6 Suppl):475–481
Warf BC (2005) Hydrocephalus in Uganda: the predominance of infectious origin and primary management with endoscopic third ventriculostomy. J Neurosurg 102(1 Suppl):1–15
Stagno V, Navarrete EA, Mirone G, Esposito F (2013) Management of hydrocephalus around the world. World Neurosurg 79(2 Suppl):S23.e17–20
Kulkarni AV (2016) First treatment in infants with hydrocephalus: the case for shunt. Neurosurgery 63(Suppl 1):73–77
Kulkarni AV, Drake JM, Kestle JR, Mallucci CL, Sgouros S, Constantini S (2010) Endoscopic third ventriculostomy vs cerebrospinal fluid shunt in the treatment of hydrocephalus in children: a propensity score-adjusted analysis. Neurosurgery 67(3):588–593
Baird LC (2016) First treatment in infants with hydrocephalus: the case for endoscopic third ventriculostomy/choroid plexus cauterization. Neurosurgery 63(Suppl 1):78–82
Kulkarni AV, Riva-Cambrin J, Butler J et al (2013) Outcomes of CSF shunting in children: comparison of Hydrocephalus Clinical Research Network cohort with historical controls: clinical article. J Neurosurg Pediatr 12(4):334–338
Anderson IA, Saukila LF, Robins JMW et al (2018) Factors associated with 30-day ventriculoperitoneal shunt failure in pediatric and adult patients. J Neurosurg 130(1):145–153
Stone JJ, Walker CT, Jacobson M, Phillips V, Silberstein HJ (2013) Revision rate of pediatric ventriculoperitoneal shunts after 15 years. J Neurosurg Pediatr 11(1):15–19
Drake JM, Kulkarni AV, Kestle J (2009) Endoscopic third ventriculostomy versus ventriculoperitoneal shunt in pediatric patients: a decision analysis. Childs Nerv Syst 25(4):467–472
Kulkarni AV, Drake JM, Mallucci CL, Sgouros S, Roth J, Constantini S (2009) Endoscopic third ventriculostomy in the treatment of childhood hydrocephalus. J Pediatr 155(2):254–9.e1
Pindrik J, Jallo GI, Ahn ES (2013) Complications and subsequent removal of retained shunt hardware after endoscopic third ventriculostomy: case series. J Neurosurg Pediatr 11(6):722–726
Limbrick DD Jr, Baird LC, Klimo P Jr, Riva-Cambrin J, Flannery AM (2014) Pediatric hydrocephalus: systematic literature review and evidence-based guidelines. Part 4: cerebrospinal fluid shunt or endoscopic third ventriculostomy for the treatment of hydrocephalus in children. J Neurosurg Pediatr 14(Suppl 1):30–4
Kulkarni AV, Riva-Cambrin J, Browd SR et al (2014) Endoscopic third ventriculostomy and choroid plexus cauterization in infants with hydrocephalus: a retrospective Hydrocephalus Clinical Research Network study. J Neurosurg Pediatr 14(3):224–229
Marques F, Sousa JC, Brito MA et al (2017) The choroid plexus in health and in disease: dialogues into and out of the brain. Neurobiol Dis 107:32–40
Tirado-Caballero J, Rivero-Garvia M, Arteaga-Romero F, Herreria-Franco J, Lozano-Gonzalez A, Marquez-Rivas J (2020) Neuroendoscopic lavage for the management of posthemorrhagic hydrocephalus in preterm infants: safety, effectivity, and lessons learned. J Neurosurg Pediatr 1–10
Schulz M, Buhrer C, Pohl-Schickinger A, Haberl H, Thomale UW (2014) Neuroendoscopic lavage for the treatment of intraventricular hemorrhage and hydrocephalus in neonates. J Neurosurg Pediatr 13(6):626–635
Qin G, Liang Y, Xu K et al (2020) Neuroendoscopic lavage for ventriculitis: Case report and literature review. Neurochirurgie 66(2):127–132
Larroche JC (1972) Post-haemorrhagic hydrocephalus in infancy. Anatomical study. Biol Neonate 20(3):287–299
Omar AT II, Bagnas MAC, Del Rosario-Blasco KAR, Diestro JDB, Khu KJO (2018) Shunt surgery for neurocutaneous melanosis with hydrocephalus: case report and review of the literature. World Neurosurg 120:583–589
Whitelaw A (2001) Intraventricular haemorrhage and posthaemorrhagic hydrocephalus: pathogenesis, prevention and future interventions. Semin Neonatol 6(2):135–146
Lategan B, Chodirker BN, Del Bigio MR (2010) Fetal hydrocephalus caused by cryptic intraventricular hemorrhage. Brain Pathol 20(2):391–398
Hill A, Shackelford GD, Volpe JJ (1984) A potential mechanism of pathogenesis for early posthemorrhagic hydrocephalus in the premature newborn. Pediatrics 73(1):19–21
Gram M, Sveinsdottir S, Cinthio M et al (2014) Extracellular hemoglobin - mediator of inflammation and cell death in the choroid plexus following preterm intraventricular hemorrhage. J Neuroinflammation 11:200
Gram M, Sveinsdottir S, Ruscher K et al (2013) Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J Neuroinflammation 10:100
Simard PF, Tosun C, Melnichenko L, Ivanova S, Gerzanich V, Simard JM (2011) Inflammation of the choroid plexus and ependymal layer of the ventricle following intraventricular hemorrhage. Transl Stroke Res 2(2):227–231
Barichello T, Fagundes GD, Generoso JS, Elias SG, Simoes LR, Teixeira AL (2013) Pathophysiology of neonatal acute bacterial meningitis. J Med Microbiol 62(Pt 12):1781–1789
Bateman GA, Brown KM (2012) The measurement of CSF flow through the aqueduct in normal and hydrocephalic children: from where does it come, to where does it go? Child’s nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery 28(1):55–63
Oi S, Di Rocco C (2006) Proposal of “evolution theory in cerebrospinal fluid dynamics” and minor pathway hydrocephalus in developing immature brain. Child’s nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery 22(7):662–669
Oreskovic D, Rados M, Klarica M (2017) Role of choroid plexus in cerebrospinal fluid hydrodynamics. Neuroscience 354:69–87
Miyajima M, Arai H (2015) Evaluation of the production and absorption of cerebrospinal fluid. Neurol Med Chir 55(8):647–656
Lohrberg M, Wilting J (2016) The lymphatic vascular system of the mouse head. Cell Tissue Res 366(3):667–677
Brinker T, Stopa E, Morrison J, Klinge P (2014) A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 11:10
Keep RF, Jones HC (1990) A morphometric study on the development of the lateral ventricle choroid plexus, choroid plexus capillaries and ventricular ependyma in the rat. Brain Res Dev Brain Res 56(1):47–53
Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, Lancaster MA (2020) Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 369(6500)
Bothwell SW, Janigro D, Patabendige A (2019) Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS 16(1):9
Buhrley LE, Reed DJ (1972) The effect of furosemide on sodium-22 uptake into cerebrospinal fluid and brain. Exp Brain Res 14(5):503–510
Stodberg T, Magnusson M, Lesko N et al (2020) SLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia. Neurol Genet 6(4):e478
Steffensen AB, Oernbo EK, Stoica A et al (2018) Cotransporter-mediated water transport underlying cerebrospinal fluid formation. Nat Commun 9(1):2167
Gregoriades JMC, Madaris A, Alvarez FJ, Alvarez-Leefmans FJ (2019) Genetic and pharmacological inactivation of apical Na(+)-K(+)-2Cl(-) cotransporter 1 in choroid plexus epithelial cells reveals the physiological function of the cotransporter. Am J Physiol Cell Physiol 316(4):C525–C544
Strominger I, Elyahu Y, Berner O et al (2018) The choroid plexus functions as a niche for T-cell stimulation within the central nervous system. Front Immunol 9:1066
Engelhardt B, Vajkoczy P, Weller RO (2017) The movers and shapers in immune privilege of the CNS. Nat Immunol 18(2):123–131
Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B (2018) Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol 135(3):337–361
Van Hove H, Martens L, Scheyltjens I et al (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22(6):1021–1035
Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18(4):225–242
Konishi H, Kobayashi M, Kunisawa T et al (2017) Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia 65(12):1927–1943
Ivan DC, Walthert S, Berve K, Steudler J, Locatelli G (2020) Dwellers and trespassers: mononuclear phagocytes at the borders of the central nervous system. Front Immunol 11:609921
Kierdorf K, Masuda T, Jordao MJC, Prinz M (2019) Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci 20(9):547–562
Goldmann T, Wieghofer P, Jordao MJ et al (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17(7):797–805
Rodriguez-Lorenzo S, Konings J, van der Pol S et al (2020) Inflammation of the choroid plexus in progressive multiple sclerosis: accumulation of granulocytes and T cells. Acta Neuropathol Commun 8(1):9
Serot JM, Foliguet B, Bene MC, Faure GC (1997) Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. NeuroReport 8(8):1995–1998
Kaur C, Rathnasamy G, Ling EA (2016) The choroid plexus in healthy and diseased brain. J Neuropathol Exp Neurol 75(3):198–213
Praetorius J, Damkier HH (2017) Transport across the choroid plexus epithelium. Am J Physiol Cell Physiol 312(6):C673–C686
Marchetti L, Engelhardt B (2020) Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc Biol 2(1):H1–H18
Strazielle N, Creidy R, Malcus C, Boucraut J, Ghersi-Egea JF (2016) T-lymphocytes traffic into the brain across the blood-CSF barrier: evidence using a reconstituted choroid plexus epithelium. PLoS One 11(3):e0150945
Schwerk C, Tenenbaum T, Kim KS, Schroten H (2015) The choroid plexus-a multi-role player during infectious diseases of the CNS. Front Cell Neurosci 9:80
Cui J, Shipley FB, Shannon ML et al (2020) Inflammation of the embryonic choroid plexus barrier following maternal immune activation. Dev Cell 55(5):617–28 e6
Engelhardt B (2020) Maternal infection impairs fetal brain development via choroid plexus inflammation. Dev Cell 55(5):519–521
Thompson D, Sorenson J, Greenmyer J, Brissette CA, Watt JA (2020) The Lyme disease bacterium, Borrelia burgdorferi, stimulates an inflammatory response in human choroid plexus epithelial cells. PLoS One 15(7):e0234993
Ge R, Tornero D, Hirota M et al (2017) Choroid plexus-cerebrospinal fluid route for monocyte-derived macrophages after stroke. J Neuroinflammation 14(1):153
Rayasam A, Faustino J, Lecuyer M, Vexler ZS (2020) Neonatal stroke and TLR1/2 ligand recruit myeloid cells through the choroid plexus in a CX3CR1-CCR2- and context-specific manner. J Neurosci 40(19):3849–3861
Demeestere D, Libert C, Vandenbroucke RE (2015) Therapeutic implications of the choroid plexus-cerebrospinal fluid interface in neuropsychiatric disorders. Brain Behav Immun 50:1–13
Shimada A, Hasegawa-Ishii S (2021) Increased cytokine expression in the choroid plexus stroma and epithelium in response to endotoxin-induced systemic inflammation in mice. Toxicol Rep 8:520–528
Balusu S, Van Wonterghem E, De Rycke R et al (2016) Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol Med 8(10):1162–1183
Marques F, Sousa JC (2015) The choroid plexus is modulated by various peripheral stimuli: implications to diseases of the central nervous system. Front Cell Neurosci 9:136
Simpson S, Preston D, Schwerk C, Schroten H, Blazer-Yost B (2019) Cytokine and inflammatory mediator effects on TRPV4 function in choroid plexus epithelial cells. Am J Physiol Cell Physiol 317(5):C881–C893
Medzhitov R (2007) TLR-mediated innate immune recognition. Semin Immunol 19(1):1–2
Coorens M, Schneider VAF, de Groot AM et al (20) Cathelicidins inhibit Escherichia coli-induced TLR2 and TLR4 activation in a viability-dependent manner. J Immunol (Baltimore, Md : 1950) 199(4):1418–1428
Marques F, Sousa JC, Coppola G et al (2009) Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab 29(5):921–932
Mottahedin A, Joakim Ek C, Truve K, Hagberg H, Mallard C (2019) Choroid plexus transcriptome and ultrastructure analysis reveals a TLR2-specific chemotaxis signature and cytoskeleton remodeling in leukocyte trafficking. Brain Behav Immun 79:216–227
Miyake K (2007) Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol 19(1):3–10
Ehrchen JM, Sunderkotter C, Foell D, Vogl T, Roth J (2009) The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol 86(3):557–566
Yang B, Zhou Z, Li X, Niu J (2016) The effect of lysophosphatidic acid on Toll-like receptor 4 expression and the nuclear factor-κB signaling pathway in THP-1 cells. Mol Cell Biochem 422(1–2):41–49
Kwon MS, Woo SK, Kurland DB et al (2015) Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int J Mol Sci 16(3):5028–5046
Fang H, Wu Y, Huang X et al (2011) Toll-like receptor 4 (TLR4) is essential for Hsp70-like protein 1 (HSP70L1) to activate dendritic cells and induce Th1 response. J Biol Chem 286(35):30393–30400
Tsan MF, Gao B (2004) Endogenous ligands of Toll-like receptors. J Leukoc Biol 76(3):514–519
Gao C, Du H, Hua Y, Keep RF, Strahle J, Xi G (2014) Role of red blood cell lysis and iron in hydrocephalus after intraventricular hemorrhage. J Cereb Blood Flow Metab 34(6):1070–1075
Fejes Z, Erdei J, Pocsi M et al (2020) Elevated pro-inflammatory cell-free microRNA levels in cerebrospinal fluid of premature infants after intraventricular hemorrhage. Int J Mol Sci 21(18)
Berkes J, Viswanathan VK, Savkovic SD, Hecht G (2003) Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut 52(3):439–451
Wilson R, Alton E, Rutman A et al (1987) Upper respiratory tract viral infection and mucociliary clearance. Eur J Respir Dis 70(5):272–279
Doyle WJ, Skoner DP, Hayden F, Buchman CA, Seroky JT, Fireman P (1994) Nasal and otologic effects of experimental influenza A virus infection. Ann Otol Rhinol Laryngol 103(1):59–69
Karimy JK, Kahle KT, Kurland DB, Yu E, Gerzanich V, Simard JM (2015) A novel method to study cerebrospinal fluid dynamics in rats. J Neurosci Methods 241:78–84
Liu G, Mestre H, Sweeney AM et al (2020) Direct Measurement of Cerebrospinal Fluid Production in Mice. Cell Rep 33(12):108524
Chen Z, Jalabi W, Shpargel KB et al (2012) Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 32(34):11706–11715
Demeestere D, Libert C, Vandenbroucke RE (2015) Clinical implications of leukocyte infiltration at the choroid plexus in (neuro)inflammatory disorders. Drug Discov Today 20(8):928–941
Kleine TO, Benes L (2006) Immune surveillance of the human central nervous system (CNS): different migration pathways of immune cells through the blood-brain barrier and blood-cerebrospinal fluid barrier in healthy persons. Cytometry A 69(3):147–151
Cox KH, Cox ME, Woo-Rasberry V, Hasty DL (2012) Pathways involved in the synergistic activation of macrophages by lipoteichoic acid and hemoglobin. PLoS One 7(10):e47333
Wang YC, Zhou Y, Fang H et al (2014) Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol 75(6):876–889
Alessi DR, Zhang J, Khanna A, Hochdorfer T, Shang Y, Kahle KT (2014) The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7(334):re3
Thastrup JO, Rafiqi FH, Vitari AC et al (2012) SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J 441(1):325–337
Yan Y, Nguyen H, Dalmasso G, Sitaraman SV, Merlin D (2007) Cloning and characterization of a new intestinal inflammation-associated colonic epithelial Ste20-related protein kinase isoform. Biochim Biophys Acta 1769(2):106–116
Yan Y, Merlin D (2008) Ste20-related proline/alanine-rich kinase: a novel regulator of intestinal inflammation. World J Gastroenterol 14(40):6115–6121
Yan Y, Dalmasso G, Nguyen HT, Obertone TS, Sitaraman SV, Merlin D (2009) Ste20-related proline/alanine-rich kinase (SPAK) regulated transcriptionally by hyperosmolarity is involved in intestinal barrier function. PLoS One 4(4):e5049
Yan Y, Laroui H, Ingersoll SA et al (2011) Overexpression of Ste20-related proline/alanine-rich kinase exacerbates experimental colitis in mice. J Immunol 187(3):1496–1505
Zhang Y, Viennois E, Xiao B et al (2013) Knockout of Ste20-like proline/alanine-rich kinase (SPAK) attenuates intestinal inflammation in mice. Am J Pathol 182(5):1617–1628
Lin TJ, Yang SS, Hua KF, Tsai YL, Lin SH, Ka SM (2016) SPAK plays a pathogenic role in IgA nephropathy through the activation of NF-kappaB/MAPKs signaling pathway. Free Radic Biol Med 99:214–224
Polek TC, Talpaz M, Spivak-Kroizman T (2006) The TNF receptor, RELT, binds SPAK and uses it to mediate p38 and JNK activation. Biochem Biophys Res Commun 343(1):125–134
Wu CP, Huang KL, Peng CK, Lan CC (2020) Acute Hyperglycemia Aggravates Lung Injury via Activation of the SGK1-NKCC1 Pathway. Int J Mol Sci 21(13)
Hung CM, Peng CK, Yang SS, Shui HA, Huang KL (2020) WNK4-SPAK modulates lipopolysaccharide-induced macrophage activation. Biochem Pharmacol 171:113738
Piechotta K, Lu J, Delpire E (2002) Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277(52):50812–50819
Xu H, Fame RM, Sadegh C et al (2021) Choroid plexus NKCC1 mediates cerebrospinal fluid clearance during mouse early postnatal development. Nat Commun 12(1):447
Walcott BP, Iorgulescu JB, Stapleton CJ, Kamel H (2015) Incidence, Timing, and Predictors of Delayed Shunting for Hydrocephalus After Aneurysmal Subarachnoid Hemorrhage. Neurocrit Care 23(1):54–58
Sharma D, Shah I, Patel S (2016) Late onset hydrocephalus in children with tuberculous meningitis. J Family Med Prim Care 5(4):873–874
Schiefenhövel F, Immig K, Prodinger C, Bechmann I (2017) Indications for cellular migration from the central nervous system to its draining lymph nodes in CD11c-GFP+ bone-marrow chimeras following EAE. Exp Brain Res 235(7):2151–2166
Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J (2017) Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest 127(9):3210–3219
Louveau A, Smirnov I, Keyes TJ et al (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523(7560):337–341
Eming SA, Hammerschmidt M, Krieg T, Roers A (2009) Interrelation of immunity and tissue repair or regeneration. Semin Cell Dev Biol 20(5):517–527
Liu Q, Zhou YH, Yang ZQ (2016) The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol 13(1):3–10
Lawrence SM, Corriden R, Nizet V (2020) How Neutrophils Meet Their End. Trends Immunol 41(6):531–544
McAllister JP, Guerra MM, Ruiz LC et al (2017) Ventricular zone disruption in human neonates with intraventricular hemorrhage. J Neuropathol Exp Neurol 76(5):358–375
Lewin JJ 3rd, Cook AM, Gonzales C et al (2019) Current practices of intraventricular antibiotic therapy in the treatment of meningitis and ventriculitis: results from a multicenter retrospective cohort study. Neurocrit Care 30(3):609–616
Rice TW, Wheeler AP, Bernard GR et al (2010) A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med 38(8):1685–1694
Liu SF, Ye X, Malik AB (1999) Inhibition of NF- B activation by pyrrolidine dithiocarbamate prevents in vivo expression of proinflammatory genes. Circulation 100(12):1330–1337
Hu Y, Wang Z, Pan S et al (2017) Melatonin protects against blood-brain barrier damage by inhibiting the TLR4/ NF-κB signaling pathway after LPS treatment in neonatal rats. Oncotarget 8(19):31638–31654
Robinson S, Conteh FS, Oppong AY et al (2018) Extended combined neonatal treatment with erythropoietin plus melatonin prevents posthemorrhagic hydrocephalus of prematurity in rats. Front Cell Neurosci 12:322
Gu C, Hao X, Li J, Hua Y, Keep RF, Xi G (2019) Effects of minocycline on epiplexus macrophage activation, choroid plexus injury and hydrocephalus development in spontaneous hypertensive rats. J Cereb Blood Flow Metab 39(10):1936–1948
Erker T, Brandt C, Tollner K et al (2016) The bumetanide prodrug BUM5, but not bumetanide, potentiates the antiseizure effect of phenobarbital in adult epileptic mice. Epilepsia
Pressler RM, Boylan GB, Marlow N et al (2015) Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): an open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol 14(5):469–477
Lemonnier E, Degrez C, Phelep M et al (2012) A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry 2:e202
Lemonnier E, Ben-Ari Y (2010) The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr 99(12):1885–1888
Lemonnier E, Villeneuve N, Sonie S et al (2017) Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry 7(3):e1056
Zhang J, Bhuiyan MIH, Zhang T et al (2020) Modulation of brain cation-Cl(-) cotransport via the SPAK kinase inhibitor ZT-1a. Nat Commun 11(1):78
Funding
KTK is supported by the NIH (RO1NS109358-04) and the Hydrocephalus Association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Robert, S.M., Reeves, B.C., Marlier, A. et al. Inflammatory hydrocephalus. Childs Nerv Syst 37, 3341–3353 (2021). https://doi.org/10.1007/s00381-021-05255-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-021-05255-z