
Abstract
Introduction
Arachnoid cysts are commonly considered congenital lesions, but this has not been proven. With the development of neuroimaging and DNA testing technology, more cases of familial arachnoid cysts have been reported. Herein, we review such cases.
Materials and methods
The PubMed, Embase, and Web of Science databases were searched for case reports of arachnoid cysts published through April 2018. Case reports were included only if two or more related patients were diagnosed with an arachnoid cyst by neuroimaging or intraoperatively. For each report, the following data were extracted: first author name, date of publication, number of families, number of patients, location of the arachnoid cysts, patient age, patient sex, and genetic mutations and associated disease.
Results
Our searches identified 33 case reports involving 35 families and 115 patients. The locations of arachnoid cysts were similar in 25 of the 35 families. Spinal extradural arachnoid cysts were reported most often, followed by arachnoid cysts in the middle fossa and posterior fossa. A left-sided predominance was noticed for arachnoid cysts of the middle fossa. Mutation of the FOXC2 gene was reported most often, and arachnoid cysts may be associated with mutations on chromosome 16.
Conclusions
Although the origin of arachnoid cysts is believed to have a genetic component by some researchers, the genes associated with arachnoid cysts remain unknown. Unfortunately, the evidence remains insufficient.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
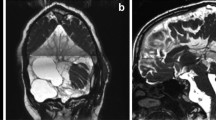
Arachnoid cysts (ACs) are collections of fluid similar to cerebrospinal fluid within the arachnoid membranes. The detection of asymptomatic ACs has increased with the development of neuroimaging techniques [1]. The most common location for ACs is the middle fossa, although they may occur in any part of the nervous system where arachnoid exists [2, 3]. Headache and epilepsy are reported as the main symptoms in pediatric patients, and vertigo/nausea is the second most common complaint [4]. Computed tomography and magnetic resonance imaging findings are consistent with the density mass of cerebrospinal fluid (CSF), without enhancement [5]. ACs are often found incidentally on radiological examination and can be treated conservatively. However, cyst excision, fenestration, or shunting has been applied in symptomatic cases [6].
ACs are also considered congenital by most authors [3, 7]. Intracranial ACs specifically are most commonly found in the temporal fossa and in male patients, while a left side predominance of ACs in the middle fossa has been reported. These findings have been interpreted as indicating a genetic component in the origin of ACs [3]. Cases of familial ACs also have been reported [8, 9] with the development of genetic testing technology. We therefore prepared this review to summarize new findings related to familial ACs.
Materials and methods
We performed a review of the English literature published before April 2018 by searching the PubMed, Embase, and Web of Science databases. There was no lower date limit. The primary literature search was performed using the following terms: “arachnoid cyst,” “leptomeningeal cyst,” “arachnoid diverticula,” and one of the “twin,” “sibling,” “brother,” “sister,” “father,” “mother,” “family,” “familial,” “gene,” “karyotype,” and “mutation.” We also manually reviewed the reference lists of the articles included in the analysis to identify additional relevant studies. Searches were performed independently by two members of the study team. We included all case reports in English whether involving intracranial or spinal ACs, and duplicate cases were excluded. The report was excluded if the diagnosis of AC was not confirmed by neuroimaging or intraoperatively. For each report included, data including the first author’s name, date of publication, number of families, number of patients, location of ACs, patient age, patient sex, associated gene mutations, and associated diseases were extracted.
Results
A total of 546 articles were identified through database searches, and 170 reports were excluded as duplicates. Upon review of the titles and abstracts, another 322 reports were excluded for not fulfilling the inclusion criteria. Finally, 23 more reports were excluded after review of the full text because they did not report familial ACs. Two reports were added after review of the reference lists of the remaining reports, and thus, a total of 33 reports were included in the present analysis.
Epidemiological features
The 33 included reports were published between 1968 and 2017 and involved 35 families and 115 patients. 32 reports noted the patients’ gender, and among these reports, there were 46 males and 51 females. Only 30 reports provided the patients’ ages, and among these, there were 57 children (< 18 years) and 45 adults (≥ 18 years) (age at onset was analyzed, but if not reported, then age at examination was used). The specific data for each included study are listed in Table 1.
The locations of the ACs were also extracted for all patients from the 35 families (Table 2). The ACs of patients within the same family were in similar locations (some in the same locations, some on different sides, etc.) for the 25 families. Specifically, 9 families had spinal extradural ACs, 8 families had middle fossa ACs, and another 7 families had posterior fossa ACs. Among all 115 patients, 26 had middle fossa ACs (13 (50%) on the left side, 10 (38%) on both sides, and only 3 (12%) on the right side), 10 had bilateral middle fossa ACs, 13 had left middle fossa ACs, and only 3 had right fossa ACs.
Genetics of ACs
The development of gene technology has resulted in the increased application of DNA testing in cases of familial conditions. Ten of the 33 reports provided findings of genetic testing (Table 3). A FOXC2 mutation was reported in two studies for three families with ACs, and all of the ACs in these patients were located in the spine. Some authors reported other syndromes in cases of ACs, but further karyotyping was not performed. The findings along with the location of the ACs are listed in Table 4. Specifically, lymphedema-distichiasis syndrome, glutaric aciduria type-1, autosomal dominant polycystic kidney disease, and oculopharyngeal muscular dystrophy were reported to co-exist with ACs.
It is assumed that formation of an AC somehow is governed by some genetic mechanisms, based on the sidedness and gender preponderances [2, 3, 41]. Aarhus et al. [42] performed a high-resolution mRNA microarray analysis to identify differences in gene expression between the normal arachnoid membrane and the cyst membrane. They only found that 9 of 33,096 genes showed differential expression between the two tissues: ASGR1, DPEP2, SOX9, SHROOM3, A2BP1, ATP10D, TRIML1, NMU, and BEND5. Future genetic studies of patients with familial ACs, even monozygotic twins with ACs, would provide more evidence for the genetic basis of this condition.
Discussion
The arachnoid cyst was first described by Bright in 1829 [43]. Likely, the first report of familial ACs was published in 1967 by Chynn et al. [40] who described congenital spinal extradural cysts in two siblings. The first four reports [37,38,39,40] found in the literature all involved cases of spinal extradural cysts, because modern neuroimaging technology was not yet available. Increasing numbers of familial cases of intracranial ACs have been reported in the last 30 years with the development of computed tomography (CT) and magnetic resonance imaging (MRI), and reports in recent years have begun to reveal relevant gene mutations.
Some authors reported a greater prevalence of ACs in males versus females and a greater prevalence of ACs in the middle fossa versus other locations [1,2,3, 44]. In our review of familial AC cases, we did not find a significant male predilection, and the middle fossa was not the most common location of ACs in this series. The discrepancies between our findings and those of previous studies may be due to several factors. Notably, the numbers of affected patients in different families have varied considerably, with Orlacchio et al. [9] reporting a family in which 18 members had ACs located at the cerebellopontine angle compared to fewer than five affected patients in most families described in other reports. At the same time, in almost 76% of families, ACs were located at similar sites among family members. After reviewing the families for which this was true, we found that spinal extradural cysts have been reported most, followed by ACs in the middle fossa and posterior fossa. A left-sided predominance in ACs of the middle fossa was also noticed, which was consistent with previous studies [1,2,3, 45].
Only 10 of the 33 included reports (11 of 35 families) clearly identified an associated genetic mutation. Notably, a FOXC2 mutation was reported twice in three families, although the authors of one study reported the mutation as a nonsense mutation [18] while the others reported a heterozygous FOXC2 loss-of-function mutation [14]. Also, lymphedema-distichiasis syndrome was believed to be associated with ACs in two reports [22, 37], and this syndrome is associated with the mutation of FOXC2 [18]. The FOXC2 gene is located at 16q24.1 and belongs to the forkhead family of transcription factors, which is characterized by a distinct DNA-binding forkhead domain. The specific function of the FOXC2 protein has yet to be determined, although several studies have shown that it may play a role in the development of mesenchymal tissues [46]. Arriola et al. [23] reported two brothers with intracranial ACs, for which karyotyping revealed a deletion in the pericentromeric heterochromatic region of the long arm of chromosome 16 (16qh-) in both brothers. Alehan et al. [26] reported cases of a father and daughter who each had a posterior fossa AC and asymptomatic autosomal dominant polycystic kidney disease (ADPKD), but they did not clearly determine the type of ADPKD. However, mutations in PKD1 can cause ADPKD type 1, and the PKD1 gene is located on chromosome 16 [47, 48]. Together, these findings indicate that we should pay attention to mutations of chromosome 16 in cases of ACs in the future. Koenigstein et al. [13] reported a pair of twins with a novel homozygous mutation in the GPSM2 gene. Both the girl and boy had an interhemispheric AC, but the patients were dizygotic twins. Mirror-image ACs in a pair of monozygotic twins were reported by Helland and Wester [21] and Zhou et al. [17], but unfortunately, karyotyping was not performed.
Conclusion
Although the exact gene(s) responsible for familial cases of ACs remain unclear at present, the continued development and use of neuroimaging and new genetic testing techniques will provide further insight into the mechanisms underlying this condition.
References
Al-Holou WN, Yew AY, Boomsaad ZE, Garton HJ, Muraszko KM, Maher CO (2010) Prevalence and natural history of arachnoid cysts in children. J Neurosurg Pediatr 5:578–585
Al-Holou WN, Terman S, Kilburg C, Garton HJ, Muraszko KM, Maher CO (2013) Prevalence and natural history of arachnoid cysts in adults. J Neurosurg 118:222–231
Helland CA, Lund-Johansen M, Wester K (2010) Location, sidedness, and sex distribution of intracranial arachnoid cysts in a population-based sample. J Neurosurg 113:934–939
Mazurkiewicz-Beldzinska M, Dilling-Ostrowska E (2002) Presentation of intracranial arachnoid cysts in children: correlation between localization and clinical symptoms. Med Sci Monit 8:CR462–CR465
Wikström J (2018) Chapter 14 - Radiological workup, CT, MRI A2 - Western, Knut. Arachnoid cysts. Academic Press, pp 173–185
Gangemi M, Seneca V, Colella G, Cioffi V, Imperato A, Maiuri F (2011) Endoscopy versus microsurgical cyst excision and shunting for treating intracranial arachnoid cysts. J Neurosurg Pediatr 8:158–164
Cress M, Kestle JR, Holubkov R, Riva-Cambrin J (2013) Risk factors for pediatric arachnoid cyst rupture/hemorrhage: a case-control study. Neurosurgery 72:716–722 discussion 722
Furey CG, Timberlake AT, Nelson-Williams C, Duran D, Li P, Jackson EM, Kahle KT (2017) Xp22.2chromosomal duplication in familial intracranial arachnoid cyst. JAMA Neurology 74:1503–1504
Orlacchio A, Gaudiello F, Totaro A, Floris R, St George-Hyslop PH, Bernardi G, Kawarai T (2004) A new SPG4 mutation in a variant form of spastic paraplegia with congenital arachnoid cysts. Neurology 62:1875–1878
Menezes AH, Hitchon PW, Dlouhy BJ (2017) Symptomatic spinal extradural arachnoid cyst with cord compression in a family: case report. J Neurosurg Spine 27:341–345
Cuny ML, Pallone M, Piana H, Boddaert N, Sainte-Rose C, Vaivre-Douret L, Piolino P, Puget S (2017) Neuropsychological improvement after posterior fossa arachnoid cyst drainage. Child's Nerv Syst 33:135–141
Kurt S, Cevik B, Aksoy D, Sahbaz EI, Gundogdu Eken A, Basak AN (2016) Atypical features in a large Turkish family affected with Friedreich ataxia. Case Rep Neurol Med 2016:4515938
Koenigstein K, Gramsch C, Kolodziej M, Neubauer BA, Weber A, Lechner S, Hahn A (2016) Chudley-McCullough syndrome: variable clinical picture in twins with a novel GPSM2 mutation. Neuropediatrics 47:197–201
Ogura Y, Yabuki S, Iida A, Kou I, Nakajima M, Kano H, Shiina M, Kikuchi S, Toyama Y, Ogata K, Nakamura M, Matsumoto M, Ikegawa S (2013) FOXC2 mutations in familial and sporadic spinal extradural arachnoid cyst. PLoS One 8:e80548
Degerliyurt A, Ceylaner G, Kocak H, Bilginer Gurbuz B, Cihan BS, Rizzu P, Ceylaner S (2012) A new family with autosomal dominant porencephaly with a novel Col4A1 mutation. Are arachnoid cysts related to Col4A1 mutations? Genet Counsel (Geneva Switzerland) 23:185–193
Bayrakli F, Okten AI, Kartal U, Menekse G, Guzel A, Oztoprak I, Pinarbasi E, Kars HZ (2012) Intracranial arachnoid cyst family with autosomal recessive trait mapped to chromosome 6q22.31-23.2. Acta Neurochir 154:1287–1292
Zhou J-Y, Pu J-L, Chen S, Hong Y, Ling C-H, Zhang J-M (2011) Mirror-image arachnoid cysts in a pair of monozygotic twins: a case report and review of the literature. Int J Med Sci 8:402–405
Sanchez-Carpintero R, Dominguez P, Nunez MT, Patino-Garcia A (2010) Spinal extradural arachnoid cysts in lymphedema-distichiasis syndrome. Genet Med 12:532–535
Bilguvar K, Ozturk AK, Bayrakli F, Guzel A, DiLuna ML, Bayri Y, Tatli M, Tekes S, Arlier Z, Yasuno K, Mason CE, Lifton RP, State MW, Gunel M (2009) The syndrome of pachygyria, mental retardation, and arachnoid cysts maps to 11p15. Am J Med Genet A 149a:2569–2572
Guzel A, Tatli M, Bilguvar K, Diluna ML, Bakkaloglu B, Ozturk AK, Bayrakli F, Gunel M (2007) Apparently novel genetic syndrome of pachygyria, mental retardation, seizure, and arachnoid cysts. Am J Med Genet A 143a:672–677
Helland CA, Wester K (2007) Monozygotic twins with mirror image cysts: indication of a genetic mechanism in arachnoid cysts? Neurology 69:110–111
Yabuki S, Kikuchi S, Ikegawa S (2007) Spinal extradural arachnoid cysts associated with distichiasis and lymphedema. Am J Med Genet A 143a:884–887
Arriola G, de Castro P, Verdu A (2005) Familial arachnoid cysts. Pediatr Neurol 33:146–148
Sinha S, Brown JI (2004) Familial posterior fossa arachnoid cyst. Child's Nerv Syst 20:100–103
Jadeja KJ, Grewal RP (2003) Familial arachnoid cysts associated with oculopharyngeal muscular dystrophy. J Clin Neurosci 10:125–127
Alehan FK, Gurakan B, Agildere M (2002) Familial arachnoid cysts in association with autosomal dominant polycystic kidney disease. Pediatrics 110:e13
Suzuki H, Takanashi J, Sugita K, Barkovich AJ, Kohno Y (2002) Retrocerebellar arachnoid cysts in siblings with mental retardation and undescended testis. Brain Dev 24:310–313
Hendriks YM, Laan LA, Vielvoye GJ, van Haeringen A (1999) Bilateral sensorineural deafness, partial agenesis of the corpus callosum, and arachnoid cysts in two sisters. Am J Med Genet 86:183–186
Tolmie JL, Day R, Fredericks B, Galea P, Moffett AW (1997) Dominantly inherited cerebral dysplasia: arachnoid cyst associated with mild mental handicap in a mother and her son. J Med Genet 34:1018–1020
Jamjoom ZA, Okamoto E, Jamjoom AH, al-Hajery O, Abu-Melha A (1995) Bilateral arachnoid cysts of the sylvian region in female siblings with glutaric aciduria type I. Report of two cases. J Neurosurg 82:1078–1081
Ferlini A, Ragno M, Gobbi P, Marinucci C, Rossi R, Zanetti A, Milan M, Camera G, Calzolari E (1995) Hydrocephalus, skeletal anomalies, and mental disturbances in a mother and three daughters: a new syndrome. Am J Med Genet 59:506–511
Aiba T, Koike T, Takeda N, Tanaka R (1995) Intracranial cavernous malformations and skin angiomas associated with middle fossa arachnoid cyst: a report of three cases. Surg Neurol 43:31–33 discussion 34
Martinezlage JF, Casas C, Fernandez MA, Puche A, Costa TR, Poza M (1994) Macrocephaly, dystonia, and bilateral temporal arachnoid cysts - glutaric aciduria type-1. Child's Nerv Syst 10:198–203
Pomeranz S, Constantini S, Lubetzki-Korn I, Amir N (1991) Familial intracranial arachnoid cysts. Child's Nerv Syst 7:100–102
Wilson WG, Deponte KA, McIlhenny J, Dreifuss FE (1988) Arachnoid cysts in a brother and sister. J Med Genet 25:714–715
Handa J, Okamoto K, Sato M (1981) Arachnoid cyst of the middle cranial fossa: report of bilateral cysts in siblings. Surg Neurol 16:127–130
Schwartz JF, O'Brien MS, Hoffman JC Jr (1980) Hereditary spinal arachnoid cysts, distichiasis, and lymphedema. Ann Neurol 7:340–343
Aarabi B, Pasternak G, Hurko O, Long DM (1979) Familial intradural arachnoid cysts. Report of two cases. J Neurosurg 50:826–829
Bergland RM (1968) Congenital intraspinal extradural cyst. Report of three cases in one family. J Neurosurg 28:495–499
Chynn KY (1967) Congenital spinal extradural cyst in two siblings. Am J Roentgenol Radium Therapy, Nucl Med 101:204–215
Wester K (1999) Peculiarities of intracranial arachnoid cysts: location, sidedness, and sex distribution in 126 consecutive patients. Neurosurgery 45:775–779
Aarhus M, Helland CA, Lund-Johansen M, Wester K, Knappskog PM (2010) Microarray-based gene expression profiling and DNA copy number variation analysis of temporal fossa arachnoid cysts. Cerebrospinal Fluid Res 7:6
Wester K (2018) Arachnoid cysts—historical perspectives and controversial aspects. Arachnoid cysts. Academic press, pp 3–16
Spacca B, Kandasamy J, Mallucci CL, Genitori L (2010) Endoscopic treatment of middle fossa arachnoid cysts: a series of 40 patients treated endoscopically in two centres. Child's Nerv Syst 26:163–172
Weber F, Knopf H (2006) Incidental findings in magnetic resonance imaging of the brains of healthy young men. J Neurol Sci 240:81–84
Kume T (2012) The role of FoxC2 transcription factor in tumor angiogenesis. J Oncol 2012:204593
Simms RJ (2016) Autosomal dominant polycystic kidney disease. BMJ 352:i679
Grantham JJ, Mulamalla S, Swenson-Fields KI (2011) Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol 7:556–566
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Qin, X., Wang, Y., Xu, S. et al. Familial arachnoid cysts: a review of 35 families. Childs Nerv Syst 35, 607–612 (2019). https://doi.org/10.1007/s00381-019-04060-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-019-04060-z