
Abstract
Introduction
Congenital infantile fibrosarcoma (CIFS) is a soft tissue sarcoma of infants mainly involving lower extremities usually presenting during the first year of life. A subset of cases occur in the head and neck, but scalp involvement is exceptionally rare.
Patients and Methods
We report clinicopathological features of three cases of CIFS involving the scalp diagnosed between 2011 and 2012.
Results
The ages of the three patients at the time of diagnosis ranged from 12 to 90 days (mean 48 days). All were males and presented with scalp swelling at birth which grew rapidly in size. The tumor was located in the left temporal region in two cases and the right temporoparietal region in one case. On imaging, underlying bone involvement was noted in two cases. The mean size of the resected tumors was 8 cm. All cases exhibited a cellular tumor arranged in sheets of uniform oval to spindle cells, increased mitosis, and hemangiopericytoma-like vessels. All patients are alive after a mean follow-up of 39.6 months. Recurrence was seen in one case due to incomplete excision. No metastasis was seen in any of the cases.
Conclusion
CIFS of the scalp is rare and carries a good prognosis. Underlying bone erosion is rare but was noted seen in two of our cases. A male predominance was seen in our cases.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Congenital infantile fibrosarcoma (CIFS) is a soft tissue sarcoma which usually involves the extremities and typically occurs in infants, presenting during the first year of life as a rapidly growing mass [1]. Almost half of all infantile fibrosarcoma cases are congenital [1]. CIFS of the scalp is very rare and only three cases have been reported in the literature [2–4]. Herein, we report three cases of congenital fibrosarcoma presenting as scalp mass. One case in the current series was previously reported by Brohi et al. [5].
Material and methods
Three cases of CIFS of the scalp, reported between 2011 and 2012, were retrieved from the surgical pathology files of the Department of Pathology and Laboratory Medicine, Aga Khan University (AKU) Hospital, Karachi, Pakistan, and were reviewed. One case was operated in our institution, while the other two cases were operated elsewhere, and specimens were sent to us for histopathological examination. Informed consent was obtained from patient’s guardian in all cases. All specimens were fixed in 10 % buffered formalin and processed routinely for paraffin embedding. Sections of 5-μm thickness were cut and stained with hematoxylin and eosin (H&E). Immunohistochemistry was performed by ready-to-use (RTU) Envision system using the following antibodies: desmin, MyoD1, CD99, LCA, Tdt, CKAE1/AE3, S100, CD31, CD34, alpha smooth muscle actin, GFAP, EMA, and vimentin.
Results
Clinical, radiological, and gross features
Case 1
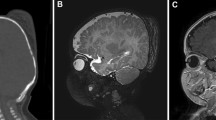
A male infant was found to have a rapidly progressive scalp swelling since birth and was seen by a neurosurgeon 2 weeks after birth. The infant was otherwise normal. The swelling was over the left temporoparietal region, firm in consistency and was immobile, with the overlying unremarkable skin inseparable from the lump. Neurological examination was unremarkable. A CT scan was carried out which showed a large extra-axial lesion eroding the bone and causing mass effect on the underlying brain (Fig. 1a). Patient was operated and the tumor was found to be moderately vascular and well circumscribed. It had eroded the bone and was involving the dura but was separable from the underlying brain by an arachnoid plane. The tumor was completely excised along with surrounding bony margins and dural substitute was used to repair the defect. Scalp was closed by a rotational flap.

a, b CT scan images (cases 1 and 2); a large mass in the left temporal region of the scalp with involvement of the underlying bone and compression of the brain parenchyma
Case 2
A newborn male child was referred to the neurosurgical service at another hospital for a large scalp lump. A CT scan of the lump showed a scalp lesion beneath the skin with involvement of the underlying bone and an intracranial component with brain tissue involvement (Fig. 1b). The child underwent local excision. Exact operative findings and details of reconstructive procedure were not available. At our institute, specimen was received as multiple soft tissue pieces and no bone was included.
Case 3
A newborn male child was referred to our institution with a large right-sided temporoparietal lump. The lump was fairly large with respect to the child’s head and appeared multilobulated, firm to hard, vascular, and fixed to the underlying bone and overlying skin. The child had no other abnormalities on either physical or neurological examination. MRI of the lump showed it to be heterogenous, within soft tissues, and not involving the bone, hypointense on T1-weighted images, hyperintense on T2-weighted images with significant flow voids and showing intense contrast enhancement. An incisional biopsy was carried out which showed a spindle cell lesion which could not be further classified as material was limited and immunohistochemical stains performed were noncontributory. While awaiting the biopsy result, the family reported significant increase in lump size and the overlying skin started to ulcerate (Fig. 2a, b). The mass was excised with the overlying scalp and multiple intra-operative samples were sent for frozen section to confirm tumor free edges of resection. Further resection had to be stopped in the depths of the infratemporal fossa, despite positive frozen sections. The underlying bone appeared intact. The large temporoparietal defect was covered with an occipital artery-based rotational flap, and the resulting occipital defect was covered with an autologous split thickness skin graft. The child made an uneventful recovery and post-operative MRI scans showed no residual tumor.

a Clinical photographs of case 3 showing massive scalp tumor, lateral view. b MRI image (case 3), demonstrating an abnormal signal intensity heterogenous lesion within the soft tissues of right side of the scalp with intact underlying bone
Histological findings
The size of the resected tumor fragments of these three cases measured in size from 6 to 9.5 cm (mean 8 cm). Case 1 is comprised of both tumor as well as bony fragments. Histological examination of all cases revealed an infiltrating cellular tumor composed of solid sheets of uniform oval to spindle cells arranged in fascicles (Fig. 3a, b). Nuclear atypia was mild, but brisk mitotic activity was seen ranging from 26/10HPF to 41/10HPF (mean 33/10HPF).

a Sheets of relatively uniform oval to spindle cells arranged in sheets with a prominent hemangiopericytoma-like vascular pattern (H&E, magnification ×40). b Cells showing mild nuclear pleomorphism along with mitoses. Sprinkling of lymphocytes is also visible (H&E, magnification ×400). c Tumor infiltrating the bone (case 1) (H&E, magnification ×40). d Tumor cells demonstrate diffuse positivity for vimentin immunostain (magnification ×100)
Focal loose cellular areas were also seen. Scattered hemangiopericytoma-like vessels were seen in all cases but were prominent in case 1 (Fig. 3a). Sprinkling of mature lymphocytes were seen in all cases (Fig. 3b). In case 1, tumor infiltration into the bone and dura was noted (Fig. 3c).
Infiltration of the skeletal muscle and adipose tissue was seen in cases 2 and 3, respectively.
Immunohistochemical stain vimentin was positive in all cases (Fig. 3d). Focal CD31 positivity was observed in case 1. Immunohistochemical stains desmin, Myo D1, CD99, LCA, Tdt, CD34, S100, actin, and GFAP were negative in all cases. LCA highlighted the scattered mature lymphocytes. Based on morphology and exclusion of all other pediatric mesenchymal tumors, a diagnosis of congenital infantile fibrosarcoma was rendered. Molecular analysis of ETV6 was not performed since this test is not available in our lab.
Follow-up
Follow-up of the patients in our series ranged from 31 to 51 months (mean, 39.6 months). All patients underwent surgical resection of the tumor. The first two patients were operated elsewhere and received no additional treatment (Table 1). Patient 1 (case 1) is alive and free of disease 51 months after surgery. In case 2, tumor was incompletely excised without removal of bone which was infiltrated by the tumor on CT scan. Patient developed a swelling at the surgical site within 3 months, which was not resected again. Patient has stable disease and has no metastasis. Patient 3 (case 3) showed no residual tumor, but due to positive surgical margins, he received 4 cycles of chemotherapy with cyclophosphamide, dactinomycin, and vincristine.
Clinical and radiological follow-up of patient 3 at 31 months did not show any local or systemic tumor recurrence. No metastases were detected in any of the other two cases.
Discussion
Infantile fibrosarcoma is a relatively rare sarcoma that histologically bears some resembles to adult fibrosarcoma. It is a distinct entity because of specific molecular translocation and different clinical course [6]. In 30 to 40 % of cases, the tumor is present at birth [7]. The tumor occurs more commonly in males with a male to female ratio of 3:2 [8]. This tumor typically presents as a painless rapidly growing large mass of distal and proximal extremities, involving the hand, wrist, forearm, feet, ankle, and knee in 60 % of cases [9]. The head and neck are involved in 10 % of cases [9]. Infantile fibrosarcoma is usually highly vascularized and occasionally causes ulceration of the overlying skin thus mimicking a vascular tumor [8]. Due to ulceration of skin and high vascularity on MRI, the tumor in case 3 in our series was radiologically interpreted as hemangioma.
Congenital infantile fibrosarcoma of the scalp is very rare, and to the best of our knowledge, excluding the cases in our series, only three cases have been previously published [2–4] (Table 1). The examples of pediatric primary scalp fibrosarcoma cited by Muzmadar et al. [3] were not true CIFS, as the youngest patient in one series was 17 years old, while age was not specified in other. Another case was an example of a scalp neurofibrosarcoma, which is the old name of malignant peripheral nerve sheath tumor. Interestingly, all the previously reported cases of CIFS were male patients with age ranging from 1 to 6 months at the time of diagnosis [2–4]. The age of the patients in our series ranged from 12 to 90 days (mean 48 days), and all patients in our series are also male. The size of tumor in previously reported cases ranged from 5 to 21 cm (mean 13.3 cm) [2–4]. Tumor size ranged from 6 to 9.5 cm (mean 8 cm) in our cases. Clinically, the mass was initially slowly growing but later rapidly growing in the case reported by Chaudhari et al. [2] and Kumar et al. [4]. Muzmadar et al. [3], however, reported rapid growth from the start. Rapid growth was seen in two of our cases and initial slow growth and later rapid growth in the third case. Skull erosion is unusual, but underlying bone infiltration was seen in two of our cases.
Histologically, CIFS is a cellular tumor composed of oval to spindle cells arranged in diffuse sheets and fascicles. Mitotic activity is usually high. Hemangiopericytoma-like vessels can be found and may lead to a mistaken diagnosis of infantile hemangiopericytoma [10].
Immunohistochemically, the tumor cells are positive for vimentin. Variable positivity for actin can be seen suggesting myofibroblastic differentiation. Desmin, Myo D1, EMA, S100, and CD34 are negative [1]. CD31 was focally positive in one case of our series which is unusual but has been reported [7]. The differential diagnosis of CIFS includes spindle cell rhabdomyosarcoma and poorly differentiated embryonal rhabdomyosarcoma, infantile fibromatosis, malignant peripheral nerve sheath tumor, synovial sarcoma, dermatofibrosarcoma protuberans, and giant cell fibroblastoma [1, 10]. Negativity for desmin and Myo D1 rules out rhabdomyosarcoma. The cellularity, atypia, and mitotic count would be higher in CIFS than in infantile fibromatosis. Dermatofibrosarcoma and giant cell fibroblastoma have storiform pattern, low mitotic activity, and CD34 positivity [1, 10].
Recently, a novel chromosomal translocation t(12;15);(p13;q25) with resultant ETV6-NTRK3 gene fusion has been identified in patients with CIFS, which is also shared by cellular mesoblastic nephroma [11]. This can be detected on frozen or paraffin-embedded tissue by reverse transcriptase polymerase chain reaction [11]. Complete excision of tumor with negative margins is the mainstay of treatment. Neoadjuvant chemotherapy has been shown effective in reducing tumor size in selected cases [8]. The chemotherapeutic options include vincristine, actinomycin D, cyclophosphamide or ifosfamide, and etoposide, and the treatment of choice is vincristine with actinomycin (VA regimen) due to its efficacy and better tolerance [9].
Chemotherapy was given to case 3 due to positive margins. The prognosis of CIFS, in general, is excellent, even though region-specific prognosis related to the scalp is not known. Overall, a 5 to 50 % local recurrence rate is seen after incomplete excision, and metastasis is seen in only 10 % of cases [10]. Tumor-associated mortality has been reported to be associated with larger tumors and those located in the head and neck region, proximal extremities, and trunk, which preclude complete excision [8]. The reported 5-year survival rate is 84 % in a large series reported by Chung and Enzinger [12]. At a mean follow-up of 39.6 months, all patients in our series are alive with no evidence of metastasis. In summary, we report three cases of CIFS of the scalp, a rare distinct sarcoma of infancy and children less than 2 years of age. A male predominance was seen in our cases. Underlying bone erosion is rare in CIFS but was seen in two of our cases.
References
Meittinen M (2010) Infantile fibrosarcoma (congenital fibrosarcoma). In: Modern soft tissue pathology tumors and non-neoplastic conditions, 1 edn. Cambridge University Press, New York, pp 301–302
Chaudhari AB, Ladapo F, Duncan JT (1978) Fibrosarcoma of scalp. Case Rep J Neurosurg 49:893–897
Muzumdar D, Michuad J, Ventureyra ECG (2006) Primary giant infantile fibrosarcoma of the scalp case report and review of literature. Childs Nerv Syst 22:300–304
Kumar R, Jagdewan A, Sharma MC (2014) Congenital infantile fibrosarcoma of scalp. Is adjuvant therapy essential? Indian J Surg Oncol 5:297–299
Brohi SR, Dilber M (2012) Congenital osteolytic fibrosarcoma of dura presenting as scalp mass. J Coll Physicians Surg Pak 22:531–532
Goldblum JR, Folpe AL, Weiss SW (2013) Congenital and infantile fibrosarcoma. In: Enzinger and Weiss’ soft tissue tumors, 6 edn. Elsevier Saunders, Philadelphia, pp. 300–304
Folpe AL, Inwards CY (2010) Infantile fibrosarcoma. In: Bone and soft tissue pathology, 1. Churchill Livingstone Saunders Elsevier, Philadelphia. 69–71
Orbach RA, Cecchetto G, et al (2010) Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 28:318–323
Ferrari A, Orbach D, Sultan I, Casanova M, Bisoqno G (2012) Neonatal soft tissue sarcomas. Semin Fetal Neonatal Med 17:231–238
Enriquez AM, Li P, Samuelson J, Reyes-Muqica M (2008) Congenital fibrosarcoma with a novel complex 3-way translocation t(12;15;19) and unusual histologic features. Hum Pathol 39:1844–1848
Bourgeois JM, Knezevich SR, Mathers JA, Sorensen PH (2000) Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol 24:937–946
Chung EB, Enzinger FM (1976) Infantile fibrosarcoma. Cancer 38:729–739
Acknowledgments
The authors thank Dr. Dickson Brendan of Mount Sinai Hospital, Toronto, Canada, for reviewing slides of case 1.
Conflict of interest
None of the authors have any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Din, N.U., Minhas, K., Shamim, M.S. et al. Congenital (infantile) fibrosarcoma of the scalp: a case series and review of literature. Childs Nerv Syst 31, 2145–2149 (2015). https://doi.org/10.1007/s00381-015-2824-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-015-2824-1