
Abstract
Case report
A primary giant infantile fibrosarcoma of the scalp in a 6-month-old boy is reported. He presented with a rapidly enlarging right paramedian parietal scalp swelling since birth. There were no signs of raised intracranial tension. Magnetic resonance (MR) imaging of the brain revealed a large vascular scalp tumor extending over the posterior frontal and parietal region bilaterally crossing the midline. There was no intracranial extension. Carotid angiography revealed an extremely vascular tumor supplied by the external carotid artery branches. The tumor was completely resected through a curvilinear scalp incision. Histology was consistent with diagnosis of an infantile fibrosarcoma. The patient had an uneventful course and received no postoperative adjuvant therapy. MR imaging of the brain at follow-up after 5 years showed no evidence of recurrence of tumor. At follow-up after 10 years, he is asymptomatic and leading an active life.
Discussion
The pathology and management of primary scalp fibrosarcoma is discussed, and the relevant literature is briefly reviewed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary fibrosarcoma of the scalp is a rare tumor. There are few anecdotal reports in the literature of primary fibrosarcomas of the scalp in the pediatric population [1, 3, 5, 8, 12]. The exact evolution of these tumors and its biological characteristics are not well defined due to the scarce literature on this subject. However, the primary treatment remains total surgical and a wide excision with adjuvant radiotherapy and chemotherapy. We report a primary fibrosarcoma of the scalp in a 6-month-old infant, who underwent total excision and has a 10-year disease-free survival without adjuvant treatment. The surgical management and prognostic indicators for long-term survival are briefly discussed.
Case report
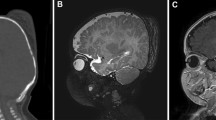
A 6-month-old boy presented with a massive scalp swelling. He was delivered as a full-term pregnancy by cesarean section due to failure of progress of labor. He was noted to have a midline parietal lemon-sized scalp swelling 3×4×6 cm in size which was diagnosed as a cephalhematoma consequent to birth trauma. There were no signs of physical or developmental delay. The scalp swelling showed a massive increase in size over the next 12 weeks, but there were no symptoms suggestive of neurological dysfunction. The mass measured 6×10×12 cm in size extending from left midparietal and temporal region coronally to the right parietal region and from the anterior fontanelle to near the posterior fontanelle (Fig. 1). The overlying skin was free and mobile. There was no increased warmth or tenderness over the swelling but had huge dilated veins coursing over it. Neurological examination did not reveal any abnormality. Magnetic resonance (MR) imaging showed a multiseptate mass extending over the posterior frontal and parietal region bilaterally crossing the midline (Fig. 2). There was erosion of the bone at places, but there was no invasion of the superior sagittal sinus. There was no intracranial extension. Right and left common carotid angiography showed an extremely vascular scalp tumor with extensive supply from external carotid arteries from both sides, including the anterior and posterior branches of superficial temporal artery, occipital arteries, and the middle meningeal arteries. There was no supply from the internal carotid artery. A tumor blush was visible on the venous phase. Although a compression of the superior sagittal sinus was noted, it was patent. The differential diagnosis included a possibility of a teratoma, sarcoma, or a vascular malformation.

Clinical photograph of the patient showing the massive midline scalp tumor. a Superior view showing the bosselated tumor with dilated veins over the stretched scalp overlying the tumor. b Lateral view showing the anteroposterior extent of the tumor

a Coronal T1-weighted contrast enhanced MR image showing the mixed intensity tumor confined within the scalp layers and its relationship with the superior sagittal sinus. b Sagittal T1-weighted contrast enhanced MR image showing the massive midline scalp tumor vividly
The patient underwent surgery through curvilinear scalp incision on the right posterior parietal region and carried towards the left across the midline. The visible arterial feeders and the draining veins were marked initially with preoperative ultrasound as well as direct palpation. The feeders were then coagulated with bipolar cautery and cut, which resulted in decrease in the vascular tension of the mass. The margin of the lesion was easily delineated above the pericranium, and adequate galeal space was present allowing subsequent reflection of the scalp flap as a single layer. The pericranium from left midparietal to right posterior parietal around the anterior circumference of the lesion was incised to a point where bone was palpable, and dissection was continued subperiosteally towards the lesion. The mass was effectively elevated above the deformed calvarium in its entirety in an anterior to posterior direction. The bleeding in the bone was effectively controlled with bone wax, gelfoam soaked in thrombin, and bipolar coagulation. There were multiple scallop deformities and spike protrusions of the bone, which were removed and remodeled with rongeurs. There was no intracranial extension of the tumor. The scalp flap was reflected anteriorly and closed in two layers. The scalp was not resected since there was adherent elasticity. The blood loss was 250 cc, which was replaced. The patient tolerated surgery well and had an uneventful recovery. The wound had healed well.
Gross specimen showed a large bosselated grayish mass measuring 14×9.5×7.2 cm and weighing 402 g (Fig. 3). It was made up of grayish fibrous tissue with areas of myxoid degeneration. There were large areas of necrosis and yellowish discoloration with focal areas of cystic degeneration. Histological examination showed a highly vascular moderately cellular fibroblastic tumor comprising of predominantly spindle-shaped cells, which formed fascicles and bundles giving an interlacing appearance (Fig. 4). The nuclei were irregular, often vesicular, containing small nucleoli, and the cytoplasm was more or less abundant. Mitotic figures, including atypia and scattered lymphocytes, were seen. The diagnosis was consistent with an infantile fibrosarcoma.

Gross specimen of the tumor showing a large bosselated grayish mass measuring 14×9.5×7.2 cm and weighing 402 g comprising of fibrous tissue with areas of myxoid degeneration. There were large areas of necrosis and yellowish discoloration with focal areas of cystic degeneration

a Low-power photomicrograph showing interlacing bundles of spindle-shaped cells lying on a moderately dense fibrous stroma (HPS stain, ×10). b Higher-power photomicrograph with a moderately dense cellularity made up of ovoid or spindle cells in a vacuolated stroma. A mitosis is seen in the center (HPS stain, ×40)
The patient did not receive any adjuvant therapy and was advised serial follow-up for neurooncologic assessment. The computed tomography of the chest/abdomen, bone scan, and bone marrow were normal. MR imaging at a follow-up after 5 years showed no evidence of a recurrence of the tumor (Fig. 5). He is asymptomatic and leading an active life at follow-up after 10 years (Fig. 6).

Postoperative T1-weighted contrast enhanced MR image at 5 years showing no evidence of recurrence of the tumor

a Clinical photograph of the patient 2 years following surgery. b Clinical photograph of the patient 10 years following surgery
Discussion
The site of origin of primary sarcomas of the central nervous system may be primarily from the soft tissue of the scalp, meninges, bone, or the intracranial structures. The primary mesenchymal tumors of the scalp include dermatofibroma, plexiform neurofibroma,dermatofibrosarcoma protuberans, fibrosarcoma, osterosarcoma, malignant fibrous histiocytoma, malignant angioendothelioma, and angiosarcoma. Fibrosarcomas and plexiform neurofibromas are known to attain massive dimensions [13]. Soft tissue sarcomas metastasizing to the brain include chondorsarcomas, osteosarcoma, leiomyosarcoma, Kaposi’s sarcoma, malignant mesothelioma, alveolar soft part sarcomas, fibrosarcomas, and rhabdomyosarcomas. They have increased incidence presumably due to the prolonged survival following remission post radiation and cancer chemotherapy. Fibrosarcomas of the scalp have been reported to develop after cranial irradiation, burn scar, trauma, and surgery for benign scalp tumors [2, 10, 11]. Patients with neurofibromatosis have a higher predilection [8].
The evolution of the tumor in the temporal course in the present case was quite interesting. At the time of labor, the patient’s mother had a vertex presentation. The tumor was palpated through the dilated os and was mistaken for a breech presentation. The baby was then promptly delivered through a cesarean section. At birth, the swelling was small and assumed to be a cephalhematoma. The slow growth of the swelling in the initial period raised the possibility of a hemangioma and the rapid increase in the growth over the next 2 months alerted to the probability of a sarcoma or teratoma. This suggests that the development of a malignant neoplasm can mimic trauma or a relatively benign lesion. A high index of suspicion is warranted for early recognition and institution of treatment, which may have ultimate bearing on the prognosis. They tend to be localized within the fascia but can be focally aggressive.
The exact etiology of the scalp tumor in the present case was not discernible. Although, trauma, chronic irritation, and chronic inflammation have been defined to play a role in some cases, there is no convincing evidence in the literature [1, 3]. The neoplastic transformation and secondary mesenchymal differentiation following soft tissue trauma is rare. The congenital nature of the tumor and its exponential growth early in life suggest a genetic influence with a strong expressivity. Radiation exposure is thought to evoke tumors such as fibrosarcomas, osteosarcomas, and leukemias [11]. There was no exposure to radiation or administration of any pharmacological agent with carcinogenic potential during the entire gestational period in the present case. A malignant transformation into a preexisting benign lesion is also a possibility. Romieu et al. [12] report a fibrosarcoma in a preexisiting neurofibroma of the scalp, while Chaudhari et al. [3] report a sarcomatous transformation over a 2-year period following operative trauma in a 17-year-old Nigerian boy who probably had a preexisting fibroma since birth. They postulate that the operative trauma resulted in an overwhelming chronic inflammation and reparative process, which ultimately lead to malignant transformation. However, the incidence of such a transformation is very rare, and it suggests a preexisting tendency for neoplastic attributes.
The fibrosarcoma of the scalp in the present case had unique features. It was a de novo neoplasm, which had a congenital origin. It demonstrated rapid increase in size following birth and attained a massive size over a short period. Although skull erosion is not a classical feature of fibrosarcoma, the minimal erosion of the skull at places in our case suggests the increased thinning of the bone due to the weight of the tumor. Although the middle and posterior third of the superior sagittal sinus were compressed, there was no invasion of the tumor into the sinus. The tumor was entirely fed by the external carotid artery circulation. There was no communication with the intracranial circulation. The tumor was well defined and confined within the scalp layers. The vascular feeders were secured early in the operation, thus assisting the surgeon to minimize the blood loss and reduce the overall time of surgery necessary for the ultimate outcome in a 6-month-old boy.
The typical fibrosarcoma differs from other mesenchymal tumors histologically [3]. The microscopic appearance of the tumor in the present case was suggestive of a highly vascular fibroblastic tumor with large areas of hemorrhage and necrosis with hyalinization of the stroma associated with calcification and foreign body giant cells. There was a widespread moderate to high mitotic index throughout the tumor. It was not characteristic of dermatofibrosarcoma protuberans, which has dense collagen around the spindle cells and histiocytic predominance in a cartwheel pattern. It was not as high vascular as a malignant angioendothelioma.
The management of fibrosarcoma of the scalp is essentially surgical [1, 3–6, 8, 9, 11, 14]. A complete surgical excision is the gold standard. A subtotal excision is fraught with a high risk of recurrence, thus increasing the morbidity and mortality. They rarely metastasize except in advanced cases [9]. The distant metastases rate is about 20%. Unlike the primary meningeal or brain sarcomas, they tend to be less infiltrating but can achieve massive proportions as seen in the present case. Skull erosion is not a classical feature, and they remain localized to the fascia and remain amenable to total resection provided they are treated early in the tumor evolution. The high index of suspicion leading to an early recognition, tumor remaining predominantly within the confines of the scalp layers with minimal erosion of the underlying skull, no involvement of the superior sagittal sinus, and being fed entirely by the external carotid circulation were the key factors leading to the complete extirpation of the lesion in the present case. Fibrosarcomas are known to be relatively radiosensitive. However, in the present case, radiotherapy was not administered since there was no clinical and radiological evidence of recurrence on serial follow-up for the past 10 years, indicating that radicality of tumor excision is a predominant indicator of long-term disease-free survival in soft tissue fibrosarcomas and radiotherapy can be completely avoided in such cases. Radiotherapy should be used as an adjuvant for uncontrollable recurrences and advanced cases. Repeated incomplete surgeries for fibrosarcoma may increase the propensity for further tumor growth, leading to fungation, maceration and ulceration of the mass, and susceptibility to frequent hemorrhages and may require a major plastic reconstructive procedure [1–3, 7, 11, 14]. Arigbabu et al. [1] have reviewed five cases of primary fibrosarcoma of the scalp seen over a 10-year period. They report an 80% recurrence-free interval obtained at 2 years with surgery and radiotherapy. Greager et al. [6] report a 28% 10-year survival rate in the 53 adult head and neck soft tissue sarcomas in which fibrosarcoma was the most common histologic type. Wide excision with adjuvant radiotherapy and chemotherapy or both was used in selected patients. They conclude that the long-term surviors either had well-differentiated tumors or less than or equal to 5-cm-diameter tumors. Wanebo et al. [14] report a 5-year survival of 60–70% for fibrosarcomas in their series of 214 head and neck sarcomas.
To conclude, giant primary fibrosarcoma of the scalp in an infant is a rare tumor. A high index of suspicion is necessary for early recognition of the disease, ensuring long-term disease-free survival. Total surgical excision of the tumor is the gold standard and it is possible to attain “cure”. Radiotherapy can be completely avoided if there is no clinical and radiological progression of the disease on follow-up.
References
Arigbabu SO, Durosinmi-Etti FA, Oyeneyin JO, Ojikutu NA (1985) Primary fibrosarcoma of the scalp: a review of a ten-year record from Lagos, Nigeria. Clin Radiol 36L:465–466
Chang SM, Barker FG II, Larson DA, Bollen AW, Prados MD (1995) Sarcomas subsequent to cranial irradiation. Neurosurgery 36:685–690
Chaudhari AB, Ladapo F, Duncan JT (1978) Fibrosarcoma of the scalp. Case report. J Neurosurg 49:893–897
Dijkstra MD, Balm AJ, Coevorden FV, Gregor RT, Hart AA, Hilgers FJ, Keus RB, Loftus BM (1996) Survival of adult patients with head and neck soft tissue sarcomas. Clin Otolaryngol 21:66–71
Fong PH, Lee ST, Lim Tan SK (1986) Primary scalp cancer in Singapore. Ann Acad Med Singapore 15:67–70
Greager JA, Patel MK, Briele HA, Walker MJ, Das Gupta TK (1985) Soft tissue sarcomas of the adult head and neck. Cancer 56:820–824
Koshima I, Inagawa K, Jitsuiki Y, Tsuda K, Moriguchi T, Watanabe A (1996) Scarpa’s adipofascial flap for repair of wide scalp defects. Ann Plast Surg 36:88–92
Niijima K (2002) Scalp neurofibrosarcoma of a von Recklinghausen’s disease patient. No To Shinkei 54:730–731 (in Japanese)
Ou SM (1988) Soft tissue sarcoma in the head and neck—analysis of 87 patients. Zhonghua Zhongliu Zazhi 10:289–292 (in Chinese)
Ozyazgan I, Kontas O (1999) Burn scar sarcoma. Burns 25:455–458
Prasad ML, Mahapatra AK, Dinda AK, Sarkar C, Roy S (1991) Fibrosarcoma of the scalp following postoperative radiotherapy for medulloblastoma. Acta Neurochir (Wien) 109:145–149
Romieu C, Pichaud A, Vlahovitch B (1968) History of a fibrosarcoma of the scalp. Ann Chir Plast 13:157–161 (French)
Scott M (1966) Massive plexiform neurofibroma of occipital scalp. J Neurosurg 25:81–82
Wanebo HJ, Koness RJ, MacFarlane JK, Eilber FR, Byers RM, Elias EG, Spiro RH (1992) Head and neck sarcoma: report of the Head and Neck Sarcoma Registry. Society of Head and Neck Surgeons Committee on Research. Head Neck 14:1–7
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Muzumdar, D., Michaud, J. & Ventureyra, E.C.G. Primary giant congential infantile fibrosarcoma of the scalp. Childs Nerv Syst 22, 300–304 (2006). https://doi.org/10.1007/s00381-005-1199-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-005-1199-0