
Abstract
Purpose
Cerebral arteriovenous malformation (AVM) is traditionally thought of as a congenital diagnosis. In recent years, there has been infrequent reports of this neurovascular condition presenting as de novo entities.
Methods
The authors report two cases of pediatric patients who present with de novo cerebral AVMs. In both cases, the patients had magnetic resonance imaging (MRI) of the brain done as part of the work-up for first-onset seizures. At that point in time, the scans were unremarkable. After a latent period of approximately 6 and 9 years respectively, a repeated MRI brain scan showed evidence of de novo AVM for each patient.
Results
Both patients did not have radiological evidence of cerebral AVM during their first presentation of seizures. However, a repeated MRI brain scan after a period of 6 and 9 years demonstrated new findings of cerebral AVM for each patient.
Conclusions
Currently, the disease of cerebral de novo AVM remain as an ambiguous condition that is poorly understood. With the advances in molecular diagnostics, there are possibilities of exploring biochemical profiles for better understanding of the origin of cerebral AVMs. However, in the meantime, owing to the unpredictable nature of cerebral AVMs, clinicians should have increased awareness of this unique condition. This is especially important, as definitive treatment is available to prevent devastating neurological sequelae from cerebral AVM rupture.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral arteriovenous malformation (AVM) is traditionally thought of as a congenital diagnosis. They are postulated to be caused by defects during the development of blood vessels and are characterized by an arteriovenous shunt without a capillary bed, but with the presence of an arterial nidus [5, 10]. In recent years, there has been scattered reports of this neurovascular condition presenting as de novo entities. However, owing to its scarcity, this concept of de novo cerebral AVMs remains as a poorly understood condition. Here, the authors present two independent cases of de novo AVM development years after an initial radiologically normal brain scan. In view of the uniqueness of this condition, the authors discuss the two patients’ cases and review the relevant literature in detail.
Case report 1
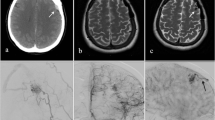
A previously well 1-year-old boy with normal developmental milestones was admitted to the hospital for first onset of non-febrile seizures that self-resolved. The patient had neither other constitutional symptoms nor significant family history that may have possibly contributed to his seizure. An electroencephalogram (EEG) did not show seizure foci. Furthermore, a magnetic resonance imaging (MRI) scan of the brain demonstrated no structural abnormalities. He was managed conservatively with anti-epileptics and discharged home stable. During the 3 years of outpatient follow-up, the patient was successfully weaned off his anti-epileptic medications. However, at 7 years of age, he was admitted again for seizures. A repeated MRI brain scan showed a left cerebellar arteriovenous malformation (AVM). No focus of restricted diffusion was present to suggest an acute infarct, nor any susceptibility artifact that would suggest previous hemorrhage. This investigation was followed up with a four-vessel cerebral angiogram that confirmed the MRI brain findings. Overall, the radiological results was suggestive of a Spetzler-Martin grade III [30] AVM (Fig. 1). For this case, the patient’s parents opted for him to undergo stereotactic radiosurgery (Novalis) for his lesion.

a Axial T2-weighted MRI brain at 1 year old showing no obvious vascular anomaly. b Axial T2-weighted MRI brain at 7 years old demonstrating an AVM consisting of a cluster of fine vessels seen in the superior left cerebellar hemisphere and a prominent serpiginous vein draining into the torcula. c A follow-up DSA after the MRI brain (b) cerebral angiogram outlining the vascular details of the left cerebellar AVM
Case report 2
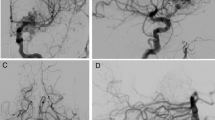
A previously well 7-year-old female presented with a single episode of seizures associated with a fever that self-resolved. Similarly, this patient had no other constitutional symptoms or family history that may have possibly contributed to her presentation. As part of the work-up investigations, a MRI brain scan was performed which showed no obvious intracranial abnormality. Her remaining tests were correspondingly unremarkable. The patient remained asymptomatic until the age of 16, during which she complained of intermittent headaches. A repeated MRI brain scan reported a small parenchymal AVM in the left anterior temporal lobe. There was no associated intra-parenchymal hemorrhage, atrophy, or gliosis detected. Based on these findings, the diagnosis was that of a Spetzler-Martin grade II [30] AVM (Fig. 2). In this case, the patient’s parents decided to proceed with surgical excision of her cerebral AVM.

a Axial T2-weighted MRI brain at 7 years old showing no obvious vascular anomaly. b Axial T2-weighted MRI brain at 16 years old showing a small parenchymal AVM in the left anterior temporal lobe. c MRA image demonstrating the left temporal AVM, where the feeding artery appears to arise from the M2 branch of the left middle cerebral artery
Discussion
Based on current understanding, an AVM is a lesion involving an abnormal connection between the arteries and veins due to the lack of capillary beds. Intracranially, it appears as a macroscopic tangle of blood vessels, consisting of a relatively well-circumscribed center, known as the nidus. The nidus is supplied by a varying number of feeding arteries and draining veins [7]. This unique vascular anatomy results in altered cerebral hemodynamics, forming a low resistance and high and, or low flow arteriovenous shunt [17]. At a microscopic level, blood vessels in the nidus are usually characterized by thin and or irregular muscularis and elastica islands of sclerotic tissue, endothelial thickening, and media hypertrophy [7]. Globally, the number of reported de novo intracranial AVMs is infrequent. To our knowledge, there have been only 12 previously published cases of de novo AVM formation in patients in the past two decades (Table 1) [1, 3, 5, 10, 18, 19, 22, 24, 29, 31].
From these cases, it was observed that there was equivocal distribution of occurrence between both genders. Interestingly, majority of de novo AVMs were found in the supratentorial region, with only one case reporting the infratentorial region. In congruency with the literature, our two patients share the pediatric range as most of the reported patients, when they were initially found to have no evidence of AVMs. Only one patient was reported to be free of AVM presentation at 26 years of age, which is well past the pediatric age range [5].
Over time, the knowledge of AVMs has considerably evolved. Once considered to be static lesions, the discovery of enlargement or regrowth of AVMs observed in postoperative angiograms suggests the lesions to be dynamic in nature instead [18, 9, 14]. Also, the origin of AVMs is still elusive to date. Traditionally, AVMs have been believed to be strictly congenital in nature [23, 26]. One common theory postulates that an early trigger prenatally in the genetic pathway for vascular development causes a multifocal vascular abnormality in the form of an AVM [21]. However, there has been little evidence for the existence of AVMs in utero, at birth, and even in infants [8, 2]. Concordantly, there is growing evidence that AVM development is a postnatal event. The pathogenesis of postnatal AVMs has been attributed to the expression of angiogenic factors, such as vascular endothelial growth factor (VEGF). Elevated expression of these factors induces abnormal endothelial proliferation, which then encourages abnormal vasculature development and AVM formation [28]. For example, patients with AVMs have been found to have both local and systemic elevations of VEGF [28, 20]. Vascular endothelial growth factor has been typically associated with vascular disorders, especially in the processes that involve hypoxia, hemorrhage, and inflammation [4, 28, 11, 27]. In addition to VEGF, platelet-derived growth factor (PDGF) ligand A and receptor α have also been found to be raised in AVMs. These are thought to rise either with increased blood flow in the lesion or with ischemia. They are involved in signal transmission into the cell, which results in expression of angiogenesis-related genes [13]. To summarize, elevated angiogenic factors may add insight into these cited cases. Interestingly, some of these biochemical factors, such as VEGF, were also implicated from the cases highlighted in Table 1. Furthermore, corresponding to the features of molecular involvement, the influence of genetics has also been found to contribute to AVM pathogenesis as well. For instance, haploinsufficiency of either activin receptor-like kinase 1 (ALK1) or endoglin (ENG) gene in endothelial cells have been observed to be associated with increased risk of AVM formation in animal studies [12, 6]. In these animal studies, it has also been demonstrated that angiogenic stimulation (such as VEGF) was necessary to trigger the start of AVM formation and that genetic mutation alone was unable to do the same [6]. Building on these observations, it is highly suggestive that AVM pathogenesis is likely to require a complex interplay of genetic and biochemical mechanisms that are uncertain at this stage. Overall, there is growing clinical evidence and scientific results to indicate that de novo development of cerebral AVMs should be reconsidered as acquired postnatal lesions.
Another notable feature of our cases is that both patients had seizures at first presentation. Seizures have been long accepted to be important manifestations of cerebral AVMs [32]. From the literature, the incidence of patients with seizures and without clinical evidence of hemorrhage has been cited to be between 17 and 30 % [25, 15]. Despite this well-known association, the cause of epileptogenesis from cerebral AVMs is still unclear [16]. Providentially, through the appreciation of improved research techniques, we are now aware that detailed analysis of cerebral AVMs’ angioarchitecture has shed insight into features that strongly correlate with epilepsy [32]. Building on the scientific evidence of AVM pathogenesis, there is a role to explore the similar molecular factors to bridge the current gaps in knowledge between seizures and cerebral AVMs.
Conclusion
Owing to the infrequent number of de novo intracranial AVMs, the traditional view of AVMs as a congenital entity still holds. Here, both of our cases add to the limited but growing pool of clinical evidence that questions the congenital origin of cerebral AVMs. Based on preliminary results of scientific research in this subject, there is a strong likelihood that there are complex biological elements underlying the formation of AVMs. However, in this stage, the details of the underlying biology have yet to be elucidated. In the meantime, due to the unpredictable nature of cerebral AVMs, clinicians should have heightened awareness of this unique condition. This is especially important, as definitive treatment is available to prevent devastating neurological sequelae from cerebral AVM rupture. Nevertheless, owing to the exponential advancements in molecular and genetic investigations, the authors foresee an equivocal leap in the understanding of cerebral AVMs’ pathogenesis. With the increment of such valuable knowledge, the neurosurgical community can look forward to a future whereby the clinical management of this condition can be rampantly improved.
References
Akimoto H, Komatsu K, Kubota Y (2003) Symptomatic de novo arteriovenous malformation appearing 17 years after the resection of two other arteriovenous malformations in childhood: case report. Neurosurgery 52(1):228–232
Al-Rodhan NR, Al-Mefty O, Rifai A, Fox JL (1986) Persistence of primitive cerebral vasculature in a newborn. A case report of whole brain AVM. Clin Neurol Neurosurg 88(4):283–287
Alvarez H, Perry V, Solle M, Castillo M (2012) De novo cerebral arteriovenous malformation in a child with previous cavernous malformation and developmental venous anomaly. J Neurosurg Pediatr 9(3):327–330. doi:10.3171/2011.12.peds11312
Arisato T, Hashiguchi T, Sarker KP, Arimura K, Asano M, Matsuo K, Osame M, Maruyama I (2003) Highly accumulated platelet vascular endothelial growth factor in coagulant thrombotic region. J thromb haemost : JTH 1(12):2589–2593
Bulsara KR, Alexander MJ, Villavicencio AT, Graffagnino C (2002) De novo cerebral arteriovenous malformation: case report. Neurosurgery 50(5):1137–1141
Chen W, Sun Z, Han Z, Jun K, Camus M, Wankhede M, Mao L, Arnold T, Young WL, Su H (2014) De novo cerebrovascular malformation in the adult mouse after endothelial alk1 deletion and angiogenic stimulation. Stroke 45(3):900–902
Choi JH, Mohr JP (2005) Brain arteriovenous malformations in adults. Lancet Neurol 4(5):299–308
DeCesare B, Omojola MF, Fogarty EF, Brown JC, Taylon C (2006) Spontaneous thrombosis of congenital cerebral arteriovenous malformation complicated by subdural collection: in utero detection with disappearance in infancy. Br J radiol 79(946):e140–144. doi:10.1259/bjr/44174031
Delitala A, Delfini R, Vagnozzi R, Esposito S (1982) Increase in size of cerebral angiomas: case report. J Neurosurg 57(4):556–558
Gonzalez LF, Bristol RE, Porter RW, Spetzler RF (2005) De novo presentation of an arteriovenous malformation: Case report and review of the literature. J Neurosurg 102(4):726–729
Gunsilius E, Petzer A, Stockhammer G, Nussbaumer W, Schumacher P, Clausen J, Gastl G (2000) Thrombocytes are the major source for soluble vascular endothelial growth factor in peripheral blood. Oncology 58(2):169–174
Hao Q, Zhu Y, Su H, Shen F, Yang GY, Kim H, Young WL (2010) VEGF Induces More Severe Cerebrovascular Dysplasia in Endoglin than in Alk1 Mice. Transl stroke res 1(3):197–201. doi:10.1007/s12975-010-0020-x
Heidaran MA, Pierce JH, Yu JC, Lombardi D, Artrip JE, Fleming TP, Thomason A, Aaronson SA (1991) Role of alpha beta receptor heterodimer formation in beta platelet-derived growth factor (PDGF) receptor activation by PDGF-AB. J biol chem 266(30):20232–20237
Hladky J-P, Lejeune J-P, Blond S, Pruvo J-P, Dhellemmes P (1994) Cerebral arteriovenous malformations in children: report on 62 cases. Childs Nerv Syst 10(5):328–333
Hofmeister C, Stapf C, Hartmann A, Sciacca RR, Mansmann U, terBrugge K, Lasjaunias P, Mohr JP, Mast H, Meisel J (2000) Demographic, morphological, and clinical characteristics of 1289 patients with brain arteriovenous malformation. Stroke; J cereb circ 31(6):1307–1310
Hoh BL, Chapman PH, Loeffler JS, Carter BS, Ogilvy CS (2002) Results of multimodality treatment for 141 patients with brain arteriovenous malformations and seizures: factors associated with seizure incidence and seizure outcomes. Neurosurgery 51(2):303–309, discussion 309-311
Iwama T, Hayashida K, Takahashi JC, Nagata I, Hashimoto N (2002) Cerebral hemodynamics and metabolism in patients with cerebral arteriovenous malformations: an evaluation using positron emission tomography scanning. J Neurosurg 97(6):1314–1321. doi:10.3171/jns.2002.97.6.1314
Jeffree RL, Stoodley MA (2009) Postnatal development of arteriovenous malformations. Pediatr Neurosurg 45(4):296–304
Kilbourn KJ, Spiegel G, Killory BD, Kureshi I (2014) Case report of a de novo brainstem arteriovenous malformation in an 18-year-old male and review of the literature. Neurosurgical review:1-7
Kilic T, Pamir MN, Kullu S, Eren F, Ozek MM, Black PM (2000) Expression of structural proteins and angiogenic factors in cerebrovascular anomalies. Neurosurgery 46(5):1179–1191, discussion 1191-1172
Lasjaunias P (1997) A revised concept of the congenital nature of cerebral arteriovenous malformations. Interv Neuroradiol : J peritherapeutic neuroradiol, surg proced relat neurosci 3(4):275–281
Mahajan A, Manchandia TC, Gould G, Bulsara KR (2010) De novo arteriovenous malformations: case report and review of the literature. Neurosurg Rev 33(1):115–119
Mullan S, Mojtahedi S, Johnson DL, Macdonald RL (1996) Embryological basis of some aspects of cerebral vascular fistulas and malformations. J Neurosurg 85(1):1–8. doi:10.3171/jns.1996.85.1.0001
O’Shaughnessy BA, DiPatri AJ Jr, Parkinson RJ, Batjer HH (2005) Development of a de novo cerebral arteriovenous malformation in a child with sickle cell disease and moyamoya arteriopathy: case report. J Neurosurg Pediatr 102(2):238–243
Perret G, Nishioka H (1966) Report on the cooperative study of intracranial aneurysms and subarachnoid hemorrhage. IV. Cerebral angiography. An analysis of the diagnostic value and complications of carotid and vertebral angiography in 5,484 patients. J Neurosurg 25(1):98–114. doi:10.3171/jns.1966.25.1.0098
Potter JM (1955) Angiomatous malformations of the brain: their nature and prognosis. Ann R Coll Surg Engl 16(4):227–243
Sakamoto S, Kiura Y, Yamasaki F, Shibukawa M, Ohba S, Shrestha P, Sugiyama K, Kurisu K (2008) Expression of vascular endothelial growth factor in duramater of patients with moyamoya disease. Neurosurg Rev 31(1):77–81. doi:10.1007/s10143-007-0102-8, discussion 81
Sandalcioglu IE, Wende D, Eggert A, Müller D, Roggenbuck U, Gasser T, Wiedemayer H, Stolke D (2006) Vascular endothelial growth factor plasma levels are significantly elevated in patients with cerebral arteriovenous malformations. Cerebrovasc Dis 21(3):154–158
Schmit BP, Burrows PE, Kuban K, Goumnerova L, Scott RM (1996) Acquired cerebral arteriovenous malformation in a child with moyamoya disease: case report. J Neurosurg 84(4):677–680
Spetzler RF, Martin NA (1986) A proposed grading system for arteriovenous malformations. J Neurosurg 65(4):476–483. doi:10.3171/jns.1986.65.4.0476
Stevens J, Leach J, Abruzzo T, Jones B (2009) De novo cerebral arteriovenous malformation: case report and literature review. Am J Neuroradiol 30(1):111–112
Turjman F, Massoud TF, Sayre JW, Vinuela F, Guglielmi G, Duckwiler G (1995) Epilepsy associated with cerebral arteriovenous malformations: a multivariate analysis of angioarchitectural characteristics. AJNR Am J Neuroradiol 16(2):345–350
Conflict of interest
We, the authors of this manuscript, report no funding, financial support, or industrial affiliations received for the writing of this article. In addition, we report no conflict of interest concerning the material or methods used in this paper. This manuscript has not been published and is not being considered for publication elsewhere.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yeo, J.J.Y., Low, S.Y.Y., Seow, W.T. et al. Pediatric de novo cerebral AVM: report of two cases and review of literature. Childs Nerv Syst 31, 609–614 (2015). https://doi.org/10.1007/s00381-014-2609-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-014-2609-y