
Abstract
Coagulation factor Xa activates the protease-activated receptor 2 (PAR2) and causes tissue fibrosis; however, the effects of Xa inhibitor edoxaban on atrial fibrosis and atrial fibrillation (AF) have not been investigated. We examined the effect of edoxaban on the progression of atrial fibrosis in a canine congestive heart failure (CHF) model. Beagle dogs were assigned to sham, placebo, and edoxaban groups (n = 6/group). Dogs of the placebo or edoxaban groups received 19 days of medication with daily oral placebo or edoxaban, respectively, followed by 14 days of ventricular tachypacing. Dogs of the sham group had no medication or pacing. Ventricular tachypacing prolonged AF duration in dogs of the placebo group (159 ± 41 s, p < 0.01 vs. sham); however, this effect was suppressed by edoxaban treatment. Compared with the sham group, tachypacing alone also significantly increased the atrial fibrotic area (2.9 ± 0.1% vs. 7.8 ± 0.4%, p < 0.01), PAR2 expression (1.0 ± 0.1 vs. 1.8 ± 0.3, p < 0.05), and atrial fibronectin expression (1.0 ± 0.2 vs. 2.0 ± 0.2, p < 0.01). These responses were suppressed by edoxaban treatment (area 5.9 ± 0.4%, p < 0.01; PAR2 1.1 ± 0.1, p < 0.05; fibronectin 1.2 ± 0.2, p < 0.05 vs. placebo). Edoxaban showed suppressive effects on atrial remodeling, AF progression, and excessive expressions of PAR2 and fibronectin in a canine CHF model. The suppression of the Xa/PAR2 pathway might be a potential pharmacological target of edoxaban.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia in clinical practice and is associated with serious complications and mortality [1, 2]. AF-induced cardiac degradation leads to structural and electrical remodeling, resulting in the progression of AF [3]. The strongest contributing factor to AF progression is atrial fibrosis. Atrial fibrosis is also caused by aging, heart failure, and ischemic heart disease, resulting in heterogeneity of conduction and facilitating reentry and AF occurrence [4,5,6].
Factor Xa is the upstream activator of thrombin. Thrombin, in turn, participates in hemostasis and thrombus formation by converting fibrinogen to fibrin, thereby playing a central role in blood coagulation. In addition, factor Xa is involved in the development of inflammatory reactions, arteriosclerosis, and fibrosis in various tissues via protease-activated receptors [7,8,9,10,11,12]. It has been suggested that Xa inhibitors inhibit myocardial fibrosis by blocking the factor Xa/PAR2 signaling pathway [13,14,15], but the contributions of factor Xa and PAR2 to atrial fibrosis and AF chronicity have not been adequately studied. Moreover, the potential benefits of edoxaban, a factor Xa inhibitor, against atrial fibrosis and AF chronicity have not been examined.
The purpose of this study was to examine possible suppression of atrial fibrosis and AF chronicity by edoxaban in a canine congestive heart failure (CHF) model.
Materials and methods
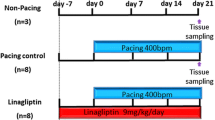
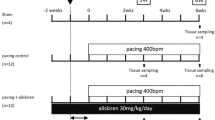
Experimental design
The CHF model was established with reference to experiments conducted previously [4, 16, 17]. All procedures performed in studies involving animals were in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996) and were approved by the Animal Research Ethics Committee of the University of Toyama. Eighteen beagles weighing 8.2–13.0 kg were assigned to the sham (n = 6), placebo (n = 6), and edoxaban (n = 6) groups. All dogs were implanted with pacemakers (detailed in the next section). Two days after implantation, dogs of the placebo and edoxaban groups started receiving a 19-day administration of medication with daily oral placebo or edoxaban, respectively. Seven days after implantation, both groups were subjected to ventricular tachypacing (VTP) of 240 beats/min for 14 days. Dogs of the sham group did not receive medication or pacing. After a total intervention period of 21 days, cardiac ultrasonography was performed and electrophysiological examinations were conducted under thoracotomy. Atrial tissues were then removed for histological and molecular biological evaluation.
Animal preparation
Under general anesthesia (isoflurane 2 vol%), a pacing lead (Cap Sure Fix 5068, Medtronic, Minneapolis, MN, USA) was inserted through the right internal jugular vein into the apex of the right ventricle, and a generator (SIP501, Star Medical, Tokyo, Japan) was implanted subcutaneously in the right neck.
After confirming that there were no hemorrhagic complications, oral placebo (placebo dogs) or edoxaban (edoxaban dogs) was administered daily from 2 days after implantation and continued for 19 days. From 7 days after implantation, the pacemaker was activated at 240 beats/min in dogs of the placebo and edoxaban groups, but not in those of the sham group; tachypacing was continued for 14 days. The dosage of edoxaban was 2.0 mg/kg/day as determined previously [18].
Echocardiographic measurements
Transthoracic echocardiography (SSA-260A; Toshiba, Tokyo, Japan) was performed in each dog just before electrophysiological study. All measurements were performed under sinus rhythm with the pacemaker deactivated. Left ventricular end-diastolic diameter (LVEDD) and left ventricular end-systolic diameter (LVESD) were measured in the parasternal long-axis view; left ventricular ejection fraction (LVEF) was calculated according to the Teichholz method. The left atrium (LA) area was measured in the two-chamber apical view at end systole.
Electrophysiological study
The ventricular pacemaker was deactivated before the electrophysiological study. After opening the chest under anesthesia, stimulation and recording were performed using an external stimulator (SEC-2102, Nihon Kohden, Tokyo, Japan), and unipolar electrode placed in the epicardium of the LA appendage coupled to a recording device (RMT-1000, Nihon Kohden, Tokyo, Japan). During examination, we evaluated atrial effective refractory period (AERP), AF inducibility, and duration of AF. AERPs (ms) were measured at the LA appendage (LAA) using a train of ten basic stimuli (S1) followed by a premature stimulus (S2) at twice the stimulation threshold. The basic cycle length (BCL) was 360, 300, 250, and 200 ms. AERP was measured at intervals of 5 ms. As an index of atrial conduction time, the width of P wave (ms) was measured at the time of sinus rhythm after stopping pacing and the average of three beats was taken. Inducibility (%) was defined as the percentage of AF events during ten inductions. For evaluating the induction rate, we delivered 20 burst pacing stimuli to the LAA with the shortest cycle length captured at 1:1 at a voltage four times the stimulation threshold. The reported duration of AF is the average of ten induced episodes. When AF lasted for more than 1200 s, electrical defibrillation was performed and the duration was counted as 1200 s.
Hemodynamic and bodyweight measurements
Hemodynamics were measured using a pressure line after the electrophysiological study, and the aortic, left ventricular, and left atrial pressures were measured. Bodyweight was measured at the end of the experiment.
Histological analysis
Atrial tissue was harvested from the LA free wall, fixed with 10% formalin, and embedded in paraffin. Sections (thickness 5-µm) were prepared, and collagen fibers were stained using Sirius Red. Observation was performed by a magnification of 400X using optical microscope (OLYMPUS, Tokyo, Japan). Microscopy images were recorded in the computer and quantitatively analyzed using the VH Analyzer software (Keyence, Osaka, Japan). The area of the connective tissue region was measured by color tone discrimination, and the target region was quantitatively evaluated as a percentage of the total area.
Western blotting
Proteins were extracted from LA free wall tissue using T-PER (Thermo Scientific Pierce Protein Research Products, Rockford, IL, USA) supplemented with a protease inhibitor cocktail kit (Thermo Scientific Pierce Protein Research Products). The protein concentration was measured using the Bio-RAD protein assay (BIO-RAD Laboratories, Inc., Hercules, CA, USA). Equal amounts of the proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes using iBlot 2 Gel Transfer Device (Life Technologies, Carlsbad, CA, USA). Blocking solution was tris-buffered saline containing 2% bovine serum albumin for PAR2 and 5% skim milk for fibronectin. Mouse anti-PAR2 monoclonal IgG (dilution 1:100, Santa Cruz Biotechnology Inc., Texas, USA), rabbit fibronectin polyclonal IgG (dilution 1:500, Abcam Plc., Cambridge, United Kingdom), and goat anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) polyclonal IgG (dilution 1:4000, Lifespan Biosciences Inc., Seattle, USA) were used as primary antibodies. In addition, anti-mouse polyclonal IgG (dilution 1:7500, Abcam Plc.), anti-rabbit polyclonal IgG (dilution 1:5000, Abcam Plc.), and anti-goat polyclonal IgG (dilution 1:22,500, Abcam Plc.) were used as secondary antibodies. Detection and quantification of proteins were performed using Odyssey (LI-COR, Lincoln, NE, USA) and Image Studio™ Lite Ver 5.2 (LI-COR), respectively. Semi-quantitative determination of target protein concentration was performed based on the band density, and GAPDH was used for loading control.
Statistical analysis
All values are expressed as the mean ± SEM. The statistical significance of a multiple-group comparison was analyzed using a one-way analysis of variance with post hoc Bonferroni-adjusted pair-wise comparisons. A p value of < 0.05 was considered statistically significant.
Results
Echocardiographic characteristics following tachypacing and effects of edoxaban
After 14 days of VTP, dogs of the edoxaban and placebo groups showed signs of left ventricular dysfunction. Specifically, LVEDD, and LVESD increased and LVEF decreased in these groups than in the sham group (p < 0.05). There were no significant differences in LVEDD, LVESD, and LVEF (Fig. 1) between dogs of the edoxaban and placebo groups. These data indicate the completion of the canine CHF model. The extent of LA area expansion was observed in the placebo group (p < 0.05 vs. sham); however, it was suppressed in the edoxaban group. As a result, there was no significant difference in the level of LA area between the edoxaban and sham groups (Fig. 2). It should be noted that LA area was evaluated by five samples of each group, because we could not obtain images that can be evaluated with each group. The other data of this paper were measured in six samples in each group.

Changes in left ventricular diameter and ejection fraction. Left ventricular (LV) echocardiographic characteristics after 19 days of medication and 14 days of tachypacing. Representative parasternal view echocardiographic images in M mode from dogs in the sham (a), placebo (b), and edoxaban (c) groups. LVEDD (d), LVESD (e), and LVEF (f). Data are presented as mean ± SEM (n = 6 per group). **p < 0.01 versus sham. There were no significant differences between dogs in the placebo and edoxaban groups. LVEDD LV end-diastolic diameter, LVESD LV end-systolic diameter, Sh sham dogs, Pl placebo dogs, Ed edoxaban dogs

Changes in left atrial area. Echocardiographic characteristics of the left atrium (LA) after 19 days of medication and 14 days of tachypacing. Representative images of the apical view echocardiogram in dogs in the sham (a), placebo (b), and edoxaban (c) groups. d End-systolic LA area. Data are presented as mean ± SEM (n = 5 per group). **p < 0.01 versus sham. There was no significant difference between dogs in the placebo and edoxaban groups. Sh sham dogs, Pl placebo dogs, Ed edoxaban dogs
Electrophysiological characteristics and AF promotion
Mean AERPs tended to be prolonged in dogs of the placebo group compared with those in the sham group (BCL 360 ms p = ns; BCL 300 ms p = 0.055; BCL 250 ms p = 0.096; BCL 200 ms p = 0.065 vs. sham); however, mean AERPs in edoxaban group had no significant differences compared with both the sham and placebo groups (Fig. 3a). Similarly, the width of P wave tended to extend in dogs of the placebo group (p = 0.08 vs. sham), but not in those of the edoxaban group (p = 0.34 vs. sham) (Fig. 3b). Alternatively, there were no significant differences in AF inducibility among the three groups (Fig. 4). The average and maximum durations of AF were significantly prolonged in dogs of the placebo groups than in those of the sham group (p < 0.01), but this prolongation in the duration of AF was completely suppressed in dogs of the edoxaban group than in those of the placebo group (p < 0.01). Indeed, duration of AF in dogs of the edoxaban group did not differ from that in dogs of the sham group (Fig. 4).

Changes in the atrial effective refractory period and P wave width. Atrial effective refractory period (AERP) (a) and P wave width during sinus rhythm (b) after 19 days of medication and 14 days of tachypacing. Data are presented as mean ± SEM (n = 6 per group). Sh sham dogs, Pl placebo dogs, Ed edoxaban dogs

Inducibility and duration of atrial fibrillation. Parameters of atrial fibrillation (AF) promotion after 19 days of medication and 14 days of tachypacing. AF inducibility (a), mean AF duration (b), maximum AF duration (c). AF was induced after burst pacing in the placebo group (d) and the edoxaban group (e). Data are presented as mean ± SEM (n = 6 per group). **p < 0.01 versus sham. ††p < 0.01 versus placebo. LAA left atrial appendage, II surface electrocardiogram lead II, Sh sham dogs, Pl placebo dogs, Ed edoxaban dogs
Hemodynamic measurement and bodyweight
End-systolic mean left atrial pressure was higher in dogs of the placebo group than those of the sham group. However, there were no significant differences in hemodynamic indices between dogs of the placebo and edoxaban groups (Table 1). Further, body weight did not differ among the groups at the end of the experiment.
Histological remodeling
Representative histological findings are shown in Fig. 5. Atrial interstitial fibrosis was significantly greater in dogs of the placebo group than that in dogs of the sham group. This fibrotic response to tachypacing was significantly reduced by edoxaban (p < 0.01 vs. placebo), although it remained higher than in the sham group (p < 0.01 vs. sham), indicating only partial mitigation.

Histological analysis. Sirius Red-stained light micrograph images (original magnification ×400) of the left atrial tissue from representative dogs in the sham (a), placebo (b), and edoxaban (c) groups. d Proportion of interstitial fibrotic tissue. Scale bar = 100 μm. Data are presented as mean ± SEM (n = 6 per group). **p < 0.01 versus sham. ††p < 0.01 versus placebo. Sh sham dogs, Pl placebo dogs, Ed edoxaban dogs
Changes in PAR2 and fibronectin expression
Compared with dogs of the sham group, PAR2 expression significantly enhanced in the placebo group (p < 0.05) but not in the edoxaban group (vs. placebo, p < 0.05. vs. sham, p = ns) (Fig. 6a). In addition, fibronectin expression was significantly enhanced in the placebo group (p < 0.01) but not in the edoxaban group (vs. placebo, p < 0.05. vs. sham, p = ns) (Fig. 6b).

Western blotting analysis. Left atrial expression levels of PAR2 (a) and fibronectin (b). Data are presented as mean ± SEM (n = 6 per group). *p < 0.05; **p < 0.01 versus sham.:†p < 0.05 versus placebo. PAR2 protease-activated receptor 2, GAPDH glyceraldehyde-3-phosphate dehydrogenase (gel loading control), Sh sham dogs, Pl placebo dogs, Ed edoxaban dogs
Discussion
Major findings
The main findings of this study are that edoxaban suppressed atrial PAR2 and fibronectin upregulations, atrial interstitial fibrosis, and AF progression induced by ventricular tachypacing in a canine CHF model.
Potential mechanisms underlying the effects of edoxaban on atrial structural remodeling
In the CHF model, VTP reduces left ventricular contractility, leading to heart failure and the progression of atrial fibrosis. VTP increases the levels of angiotensin II and transforming growth factor-β (TGF-β) in the atrium. As a result, it promotes the expression of connective tissue growth factor (CTGF), matrix metalloproteinase-2 (MMP-2), tissue inhibitor of metalloproteinase-1 (TIMP-1), and extracellular matrix proteins such as collagen and fibronectin [17, 19, 20]. In addition, VTP increases tumor necrosis factor (TNF-α) and extracellular signal-regulated kinase (ERK1/2), c-Jun N-terminal kinase, and p38 in the atrium [17, 21]. However, after 2 weeks, VTP does not increase the levels of fibrotic mediators and does not promote fibrosis in the left ventricle [17, 20].
In the present study, the progression of atrial fibrosis and upregulation of PAR2 and fibronectin were found to be induced by ventricular tachypacing for 2 weeks, but edoxaban partially inhibited these changes. The addition of factor Xa to the human atrial tissue has been reported to increase the expression of ERK1/2, whereas PAR2 antagonist inhibits this enhancement [22]. In addition, increased fibrosis and the increased gene expression of TGF-β, CTGF, TIMP1, and MMP2 have been reported in mice overexpressing α-MHC PAR2 [11]. These data suggest that the Xa/PAR2 signal is involved in myocardial fibrosis, and hypertrophy. It has been reported that coagulation activity is enhanced in individuals with CHF accompanied by left ventricular dysfunction because of stagnation of blood flow caused by low cardiac output and enhancement of sympathetic activity [23,24,25]. Therefore, there is a possibility that factor Xa increased in this model, thus increasing the level of PAR2. However, we could not evaluate Xa activity in the present study because of technical limitations. Another possibility is that there is a pathway via PAR2 for remodeling of heart failure. The Xa inhibitor, rivaroxaban, has been reported to suppress the cell proliferation and upregulation of expression of TIMP1 and TNF-α in mouse cardiac fibroblasts stimulated by angiotensin II [26]. Additionally, Xa inhibitor, rivaroxaban, and PAR2 antagonist, FSLLRY, have been reported to suppress overexpression of protein levels of PAR2, phosphorylate ERK1/2, and phosphorylate smad3 in cardiac microvascular endothelial cells induced by intermittent hypoxia [15]. Ritchie et al. [27] reported that the overexpression of PAR2 is induced by TNF-α in human endothelial cell experiment. Therefore, it is suggested that the levels of angiotensin II, cytokines, and hypoxia, which increase during heart failure, are involved in the hyper proliferation of PAR2, and it is possible that Xa inhibitor partially suppresses the PAR2 signal by inhibiting factor Xa. Therefore, we hypothesized that upregulation of PAR2 signaling was suppressed by edoxaban, ultimately inhibiting atrial fibrosis in the LA and AF progression.
Effects of edoxaban on atrial electrical remodeling
It is known that shortening of AERP facilitates easier sustenance of AF [28,29,30,31]. In our CHF model, it is experimentally proven that the (1) the downregulation of atrial transient outward (Ito), slow delayed rectifier (Iks), and L-type Ca2+; (2) the upregulation of Na+–Ca2+ exchange current; (3) increase in Cx43 dephorylation and Cx43 redistribution toward transverse cell boundaries are observed by tachypacing, changes that would prolong the AERP [4]. These biophysical changes were rapidly reversed when ventricular tachypacing was stopped; therefore, the main pathomechanism underlying persistent AF in the CHF model is thought to be structural remodeling [31, 32]. In the present study as well, tachypacing tended to prolong AERP and extend width of P wave in the placebo group, an effect that was not observed in the edoxaban group. However, because myocardial ion channels are not considered adequately in the present study, further investigation is necessary.
Study limitations
The present experiment suggested that the overexpression of PAR2 caused atrial fibrosis; however, in this experiment, it was not possible to evaluate Χa activity and the mediators downstream of PAR2. Essentially, the quantitative upregulation of PAR2 was observed. We suggest that edoxaban may suppress upregulation of PAR2, although it is unclear as to what extent PAR2 affects atrial fibrosis compared with other pathways. Thus, further studies are required to confirm this and eliminate other possible mechanisms. Second, it is unknown whether edoxaban can suppress atrial remodeling in an atrial tachypacing model of AF without CHF because patients with AF and without CHF are expected to have lower coagulation activity and reduced levels of angiotensin II and cytokines than individuals with heart failure. Lastly, similar to any animal study, relevance to human pathology is unknown. The mechanism of edoxaban inhibition against atrial fibrosis is thought to be mediated via PAR2, and the mechanism differs from angiotensin II receptor blocker and statin which have shown an inhibitory effect on AF in animal experiments but failed to exhibit so-called upstream therapy in clinical studies in the past [33, 34]. Similar to other drugs, it is unknown whether edoxaban would exhibit similar clinical effects. In particular, the dose used in this experiment is higher than the usual clinical dose, and high doses may cause serious bleeding. Therefore, further study is needed to determine whether edoxaban is a viable remedy for progression of AF.
Conclusions
Edoxaban can inhibit atrial structural remodeling and progression of AF in a canine CHF model induced by ventricular tachypacing. These findings suggested that the suppression of the Xa/PAR2 pathway is a potential pharmacological target of edoxaban.
References
Iwasaki YK, Nishida K, Kato T, Nattel S (2011) Atrial fibrillation pathophysiology: implications for management. Circulation 124:2264–2274
Nishida K, Nattel S (2014) Atrial fibrillation compendium: historical context and detailed translational perspective on an important clinical problem. Circ Res 114:1447–1452
Nattel S, Burestein B, Dobrev D (2008) Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 1:62–73
Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH, Nattel S (2009) Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res 105:1213–1222
Nishida K, Qi XY, Wakili R, Comtois P, Chaetier D, Harada M, IwasakiYK Romeo P, Maguy A, Dobrev D, Michael G, Talajic M, Nattel S (2011) Mechanisms of atrial tachyarrhythmia associated with coronary artery occlusion in a chronic canine model. Circulation 123:137–146
Burstein B, Nattel S (2008) Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol 51:802–809
Borisoff J, Spronk HM, ten Cate H (2011) The hemostatic system as a modulator of atherosclerosis. N Engl J Med 364:1746–1760
Spronk HM, de Jong AM, Crijns HJ, Schotten U, Van Gelder IC, Ten Cate H (2014) Pleiotropic effects of factor Xa and thrombin: what to expect from novel anticoagulants. Cardiovasc Res 101:344–351
Bukowska A, Zacharias I, Weinert S, Skopp K, Hartmann C, Huth C, Goette A (2013) Coagulation factor Xa induces an inflammatory signaling by activation of protease-activated receptors in human atrial tissue. Eur J Pharmacol 718:114–123
Borensztajn K, Peppelenbosch MP, Spek CA (2008) Factor Xa: at the crossroads between coagulation and signaling in physiology and disease. Trends Mol Med 14:429–440
Antoniak S, Sparkenbaugh EM, Tencati M, Rojas M, Mackman N, Pawlinski R (2013) Protease activated receptor-2 contributes to heart failure. PLoS ONE 8:e81733
Borenstajin K, Stiekema J, Nijmeijer S, Reitsma PH, Peppelenbosch MP, Spek CA (2008) Factor Xa stimulates proinflammatory and profibrotic responses in fibroblasts via protease-activated receptor-2 activation. Am J Pathol 172:309–320
Kondo H, Abe I, Fukui A, Saito S, Miyoshi M, Aoki K, Shinohara T, Teshima Y, Yufu K, Takahashi N (2018) Possible role of rivaroxaban in attenuating pressure-overload-induced atrial fibrosis and fibrillation. J Cardiol 71:310–319
Bode MF, Auriemma AC, Grove SP, Hisada Y, Rennie A, Bode WD, Vora R, Subramaniam S, Cooley B, Andrade-Gordon P, Antoniak S, Mackman N (2018) The factor Xa inhibitor rivaroxaban reduces cardiac dysfunction in a mouse model of myocardial infarction. Thromb Res 167:128–134
Imano H, Kato R, Tanikawa S, Yoshimura F, Nomura A, Ijiri Y, Yamaguchi T, Izumi Y, Yoshiyama M, Hayashi T (2018) Factor Xa inhibition by rivaroxaban attenuates cardiac remodeling due to intermittent hypoxia. J Pharmacol Sci 137:274–282
Nishida K, Michael G, Dobrev D, Nattel S (2010) Animal models for atrial fibrillation: clinical insights and scientific opportunities. Europace 12:160–172
Hanna N, Cardin S, Leung TK, Nattel S (2004) Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc Res 63:236–244
Furugohri T, Isobe K, Honda Y, Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (2008) Du-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost 6:1542–1549
Cardin S, Libby E, Pelletier P, Bouter SL, Takeshita A, Meur NL, Leger J, Demolombe S, Ponton A, Glass L, Nattel S (2007) Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res 100:425–433
Cardin S, Pelletier P, Libby E, Bouter SL, Xiao L, Kaab S, Demolombe S, Glass L, Nattel S (2008) Marked differences between atrial and ventricular gene-expression remodeling in dogs with experimental heart failure. J Mol Cell Cardiol 45:821–831
Lee KW, Everett T, Rahmutula D, Guerra JM, Wilson ED, Ding C, Olgin JE (2006) Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 114:1703–1712
Bukowska A, Zachrias I, Weinert S, Skopp K, Hartmann C, Huth C, Goette A (2013) Coagulation factor Xa induces an inflammatory signaling by activation of protease-activated receptors in human atrial tissue. Eur J Pharmacol 718:114–123
Maze SS, Kotler MN, Parry WR (1989) Flow characteristics in the dilated left ventricular with thrombus: qualitative and quantitative Doppler analysis. J Am Coll Cardiol 13:873–881
Yamamoto K, Ikeda U, Furuhashi K, Irokawa M, Nakayama T, Shimada K (1995) The coagulation system is activated in idiopathic cardiomyopathy. J Am Coll Cardiol 25:1634–1640
Gustavsson CG, Persson SU, Larsson H, Persson S (1994) Changed blood rheology in patients with idiopathic dilated cardiomyopathy. Angiology 45:107–111
Hashikata T, Tojo M, Namba S, Kitasato L, Kameda R, Murakami M, Niwano H, Shimohama T, Tojo T, Ako J (2015) Rivaroxaban inhibits fibrotic progression in vitro. Int Heart J 56:544–550
Ritchie E, Saka M, MacKenzie C, Drummond R, Wheeler-Jones C, Kanke T, Plevin R (2007) Cytokine upregulation of proteinase-activated-receptors 2 and 4 expression mediated by p38 MAP kinase and inhibitory kappa B kinase β in human endothelial cells. Br J Pharmacol 150:1044–1054
Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chaetier D, Villeneuve LR, Brundel BJ, Dobrev D, Nattel S (2008) Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res 103:845–854
Nattel S, Li D (2000) Ionic remodeling in the heart: pathophysiological significance and new therapeutic opportunities for atrial fibrillation. Circ Res 87:440–447
Allessie M, Ausma J, Schotten U (2002) Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res 54:230–246
Li D, Fareh S, Leung TK, Nattel S (1999) Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 100:87–95
Cha TJ, Ehrlich JR, Zhang L, Shi Y-F, Tardif J-C, Leung T-K, Nattel S (2004) Dissociation between ironic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation 109:412–418
Goette A, Schon N, Kirchhof P, Breithardt G, Fetsch T, Hausler KG, Klein HU, Steinbeck G, Wegscheider K, Meinertz T (2012) Angiotensin II-antagonist in paroxysmal atrial fibrillation (ANTIPAF) trial. Circ Arrythm Electrophysiol 5:43–51
Rahimi K, Emberson J, McGale P, Majoni W, Merhi A, Asselbergs FW, Krane V, Macfarlane PW (2011) Effect of statin on atrial fibrillation: collaborative meta-analysis of published and unpublished evidence from randomized controlled trials. BMJ. https://doi.org/10.1136/bmj.d1250
Acknowledgments
The authors thank Kiyomi Note for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tsujino, Y., Sakamoto, T., Kinoshita, K. et al. Edoxaban suppresses the progression of atrial fibrosis and atrial fibrillation in a canine congestive heart failure model. Heart Vessels 34, 1381–1388 (2019). https://doi.org/10.1007/s00380-019-01377-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-019-01377-2