
Abstract
Key message
Heterologous expression of a nematode-responsive promoter in tomato successfully driven the RNAi constructs to impart root-knot nematode resistance.
Abstract
The root-knot nematode Meloidogyne incognita seriously afflicts the global productivity of tomatoes. Nematode management options are extremely reliant on chemical methods, however, only a handful of nematicides are commercially available. Additionally, nematodes have developed resistance-breaking phenotypes against the commercially available Mi gene-expressing tomatoes. Nematode resistance in crop plants can be enhanced using the bio-safe RNAi technology, in which plants are genetically modified to express nematode gene-specific dsRNA/siRNA molecules. However, the majority of the RNAi crops conferring nematode tolerance have used constitutive promoters, which have many limitations. In the present study, using promoter-GUS fusion, we functionally validated two nematode-inducible root-specific promoters (pAt1g74770 and pAt2g18140, identified from Arabidopsis thaliana) in the Solanum lycopersicum-M. incognita pathosystem. pAt2g18140 was found to be nematode-responsive during 10–21 days post-inoculation (dpi) and became non-responsive during the late infection stage (28 dpi). In contrast, pAt1g74770 remained nematode-responsive for a longer duration (10–28 dpi). Next, a number of transgenic lines were developed that expressed RNAi constructs (independently targeting the M. incognita integrase and splicing factor genes) driven by the pAt1g74770 promoter. M. incognita parasitic success (measured by multiplication factor ratio) in pAt1g74770:integrase and pAt1g74770:splicing factor RNAi lines were significantly reduced by 60.83–74.93% and 69.34–75.31%, respectively, compared to the control. These data were comparable with the RNAi lines having CaMV35S as the promoter. Further, a long-term RNAi effect was evident, because females extracted from transgenic lines were of deformed shape with depleted transcripts of integrase and splicing factor genes. We conclude that pAt1g74770 can be an attractive alternative to drive localized expression of RNAi constructs rather than using a constitutive promoter. The pAt1g74770-driven gene silencing system can be expanded into different plant–nematode interaction models.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plant–parasitic root-knot nematode (RKN), Meloidogyne incognita, can infect more than 3000 plant species and cause an immense yield decline in Solanaceous and Cucurbitaceous vegetable crops (Phani et al. 2023). This sedentary endoparasitic nematode maintains an intricate and long-lasting relationship with the host plant by establishing a hypermetabolic feeding site (may contain multiple numbers of giant cells) in the vascular cylinder that serves as the continual food source for the developing nematodes (Vieira and Gleason 2019). The tissues surrounding the feeding site become hypertrophied to form the macroscopic knots or galls that hinder the normal physiological processes of plants, including water and nutrient transport across the vascular tissue (Kaloshian and Teixeira 2019). Finding sustainable RKN management tactics has been a difficult task because of the phasing out of environmentally harmful nematicides, and limitations in the wide-spectrum applicability and feasibility of cultural, physical and biological control measures. Although a number of resistance (R) genes were identified from the wild relatives of Solanaceous crops and upon introgression into the cultivated species either via grafting or molecular breeding conferred improved RKN resistance (Barbary et al. 2015), only a few of the R genes such as Mi-1.2 (in tomato) and N (in pepper) are commercially available (Phani et al. 2023).
RNA interference (RNAi) is considered a powerful reverse genetic tool, which uses double-stranded RNA (dsRNA) or small-interfering RNA (siRNA, generated upon cleavage of dsRNA by dicer enzyme) molecules to induce the degradation of cognate endogenous mRNAs and prevent the synthesis of the encoded protein (Rosso et al. 2009; Lilley et al. 2012). In the host-induced gene silencing (HIGS) strategy, plants are genetically modified to express dsRNA molecules, which correspond to the pathogen (that infects the host plant)-specific gene sequences. A number of nematode survival- and parasitism-related genes were targeted using HIGS, which conferred improved nematode resistance in different crops (Dutta et al. 2015a,b, 2020; Shivakumara et al. 2017; Chaudhary et al. 2019; Joshi et al. 2020; Mani et al. 2020; Moreira et al. 2022, 2023). Due to the ease of designing the dsRNA/siRNA molecules, target selectivity, and biodegradable nature, RNAi has a low environmental impact (Fletcher et al. 2020). In addition, negligible biosafety concerns are associated with HIGS because no foreign proteins are translated in planta (Dutta et al. 2015a). However, a slow adoption of RNAi crops is apparent, probably because HIGS-based products are regulated as genetically modified organisms (GMOs) in many countries (Papadopoulou et al. 2020). Amid this regulatory conundrum, a number of RNAi crops have already been commercialized. For example, Bayer “SmartStax Pro” maize (Mon87411) expresses a dsRNA that corresponds to the Snf7 gene of the western corn rootworm, Diabrotica virgifera virgifera (De Schutter et al. 2022). Bayer “Vistive Gold” high-oleic soybean (Mon87705) produces healthier oils by targeting a gene in the fatty acid biosynthesis pathway (De Schutter et al. 2022).
The majority of the HIGS studies targeting nematodes have used constitutive promoters including CaMV35S and pUbi1 to drive the expression of dsRNA cassettes (Sindhu et al. 2009; Dutta et al. 2015a, b; Banerjee et al. 2017; Moreira et al. 2023). Since constitutive promoters can drive target gene expression in the whole plant, the likelihood of obtaining off-target pleiotropic phenotypes (especially when plant endogenous genes have sequence homology to target dsRNA/siRNA sequences) is very obvious. This type of phenotypic variation can interfere with data interpretation in wild-type and transgenic plants (Peremarti et al. 2010). Additionally, expression of the constitutive promoter was found to be variable in the nematode feeding sites (Urwin et al. 1997; Ali et al. 2017). In many cases, downregulation of the target transcripts was observed in the feeding cells (Goddijn et al. 1993; Goverse et al. 1998; Bertioli et al. 1999). In view of this, the deployment of tissue-specific, nematode-responsive promoters appears to be an attractive alternative.
Research on nematode-inducible promoters was mostly performed in Arabidopsis thaliana, probably because of the amenability of promoter tagging and T-DNA mutant generation in this model plant. Syncytia (feeding cells induced by cyst nematodes)-specific promoters such as Pdf2.1 and MIOX5 were identified from the A. thaliana-Heterodera schachtii pathosystem (Siddique et al. 2009, 2011). A number of root-specific promoters such as Atcel1, AtWRKY23, TobRB7, LEMMI9, Hahsp17, MDK4-20, ZmRCP-1 and TUB-1 were used to drive the expression of reporter genes, proteinase inhibitors and nematode-repellent peptides in different host plants (Opperman et al. 1994; Escobar et al. 1999, 2003; Lilley et al. 2004, 2011; Sukno et al. 2006; Grunewald et al. 2008; Green et al. 2012; Papolu et al. 2016). In our earlier studies, two root-specific promoters pAt1g74770 and pAt2g18140 that regulate the expression of A. thaliana endogenous genes zinc-ion-binding protein (Gene ID: AT1G74770) and peroxidase (AT2G18140), respectively, were found to be responsive to M. incognita infection in A. thaliana (Kakrana et al. 2017; Joshi et al. 2022). A fusion with the reporter gene glucuronidase (GUS) showed root gall-specific expression of pAt2g18140, whereas pAt1g74770 exhibited strong expression across the nematode-infected root tissues (Kakrana et al. 2017). Further, pAt1g74770 and pAt2g18140 successfully driven the RNAi constructs of splicing factor (involved in nematode survival and development) and Mi-msp2 (a pioneer effector) genes, respectively, in A. thaliana, which conferred improved resistance against M. incognita, compared to the wild-type plants (Kakrana et al. 2017; Joshi et al. 2022). Considering the need for sustainable nematode management options, exploring the utility of these nematode-inducible promoters in commercial crops would have been advantageous.
In the present study, both pAt1g74770 and pAt2g18140 were heterologously expressed (with GUS reporter fusion) in a Solanaceous crop tomato (Solanum lycopersicum L.) and their nematode responsiveness was assessed upon M. incognita infection. Additionally, the RNAi constructs of M. incognita development-related genes integrase and splicing factor were expressed (driven by pAt1g74770) in tomato to examine the potential of the root-specific promoter in conferring HIGS.
Materials and methods
Maintenance of M. incognita culture
The pure culture of M. incognita (Kofoid & White) Chitwood race 1 was propagated in the roots of eggplant (Solanum melongena L.) in a greenhouse at 28ºC, 60% relative humidity and 16:8 h light–dark photoperiod. Identification of the pure culture was performed by analyzing the perineal pattern of females (Koulagi et al. 2020) and using a SCAR-PCR-based species-specific molecular marker (Adam et al. 2007). At 30 days post-infection, egg masses were hand-picked from the infected roots using sterilized forceps and kept for hatching in a modified Baermann assembly (Southey 1986) containing sterile tap water at 28ºC. Freshly hatched second-stage juveniles (J2s) were used for infection experiments.
Generation of promoter::GUS fusion constructs
The genomic DNA of A. thaliana was extracted as described previously (Dutta et al. 2023a). The promoter regions (approximately 1500 bp upstream of the start codons) of the At1g74770 and At2g18140 genes were PCR-amplified from the genomic DNA using specific primers (Supplementary Table S1) flanked by BamHI and SalI restriction enzyme recognition sites. PCR products (1.5 Kb) were resolved on 1% (w/v) agarose gel and gel-eluted DNA fragments were double-digested with BamHI and SalI (New England Biolabs). Promoter fragments were ligated into the linearized (via double digestion with BamHI and SalI) pORE-R2 vector at the upstream of the gusA gene (Coutu et al. 2007) using T4 DNA ligase (New England Biolabs) with a recommended insert-vector molar ratio of 3:1. pORE-R2:pAt1g74770 and pORE-R2:pAt2g18140 recombinant clones were initially transformed into E. coli DH5α cells via electroporation, sequence verified and eventually transformed into Rhizobium radiobacter (syn. Agrobacterium tumefaciens) strain GV3101 by the freeze–thaw method. Recombinant Rhizobium colonies were identified by colony PCR and those colonies were propagated in yeast-peptone (YEP) agar (Sigma-Aldrich) supplemented with rifampicin (25 µg mL−1), gentamicin (50 µg mL−1) and kanamycin (50 µg mL−1) antibiotics.
Transformation of tomato with promoter::GUS fusion
Tomato cv. Pusa Ruby seeds were surface-sterilized with 70% ethanol for 5 min and 10% NaOCl for 15 min followed by rinsing with sterile distilled water five to six times. Seeds were germinated on Murashige and Scoog (MS) agar medium (Sigma-Aldrich, pH 5.8) and cotyledonary leaves (~ 1 cm2 leaf discs or explants) of a fortnight-old seedlings were excised for R. radiobacter co-culture. Explants were pre-cultivated in MS agar supplemented with 0.5 mg L−1 each of indole acetic acid (IAA) and zeatin riboside (ZR). In parallel, recombinant R. radiobacter colonies were grown overnight (0.6–0.8 OD600) in YEP broth. Rhizobium culture was precipitated via centrifugation at 5000 g for 5 min at 4 °C, and pellets were suspended in MS broth. Two-day-old pre-cultivated explants were infected with R. radiobacter colonies for 15 min via shaking at 180 rpm at 28 °C in the dark. Co-cultured explants were blot-dried and placed in MS agar supplemented with 0.5 mg L−1 each of IAA and ZR. Two-day-old co-cultivated explants were rinsed with 250 mg L−1 cefotaxime antibiotic for 20 min, and then transferred onto the MS agar containing 0.5 mg L−1 each of IAA and ZR, 50 mg L−1 kanamycin, and 250 mg L−1 cefotaxime. MS agar plates were incubated at 25 °C with a 16:8 h light–dark photoperiod (light level—250 μmol m−2 s−1). After 15–20 days of culturing in regeneration media, shoots emerged from the explant were excised, and sub-cultured in a fresh medium for shoot elongation. For root induction, elongated shoots were placed in MS agar containing 0.5 mg L−1 naphthalene acetic acid (NAA), 50 mg L−1 kanamycin, and 250 mg L−1 cefotaxime. At 20–25 days after rooting, plantlets were transferred to 10 cm-diameter plastic pots containing an equal mixture of soil rite (Keltech Energies Ltd.) and autoclaved top soil. Well-established plants with hardened roots were shifted to the National Phytotron Facility, Indian Agricultural Research Institute, for flowering, fruiting and T0 seed production. T0 events (after growing in MS media containing 100 µg mL−1 kanamycin) were selfed to generate T1 seeds. Plants transformed with an empty pORE-R2 vector served as the control.
Molecular analysis of transformed tomato plants
For PCR-based detection of the transgene, genomic DNA was extracted from the leaves of T1 events using the NucleoSpin Plant II Kit (TaKaRa) as per the manufacturer’s protocol. Fragments of different target genes such as GUS, nptII (antibiotic marker), pAt1g74770 and pAt2g18140 were PCR-amplified from the genomic DNA using a standard protocol (TaKaRa) with specific primers (Supplementary Table S1). Amplified fragments were electrophoresed in a 1% (w/v) agarose gel.
For detecting the integration patterns of pAt1g74770 and pAt2g18140 in T1 events, Southern hybridization was performed. 10 µg of genomic DNA from each PCR-positive event was digested with 50U BamHI (New England Biolabs) overnight at 37 °C. Cleaved DNA was run on a 0.8% (w/v) agarose gel followed by transfer into a nitro-cellulose membrane (Pall Life Sciences) via capillary action in 10X saline sodium citrate (SSC) buffer (1.5 M NaCl, 0.15 M sodium citrate pH 7.0). For probe preparation, pAt1g74770 and pAt2g18140 DNA fragments (1.5 Kb) were biotin labelled with a chemiluminescent reagent Biotin DecaLabel DNA labelling kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. The membrane was hybridized overnight at 65 °C in a hybridization buffer (2 M Na2HPO4 pH 7.2, 10% sodium dodecyl sulfate (SDS), 0.5 M EDTA pH7.0) containing denatured probes. Next, membrane was subjected to three consecutive washes (30 min each at 65 °C) in 3X SSC + 0.1% SDS followed by 0.5X SSC + 0.1% SDS followed by 0.1X SSC + 0.1% SDS. Hybridized bands were detected using streptavidin–alkaline phosphatase conjugate that detects biotinylated antibodies (Thermo Fisher Scientific).
To analyze the GUS expression in T1 events, RT-qPCR was carried out. Total RNA was extracted from different events using the NucleoSpin RNA Plant Kit (TaKaRa) as per the manufacturer’s protocol. RNA integrity was assessed via electrophoresis in 1% (w/v) agarose gel. RNA purity and quantity were ascertained in a Nanodrop spectrophotometer (Thermo Fisher Scientific). 1 µg RNA was reverse-transcribed to cDNA using the Verso cDNA synthesis Kit (Thermo Fisher Scientific). The qPCR reaction was performed in a CFX96 thermal cycler (BioRad) with a reaction mix (10 μL) containing 1.5 ng cDNA, 750 nM each of sense and antisense primers, and 5 μL SYBR Green PCR master-mix (BioRad). Amplification conditions comprised a hot start phase of 95 °C for 30 s, then 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Amplification specificity was determined by using a melt curve program (95 °C for 15 s, 60 °C for 15 s, and finally a slow ramp from 60 to 95 °C). Quantification cycle (Cq) values were obtained from CFX Maestro software (BioRad). S. lycopersicum housekeeping genes (18S rRNA and actin) were used as internal references to normalise the target gene expression data. Fold change in gene expression was calculated using the comparative Cq method (Schmittgen and Livak 2008). The PCR efficiency of the primers was calculated by following the standard MIQE guidelines (Bustin et al. 2009). The qPCR run constituted of three biological and five technical replicates for each sample. The qPCR primer details and PCR efficiency are given in Supplementary Table S1.
Histochemical GUS assay
T1 plants growing in 500 ml pots containing an equal mixture of soil rite and top soil were inoculated with 1000 J2s per root system (@ 2 J2s per g of soil). Histochemical localisation of GUS activity was carried out using the substrate 5-bromo-4-chloro-3-indolyl-β-D glucuronide (XGluc). M. incognita-inoculated roots at different infection stages (7, 14, 21, and 28 days post-inoculation) were harvested, washed to remove adhered soil, and immersed in a readily-prepared GUS staining solution (0.5 mM X-Gluc, 0.1 M NaHPO4 pH 8.0, 0.5 mM K3Fe(CN)6, 0.5 mM K4Fe(CN)6, 0.01 M EDTA pH 8.0, 20% methanol, 0.1% Triton X-100) for overnight in the dark at 37 °C. Root segments were washed with 10% phosphate buffer to stop the reaction and cleared of stain by immersing in 70% ethanol. Photomicrographs of GUS-stained root segments were taken with a Nikon stereomicroscope. Additionally, roots were stained with acid fuchsin (the detailed method is provided in Byrd et al. 1983) to detect the endoparasites in the infected tissue.
Cloning of nematode-inducible promoter and downstream RNAi constructs in a binary vector
DsRNA molecules for integrase (624 bp, Genebank ID: AW871671) and splicing factor (349 bp, AW828516) were designed based on the greater siRNA formation probability in the targeted region compared to the no-targeted sequence of the identical gene using different in silico tools such as siDirect, Dharmacon and dsCheck (described in Dutta et al. 2023b). Additionally, target siRNA sequence homologies were analyzed with siRNAs from non-target organisms (including C. elegans) using the dsCheck tool to avert the off-target effects (Dutta et al. 2023b). Integrase and splicing factor gene fragments (in sense and antisense orientation) were PCR-amplified from the cDNA of M. incognita J2s (RNA was extracted and reverse-transcribed to cDNA as described above). The backbone of the RNAi binary vector pBC06 (Yadav et al. 2006) was used for cloning. Initially, the CaMV35S sequence was replaced with the pAt1g74770 sequence using SbfI and BamHI restriction enzymes. Next, sense (flanked by BamHI and XhoI restriction sites) and antisense (flanked by KpnI and SacI restriction sites) fragments of integrase were cloned in pBC06 to obtain the pAt1g74770::integrase RNAi construct. Similarly, sense and antisense fragments of splicing factor were cloned in pBC06 to obtain the pAt1g74770::splicing factor RNAi construct. In order to compare our nematode-inducible promoter results with that of the CaMV35S promoter, integrase and splicing factor sequences were separately cloned into the wild-type pBC06 vector. Cloned fragments were digested via selective restriction enzymes, gel-purified and Sanger sequenced to confirm their identity. RNAi constructs were transformed into S. lycopersicum cv. Pusa Ruby via the R. radiobacter co-culture method as described above. T1 plants were generated and molecular analysis including PCR, RT-qPCR and Southern hybridization were performed by following the methodologies described above.
Nematode infectivity assay
RNAi plants were infected with 1000 J2s per root system as described above. Plants were grown in a controlled environment in a greenhouse at 28 °C, 60% relative humidity and a 16:8 h light–dark photoperiod. Plants were harvested at 30 days post-inoculation (dpi), roots were carefully washed free of soil, and different infection parameters such as number of galls, egg masses, eggs per egg mass and multiplication factor (MF) ratio (Dutta et al. 2015b) were assessed per root system. Each treatment contained at least five replicates and the whole experiment was repeated at least thrice.
To verify the target gene silencing in feeding nematodes, RT-qPCR was performed. Reproducing females (at 30 dpi) feeding on the galled roots of RNAi plants were extracted via maceration of root tissues using forceps under the microscope, frozen in liquid nitrogen and stored at − 80 °C. Total RNA was extracted using the NucleoSpin RNA Plant Kit (TaKaRa) and reverse-transcribed to cDNA as described above. The transcript abundance of integrase and splicing factor genes was assessed using qPCR. Amplification conditions are described above. M. incognita 18S rRNA and actin genes were used as internal references.
Statistical analysis
Bioassays were set up as a completely randomized block design. Initially, the normality of both gene expression and bioassay data was checked via the Shapiro–Wilk test. Data were subjected to a one-way ANOVA followed by Tukey’s honest significant difference (HSD) at P < 0.01 using SAS version 14.1. Statistical comparisons were made between different treatments or between control and treatment, as stated in the figure captions.
Results
Root gall-specific expression of pAt1g74770 and pAt2g18140 upon M. incognita infection in tomato
Solanum lycopersicum cv. Pusa Ruby plants were separately transformed with pAt1g74770:GUS and pAt2g18140:GUS constructs that contained the promoter regions of At1g74770 and At2g18140, respectively, fused with the β-glucuronidase (GUS) reporter gene using the Rhizobium radiobacter co-culture method (Fig. 1a; Supplementary Figure S1). PCR amplification of the pAt1g74770 + gusA sequence (748 bp encompassing the partial sequences of pAt1g74770 and gusA) was verified in 13 out of 16 tested T1 events harboring the pAt1g74770:GUS construct (Fig. 1b). PCR amplification of the pAt2g18140 + gusA sequence (787 bp encompassing the partial sequences of pAt2g18140 and gusA) was verified in 9 out of 11 tested T1 events harboring the pAt2g18140:GUS construct (Fig. 1b). The PCR-positive events were subjected to a Southern hybridization assay to analyze the integration patterns of pAt1g74770 and pAt2g18140 in the S. lycopersicum genome. Four (event numbers 2, 3, 4, and 9) and six (event numbers 4, 5, 6, 8, 9, and 10) single-copy events harboring the independent integration of pAt1g74770 and pAt2g18140 genes, respectively, were confirmed from the blots (Fig. 1c). Event number 3 harboring the pAt2g18140 gene was a double-copy event (Fig. 1c). This event was not considered for further analysis because of the possibility of transgene co-suppression.

Generation of transformed tomato lines with promoter::GUS fusion. a The T-DNA portion of recombinant pORE-R2 is schematically represented. pAt1g74770 and pAt2g18140 drive the expression of gusA, an open reading frame (ORF) encoding β-glucuronidase. An ORF encoding neomycin phosphotransferase II (nptII) was used for selection in plants. PENTCUP2, Nicotiana tabacum cryptic constitutive promoter. TNOS—polyadenylation signal of the nopaline synthase gene. Arrows show the direction of transcription. LB, RB—left and right borders. Forward primer (FP) and reverse primer (RP) binding sites are indicated as black arrowheads. b Approximately 748 (corresponding to pAt1g74770 + gusA) and 787 (pAt2g18140 + gusA) bp fragments were PCR-amplified from different T1 events. Positive control consists of cloning vector fragments. Negative control contained no template DNA. M—1 Kb plus DNA ladder. c Southern blots showing the integration of pAt1g74770 and pAt2g18140 transgenes in different genomic locations of T1 events. Genomic DNA of untransformed control (UC) plants did not reveal any hybridization signal. Positive controls (PC) are the probes used to hybridize pAt1g74770 and pAt2g18140 fragments. M—Lambda HindIII digest marker
Histochemical GUS staining of nematode-infected T1 roots demonstrated the nematode-responsive root-specific expression of pAt1g74770 and pAt2g18140 promoters (Figs. 2, 3). No GUS activity was observed in uninfected roots, mechanically wounded roots and above-ground part of the infected roots (Figs. 2, 3). Both the promoters were unresponsive during the initial stage of nematode infection, i.e., 7 days post-inoculation (7dpi). From 10 dpi, GUS activity was detected in the infected roots. During 14 dpi, strong GUS activity was observed in and around the galled roots that was sustained till 21 dpi (Figs. 2, 3). However, during 28 dpi, although GUS activity was detectable in roots harboring the pAt1g74770:GUS construct (Fig. 2), no GUS activity was detected in roots harboring the pAt2g18140:GUS construct (Fig. 3). Our RT-qPCR-based gusA expression study supported the results of histochemical analyses. At 7 dpi, gusA expression was not significantly (P > 0.01) altered in nematode-infected roots of different T1 events ( 1 g-2, 1 g-3, 1 g-4 and 1 g-9 harboring the pAt1g74770:GUS fusion; 2 g-4, 2 g-5, 2 g-6, 2 g-8, 2 g-9 and 2 g-10 harboring the pAt2g18140:GUS fusion) compared to the uninfected roots (Supplementary Figure S2). At 14 and 21 dpi, gusA expression was differentially and significantly (P < 0.01) upregulated in nematode-infected roots of different T1 events compared to the uninfected roots (Supplementary Figure S2). At 28 dpi, compared to uninfected roots, gusA expression was significantly (P < 0.01) upregulated in nematode-infected T1 events harboring the pAt1g74770:GUS fusion, whereas gusA expression was unaltered (P > 0.01) in nematode-infected T1 events harboring the pAt2g18140:GUS fusion (Supplementary Figure S2). We hypothesized that pAt1g74770 may remain nematode-inducible for a longer duration of tomato–M. incognita interaction, in contrast to the non-responsiveness of pAt2g18140 during the late stage of nematode infection. In view of this, we selected the pAt1g74770 promoter for generating the RNAi plants.

Expression of pAt1g74770:GUS in tomato root upon M. incognita infection. a No GUS activity in uninfected roots upon mechanical wounding, b no GUS activity in nematode-infected roots at 7 dpi, c initiation of GUS activity in nematode-infected roots at 10 dpi, d Intense GUS activity in nematode-infected roots at 14 dpi, e, f GUS activity in nematode-induced galls at 21 dpi, g–i strong GUS activity in nematode-induced galls at 28 dpi. N—nematode female. Scale bar = 500 µm

Expression of pAt2g18140:GUS in tomato root upon M. incognita infection. a No GUS activity in uninfected roots, b no GUS activity in nematode-infected roots at 7 dpi, c no GUS activity in leaves of the nematode-infected plant at 10 dpi, d initiation of GUS activity in nematode-infected roots at 10 dpi, e, f intense GUS activity in nematode-induced galls at 14 dpi, g GUS activity in nematode-induced galls at 21 dpi, h no GUS activity in galls induced in secondary roots at 28 dpi, i, j no GUS activity in galls induced in primary roots at 28 dpi. Scale bar = 500 µm
pAt1g74770-driven RNAi construct expression protected tomato against M. incognita infection alike of pCaMV35S-driven RNAi construct
For generating the pAt1g74770-driven RNAi lines, the pCaMV35S sequence in the RNAi binary vector pBC06 was replaced with the pAt1g74770 sequence, followed by cloning of M. incognita integrase and splicing factor genes (separately) in sense and antisense orientations for eventual expression as dsRNA molecules (Fig. 4a). For a realistic comparison, integrase and splicing factor dsRNA cassettes were also cloned separately into the wild-type pBC06 containing the CaMV35S promoter. S. lycopersicum cv. Pusa Ruby plants were transformed with RNAi constructs using the R. radiobacter co-culture method. Sense strands of integrase and splicing factor transgenes were detected in different T1 events by PCR amplification (Supplementary Figure S3). Additionally, antisense strands and antibiotic marker fragments were detected in different events (data not shown). The PCR-positive events were subjected to a Southern hybridization assay to analyze the integration patterns of integrase and splicing factor transgenes in the S. lycopersicum genome. Five (event numbers 2, 3, 4, 6, and 10) and three (event numbers 4, 6, and 7) single-copy events harboring the independent integration of integrase and splicing factor genes, respectively, were confirmed from the blots (Fig. 4b). Event numbers 2, 3, and 5 harboring the dsRNA cassettes of the splicing factor gene showed double-copy integration of the transgene (Fig. 4b). Expression of integrase and splicing factor transgenes was detected in all these T1 events by RT-qPCR. Conversely, integrase and splicing factor expression were not detected in the RNAs isolated from control plants. Therefore, transgene expression data was represented as mean ΔCq relative to the Cq mean of S. lycopersicum 18S rRNA. Integrase transcripts were variably expressed in different single-copy events (I2, I3, I4, I6, and I10) (Fig. 4c). However, splicing factor transcripts were significantly (P < 0.01) greatly expressed (as determined via a lower ΔCq value) in single-copy events (Sf4, Sf6, and Sf7) compared to the double-copy events (Sf2, Sf3, and Sf5) (Fig. 4c). In view of this, only single-copy RNAi lines were taken up for further analysis.

Generation of specific promoter-driven RNAi tomato lines expressing the M. incognita integrase and splicing factor dsRNAs. a The T-DNA portion of recombinant pBC06 is schematically represented. pAt1g74770 drives the expression of integrase and splicing factor mRNAs in sense and antisense orientations. An ORF encoding neomycin phosphotransferase II (nptII) was used for selection in plants. PNOS, TNOS—promoter and terminator sequences of the nopaline synthase gene. Arrows show the direction of transcription. LB, RB—left and right borders. During plant transformation, dsRNAs are produced from intron-spliced hairpin RNAs. Feeding nematodes may ingest dsRNAs or Dicer-processed siRNAs. b Southern blots showing the integration of integrase and splicing factor transgenes in different genomic locations of T1 events. Genomic DNA of untransformed control (UC) plants did not reveal any hybridization signal. Probes used for hybridization correspond to the integrase (624 bp) and splicing factor (349 bp) genes. M—Lambda HindIII digest marker. c Detection of transgene expression in different T1 events via RT-qPCR. Relative transcript levels of integrase and splicing factor are expressed as ΔCq values that represent the differences in the Cq mean of transgene and a reference gene (S. lycopersicum 18S rRNA). Greater ΔCq value indicates the lower expression of the target gene in a specific event. Each bar represents the mean ΔCq value ± SE of qPCR runs in three biological and five technical replicates. Bars with different letters are significantly different at P < 0.01, Tukey’s HSD test
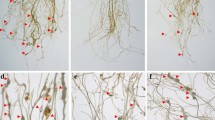
T1 plants transformed with RNAi constructs did not exhibit any pleiotropic effect when compared with the untransformed control or empty vector-transformed (UC) plants in terms of shoot length, root length, shoot weight and root weight data of 30-day-old plants (Supplementary Figure S4). Each plant was ‘challenge inoculated’ with 1000 J2s near the root, and at 30 dpi, plants were harvested to assess different infection parameters. Qualitative data showed greater galling intensity in control roots compared to the roots of RNAi plants (Fig. 5a). The number of galls per root system was significantly (P < 0.01) reduced by 42.85–53.34% in different T1 events expressing the pAt1g74770-driven integrase RNAi construct, compared to the control (Fig. 5b; Supplementary Figure S5). Similarly, the number of egg masses and the number of eggs/egg mass per root system were significantly (P < 0.01) reduced by 38.84–51.38% and 35.96–48.43%, respectively, in different T1 events expressing the pAt1g74770-driven integrase RNAi construct, compared to the control (Fig. 5b; Supplementary Figure S5). Ultimately, the nematode multiplication factor (MF) ratio [(egg mass × egg per egg mass) ÷ initial inoculum] was drastically (P < 0.01) reduced by 60.83–74.93% in T1 events compared to control (Fig. 5b; Supplementary Figure S5). Notably, the number of galls, egg masses, eggs per egg mass and MF ratio were significantly (P < 0.01) reduced by 46.06, 45.92, 44.91 and 70.21%, respectively, in a T1 event expressing the pCaMV35S-driven integrase RNAi construct, compared to the control (Fig. 5b; Supplementary Figure S5). The degree of improvement in nematode resistance was very much alike for pAt1g74770- and pCaMV35S-driven integrase RNAi plants, when compared to the control. A similar result was obtained for pAt1g74770- and pCaMV35S-driven splicing factor RNAi plants. The number of galls, egg masses, eggs per egg mass and MF ratio were significantly (P < 0.01) reduced by 47.84–59.07%, 45.83–51.33%, 43.41–49.28% and 69.34–75.31%, respectively, in different T1 events expressing the pAt1g74770-driven splicing factor RNAi construct, compared to the control (Fig. 5c; Supplementary Figure S5). A similar degree of reduction in nematode infection parameters was documented in a pCaMV35S-driven splicing factor RNAi event, compared to the control (Fig. 5c; Supplementary Figure S5).

Specific promoter-driven RNAi tomato lines affected the infectivity and reproductive success of M. incognita at 30 dpi. a Comparative galling intensity of the nematode-infected root system corresponding to the untransformed/empty vector-transformed control (UC) plant and T1 plants expressing the dsRNA constructs of integrase and splicing factor. b Relative differences in the various nematode infection parameter data in control and single-copy T1 events (I2, I3, I4, I6, and I10) expressing the integrase RNAi construct driven by the pAt1g74770 promoter. c Relative differences in the various nematode infection parameter data in control and single-copy T1 events (Sf4, Sf6, and Sf7) expressing the splicing factor RNAi construct driven by the pAt1g74770 promoter. For a realistic comparison, integrase and splicing factor RNAi constructs driven by the CaMV35S promoter were used. Each bar represents the mean ± SE (n = 10). Bars with different letters (within an identical parameter) are significantly different at P < 0.01, Tukey’s HSD test
Intriguingly, adult females recovered from RNAi lines were of deformed shape compared to the normal melon-shaped females extracted from control plants, at 30 dpi (Fig. 6a). Microscopic measurements indicated a size reduction in females feeding on the RNAi lines, compared to the control females. Although the average body length was statistically similar (P > 0.01), average greatest body width was greatly reduced (P < 0.01) in the females isolated from RNAi lines, compared to the control (Fig. 6b). This long-term RNAi effect of integrase and splicing factor was further confirmed by analyzing the expression of these genes in isolated females. Compared to control, expression of integrase was significantly (P < 0.01) downregulated by 68–73% in females extracted from different T1 events harboring the pAt1g74770-driven integrase RNAi construct (Fig. 6c). Compared to control, expression of splicing factor was significantly (P < 0.01) attenuated by 75–80% in females extracted from different T1 events harboring the pAt1g74770-driven splicing factor RNAi construct (Fig. 6d). Likewise, expression of integrase and splicing factor was significantly (P < 0.01) reduced by 55 and 50% in females isolated from T1 lines harboring the pCaMV35S-driven integrase and splicing factor RNAi constructs, respectively, compared to control (Fig. 6c, d).

Long-term effect of HIGS on M. incognita females infecting the RNAi lines at 30 dpi. a T1 plants expressing the dsRNA constructs caused morphological aberrations in nematode females. Females extracted from untransformed/empty vector-transformed control (UC) plants were of normal melon-shaped, whereas females extracted from transgenic lines were of elongated shape with a deformed neck and transparent body. b Average body length and greatest body width of females as measured under the microscope. Each bar represents the mean ± SE (n = 10). Bars with different letters (within an identical parameter) are significantly different at P < 0.01, Tukey’s HSD test. c Targeted downregulation of the integrase gene in nematode females extracted from five independent T1 lines (I2, I3, I4, I6, and I10) expressing the integrase RNAi construct driven by the pAt1g74770 promoter. For comparative analysis, a T1 line (35S-I) expressing the integrase RNAi construct driven by the CaMV35S promoter was used. d Targeted downregulation of the splicing factor gene in nematode females extracted from three independent T1 lines (Sf4, Sf6, and Sf7) expressing the splicing factor RNAi construct driven by the pAt1g74770 promoter. A T1 line (35S-Sf) expressing the splicing factor RNAi construct driven by the CaMV35S promoter was used for comparison. Fold change in gene expression was set at 1 in control and compared with other treatments. M. incognita 18S rRNA and actin were used as internal references to normalize the target gene expression. Each bar represents the mean fold change value ± SE of qPCR runs in three biological and five technical replicates. Bars with different letters are significantly different at P < 0.01, Tukey’s HSD test
Discussion
Considering that the constitutive overexpression or suppression of a specific gene can have a detrimental effect on plant growth and development (Ali et al. 2017), nematode-responsive root-specific promoters are a better choice to drive the expression of a candidate gene. In our earlier study, two root-specific promoters such as pAt1g74770 and pAt2g18140 were identified from A. thaliana that were highly expressed in the M. incognita-infected root galls (Kakrana et al. 2017). In the present study, we heterologously expressed pAt1g74770 and pAt2g18140 via GUS reporter fusion in S. lycopersicum and analyzed their induction upon M. incognita infection. BLASTn analysis showed that these promoters are exclusively present in the A. thaliana genome, because no homologous nucleotide sequences were detected in the genomes of Solanaceous plants including S. lycopersicum (Supplementary Figure S6), tobacco, potato, eggplant, pepper and petunia (data not shown). Although both of these promoters were found to be non-responsive during the early stage of nematode infection (7 dpi), their induction was detectable from 10 dpi onwards with greater GUS activity at 14 and 21 dpi in nematode-infected galls. However, a sharp contrast in their induction during the late stage of nematode infection (28 dpi) was evident because pAt1g74770 still remained nematode-inducible during that period while pAt2g18140 did not. Additionally, no leaky expression of these promoters in the shoot tissues and upon mechanical wounding was observed. Assuming that pAt1g74770 has the better potential to remain nematode-responsive for a longer duration than pAt2g18140, we selected the former to examine its efficacy in driving the RNAi constructs.
Notably, At1g74770 and At2g18140 annotate for zinc-binding and peroxidase proteins, respectively (Kakrana et al. 2017; Joshi et al. 2022). Zinc-binding proteins play a pivotal role in the plant’s response to pests and pathogens (Shirasu et al. 1999; Sharma et al. 2019; Cabot et al. 2019). They can aid in inducing plant defense as well as eliciting pathogen virulence (Cabot et al. 2019). A zinc-binding protein (HIPP3; At5g60800) in A. thaliana was specifically induced upon infection of Pseudomonas syringae pv. tomato; HIPP3 regulates the salicylate-dependent pathway of plant immune response to pathogen attack (Zschiesche et al. 2015). A number of investigations have shown that plant peroxidases are directly involved in plant–nematode interactions; peroxidases scavenge reactive oxygen molecules and are implicated in plant defense (Vercauteren et al. 2001; Jammes et al. 2005; Severino et al. 2012). The promoter element of the Coffea arabica peroxidase gene was specifically induced upon M. incognita infection (Severino et al. 2012). It was assumed that peroxidases may play a conserved role during root-knot nematode invasion in different host plants (Joshi et al. 2022). Supported by these findings, we speculate that pAt2g18140 expression in S. lycopersicum is related to the elicitor molecules produced by the early to mid-infection stages of M. incognita, i.e., the post-parasitic juvenile stage to the young female stage. On the contrary, pAt1g74770 remains responsive to the elicitor molecules produced by all the parasitic stages of M. incognita.
To date, a number of nematode-responsive promoters have been identified from different host plants, including TobRB7 from Nicotiana tabacum (Opperman et al. 1994), LEMMI9 from S. lycopersicum (Escobar et al. 1999), Hahsp17.7G4 from Helianthus annuus (Escobar et al. 2003), TUB-1 from A. thaliana and S. melongena (Lilley et al. 2004; Papolu et al. 2016), Atcel1 from A. thaliana (Sukno et al. 2006), AtWRKY23 from A. thaliana (Grunewald et al. 2008), Pdf2.1 and MIOX5 from A. thaliana (Siddique et al. 2009, 2011), MDK4-20 from A. thaliana (Lilley et al. 2011), ZmRCP-1 from maize, bananas, and plantains (Onyango et al. 2016). However, only a few of these promoters were deployed to drive the expression of RNAi constructs to improve nematode resistance in the host plant. In an isolated study, TobRB7 was deployed to drive the dsRNA cassette of a nematode gene (MjTis11 of Meloidogyne javanica) in N. tabacum. However, no significant improvement in nematode resistance was observed in RNAi plants (over control), indicating the poor efficacy of TobRB7 in driving a RNAi construct (Fairbairn et al. 2007). Notably, in our laboratory, pAt1g74770 and pAt2g18140 were used to effectively confer HIGS in A. thaliana targeting M. incognita (Kakrana et al. 2017; Joshi et al. 2022). This prompted us to further translate this approach into commercial crops such as tomatoes.
M. incognita integrase and splicing factor genes are considered as attractive targets for HIGS studies, because these genes are constitutively involved in nematode metabolism and development processes (Yadav et al. 2006; Kumar et al. 2017). Silencing the nematode metabolism and development-related genes would provide a greater degree of RNAi-mediated resistance in crop plants compared to the other target genes such as effectors (Dutta et al. 2015a, b). Moreover, nematode avirulent effectors are the targets of the plant resistance genes (as they are involved in R-Avr interaction) and therefore, would be under incessant selection pressures to diversify and generate resistance-breaking nematode phenotypes (Eves-van den Akker 2021). In earlier studies, tobacco and Arabidopsis plants independently expressing the RNAi constructs of integrase and splicing factor genes (driven by the CaMV35S promoter) conferred improved M. incognita resistance in host plants (Yadav et al. 2006; Kumar et al. 2017). In the current study, pAt1g74770 was found to be highly efficient to drive the dsRNA expression cassettes of integrase and splicing factor genes for conferring improved M. incognita resistance in tomato. Most importantly, the degree of reduction in different nematode infection parameters (such as number of galls, egg masses, eggs per egg mass, and nematode multiplication factor) in pAt1g74770-driven integrase and splicing factor RNAi plants were alike of pCaMV35S-driven integrase and splicing factor RNAi plants. The reproductive success (in terms of multiplication factor ratio) of M. incognita was profoundly reduced by 60.83–74.93% and 69.34–75.31% in pAt1g74770:integrase and pAt1g74770:splicing factor RNAi lines, respectively, compared to the control. A comparable figure was documented in pCaMV35S:integrase (70.21% reduction in MF ratio over control) and pCaMV35S:splicing factor (72.34%) RNAi plants.
Intriguingly, a long-term RNAi effect of integrase and splicing factor genes was observed in M. incognita females. Females extracted from RNAi lines were of reduced size and deformed shape compared to the females extracted from untransformed lines. Additionally, a pronounced reduction in integrase and splicing factor transcript abundance was observed in recovered females compared to the control. This exemplified the targeted delivery of dsRNA/siRNA molecules (corresponding to the integrase/splicing factor gene) into the feeding females from RNAi plants. Notably, pAt1g74770 expression remains functional during the late stage of nematode infection.
More than 2 decades of research have shown that RNAi can contribute to global food security with minimal environmental impacts (Koch and Wassenegger 2021). Regarding innovations in the field of crop protection, RNAi technology has witnessed notable development worldwide with an annual average number of inventions amounting to 22, which corresponds to 122 patent applications (Ventura and Frisio 2021). A number of RNAi crops have already been commercialized globally. The list includes Bayer “SmartStax Pro” maize (confer resistance to D. virgifera virgifera), Bayer “Vistive Gold” high-oleic soybean, “Super-High Oleic” (SHO) safflower, Simplot GM “Innate” potatoes (reduced acrylamide content and improved starch quality), “HarvXtra” alfalfa (forage cattle feed with reduced lignin content for better digestibility) (De Schutter et al. 2022). A number of crops are already in the advanced stage of regulatory pipeline for future commercialization (De Schutter et al. 2022). An independent food and feed safety assessment performed on “SmartStax Pro” maize by European Food Safety Authority concluded that the plant-produced dsRNA/siRNA molecules do not exert any toxic effects once ingested by humans and animals (Papadopoulou et al. 2020). Although CRISPR/Cas9-based genome editing technology has gained considerable momentum to confer resistance in crop plants against various biotic stresses (Wheatley and Yang 2021), its application for improving nematode resistance in plants is yet in its nascent stage. This maybe because a limited number of plant susceptibility (S) genes have yet been identified for targeted knockout experiments (Dutta et al. 2023a). In addition, complete knockout of a target gene is unwarranted or may confer pleiotropic effects (because endogenous S genes are involved in plant developmental pathways). In contrast, RNAi is a better alternative, because gene silencing is required in targeted cells and not across the organism (Jones 2021). A continued research on HIGS mechanism would further improve its applicability in sustainable agriculture.
In conclusion, our results show that the nematode-inducible root-specific promoter pAt1g74770 can be heterologously expressed to impart RNAi-based nematode tolerance in an agriculturally important crop such as tomato. In contrast to the constitutive promoter, pAt1g74770 can putatively regulate the localized expression of dsRNA constructs targeting specific nematode genes. The spatiotemporal modulation of gene expression using inducible promoters is considered as a preferable strategy to unravel specific gene functions. In addition, application of our nematode-inducible gene silencing system can circumvent lethality problems and off-target effects. This technology can further be expanded in different plant-nematode pathosystems. A number of nematode genes can also be targeted simultaneously to achieve complete resistance in crop plants alike of gene pyramiding.
Data availability
The data sets supporting this article are included in the article and in the supplemental files.
References
Adam MAM, Phillips MS, Blok VC (2007) Molecular diagnostic key for identification of single juveniles of seven common and economically important species of root-knot nematode (Meloidogyne spp.). Plant Pathol 56:190–197
Ali MA, Azeem F, Abbas A, Joyia FA, Li H, Dababat AA (2017) Transgenic strategies for enhancement of nematode resistance in plants. Front Plant Sci 8:750
Banerjee S, Banerjee A, Gill SS, Gupta OP, Dahuja A, Jain PK, Sirohi A (2017) RNA interference: a novel source of resistance to combat plant parasitic nematodes. Front Plant Sci 8:834
Barbary A, Djian-Caporalino C, Palloix A, Castagnone-Sereno P (2015) Host genetic resistance to root-knot nematodes, Meloidogyne spp., in Solanaceae: from genes to the field. Pest Manag Sci 71:1591–1598
Bertioli DJ, Smoker M, Burrows PR (1999) Nematode-responsive activity of the cauliflower mosaic virus 35S promoter and its subdomains. Mol Plant-Microbe Interact 12:189–196
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Byrd DW, Kirkpatrick JRT, Barker KR (1983) An improved technique for clearing and staining plant tissues for detection of nematodes. J Nematol 15:142–143
Cabot C, Martos S, Llugany M, Gallego B, Tolrà R, Poschenrieder C (2019) A role for zinc in plant defense against pathogens and herbivores. Front Plant Sci 10:1171
Chaudhary S, Dutta TK, Tyagi N, Shivakumara TN, Papolu PK, Chobhe KA, Rao U (2019) Host-induced silencing of Mi-msp-1 confers resistance to root-knot nematode Meloidogyne incognita in eggplant. Transgenic Res 28:327–340
Coutu C, Brandle J, Brown D, Miki B, Simmonds J (2007) PORE: a modular binary vector series suited for both monocot and dicot plant transformation. Transgenic Res 16:771–781
De Schutter K, Taning CNT, Van Daele L, Van Damme EJM, Dubruel P, Smagghe G (2022) RNAi-based biocontrol products: market status, regulatory aspects, and risk assessment. Front Insect Sci 1:818037
Dutta TK, Banakar P, Rao U (2015a) The status of RNAi-based transgenic research in plant nematology. Front Microbiol 5:760
Dutta TK, Papolu PK, Banakar P, Choudhary D, Sirohi A, Rao U (2015b) Tomato transgenic plants expressing hairpin construct of a nematode protease gene conferred enhanced resistance to root-knot nematodes. Front Microbiol 6:260
Dutta TK, Papolu PK, Singh D, Sreevathsa R, Rao U (2020) Expression interference of a number of Heterodera avenae conserved genes perturbs nematode parasitic success in Triticum aestivum. Plant Sci 301:110670
Dutta TK, Vashisth N, Ray S, Phani V, Chinnusamy V, Sirohi A (2023a) Functional analysis of a susceptibility gene (HIPP27) in the Arabidopsis thaliana-Meloidogyne incognita pathosystem by using a genome editing strategy. BMC Plant Biol 23:390
Dutta TK, Santhoshkumar K, Veeresh A, Waghmare C, Mathur C, Sreevathsa R (2023b) RNAi-based knockdown of candidate gut receptor genes altered the susceptibility of Spodoptera frugiperda and S. litura larvae to a chimeric toxin Cry1AcF. PeerJ 11:e14716
Escobar C, De Meutter J, Aristizábal FA, Sanz-Alférez S, del Campo FF, Barthels N et al (1999) Isolation of the LEMMI9 gene and promoter analysis during a compatible plant-nematode interaction. Mol Plant-Microbe Interact 12:440–449
Escobar C, Barcala M, Portillo M, Almoguera C, Jordano J, Fenoll C (2003) Induction of the Hahsp17.7G4 promoter by root-knot nematodes: involvement of heat-shock elements in promoter activity in giant cells. Mol Plant Microbe Interact 16:1062–1068
Eves-van den Akker S (2021) Plant–nematode interactions. Curr Opin Plant Biol 62:102035
Fairbairn DJ, Cavallaro AS, Bernard M, Mahalinga-Iyer J, Graham MW, Botella JR (2007) Host-delivered RNAi: an effective strategy to silence genes in plant parasitic nematodes. Planta 226:1525–1533
Fletcher SJ, Reeves PT, Hoang BT, Mitter N (2020) A perspective on RNAi-based biopesticides. Front Plant Sci 11:51
Goddijn OJ, Lindsey K, van der Lee FM, Klap JC, Sijmons PC (1993) Differential gene expression in nematode-induced feeding structures of transgenic plants harbouring promoter-gusA fusion constructs. Plant J 4:863–873
Goverse A, Biesheuvel J, Wijers GJ, Gommers FJ, Bakker J, Schots A, Helder J (1998) In planta monitoring of the activity of two constitutive promoters, CaMV 35S and TR2´, in developing feeding cells induced by Globodera rostochiensis using green fluorescent protein in combination with confocal laser scanning microscopy. Physiol Mol Plant Pathol 52:275–284
Green J, Wang D, Lilley CJ, Urwin PE, Atkinson HJ (2012) Transgenic potatoes for potato cyst nematode control can replace pesticide use without impact on soil quality. PLoS ONE 7:e30973
Grunewald W, Karimi M, Wieczorek K, Van de Cappelle E, Wischnitzki E, Grundler F, Inze D, Beeckman T, Gheysen G (2008) A role for AtWRKY23 in feeding site establishment of plant-parasitic nematodes. Plant Physiol 148:358–368
Jammes F, Lecomte P, de Almeida-Engler J, Bitton F, Martin-Magniette ML, Renou JP et al (2005) Genome-wide expression profiling of the host response to root-knot nematode infection in Arabidopsis. Plant J 44:447–458
Jones HD (2021) Gene silencing or gene editing: the pros and cons. In: RNAi for plant improvement and protection. CABI, Wallingford, pp 47–53
Joshi I, Kumar A, Kohli D, Singh AK, Sirohi A, Subramaniam K, Chaudhury A, Jain PK (2020) Conferring root-knot nematode resistance via host-delivered RNAi-mediated silencing of four Mi-msp genes in Arabidopsis. Plant Sci 298:110592
Joshi I, Kumar A, Kohli D, Bhattacharya R, Sirohi A, Chaudhury A, Jain PK (2022) Gall-specific promoter, an alternative to the constitutive CaMV35S promoter, drives host-derived RNA interference targeting Mi-msp2 gene to confer effective nematode resistance. Front Plant Sci 13:1007322
Kakrana A, Kumar A, Satheesh V, Abdin MZ, Subramaniam K, Bhattacharya RC, Srinivasan R, Sirohi A, Jain PK (2017) Identification, validation and utilization of novel nematode-responsive root-specific promoters in Arabidopsis for inducing host-delivered RNAi mediated root-knot nematode resistance. Front Plant Sci 8:2049
Kaloshian I, Teixeira M (2019) Advances in plant-nematode interactions with emphasis on the notorious nematode genus Meloidogyne. Phytopathology 109:1988–1996
Koch A, Wassenegger M (2021) Host-induced gene silencing—mechanisms and applications. New Phytol 231:54–59
Koulagi R, Banerjee S, Gawade BH, Singh AK, Jain PK, Praveen S, Subramaniam K, Sirohi A (2020) Host-delivered RNA interference in tomato for mediating resistance against Meloidogyne incognita and Tomato leaf curl virus. Plant Cell Tissue Organ Cult (PCTOC) 143:345–361
Kumar A, Kakrana A, Sirohi A, Subramaniam K, Srinivasan R, Abdin MZ, Jain PK (2017) Host-delivered RNAi-mediated root-knot nematode resistance in Arabidopsis by targeting splicing factor and integrase genes. J Gen Plant Pathol 83:91–97
Lilley CJ, Urwin PE, Johnston KA, Atkinson HJ (2004) Preferential expression of a plant cystatin at nematode feeding sites confers resistance to Meloidogyne incognita and Globodera pallida. Plant Biotechnol J 2:3–12
Lilley CJ, Wang D, Atkinson HJ, Urwin PE (2011) Effective delivery of a nematode-repellent peptide using a root-cap-specific promoter. Plant Biotechnol J 9:151–161
Lilley CJ, Davies LJ, Urwin PE (2012) RNA interference in plant parasitic nematodes: a summary of the current status. Parasitology 139:630–640
Mani V, Reddy CS, Lee SK, Park S, Ko HR, Kim DG, Hahn BS (2020) Chitin biosynthesis inhibition of Meloidogyne incognita by RNAi-mediated gene silencing increases resistance to transgenic tobacco plants. Int J Mol Sci 21:6626
Moreira VJV, Lourenço-Tessutti IT, Basso MF, Lisei-de-Sa ME, Morgante CV, Paes-de-Melo B, Grossi-de-Sa MF (2022) Minc03328 effector gene downregulation severely affects Meloidogyne incognita parasitism in transgenic Arabidopsis thaliana. Planta 255:44
Moreira VJV, Pinheiro DH, Lourenço-Tessutti IT, Basso MF, Lisei-de-Sa ME, Silva M, Grossi-de-Sa MF (2023) In planta RNAi targeting Meloidogyne incognita Minc16803 gene perturbs nematode parasitism and reduces plant susceptibility. J Pest Sci. https://doi.org/10.1007/s10340-023-01623-7
Onyango SO, Roderick H, Tripathi JN, Collins R, Atkinson HJ, Oduor RO, Tripathi L (2016) The ZmRCP-1 promoter of maize provides root tip specific expression of transgenes in plantain. J Biol Res Thessaloniki 23:4
Opperman CH, Taylor CG, Conkling MA (1994) Root-knot nematode directed expression of a plant root-specific gene. Science 263:221–223
Papadopoulou N, Devos Y, Álvarez-Alfageme F, Lanzoni A, Waigmann E (2020) Risk assessment considerations for genetically modified RNAi plants: EFSA’s activities and perspective. Front Plant Sci 11:445
Papolu PK, Dutta TK, Tyagi N, Urwin PE, Lilley CJ, Rao U (2016) Expression of a cystatin transgene in eggplant provides resistance to root-knot nematode, Meloidogyne incognita. Front Plant Sci 7:1122
Peremarti A, Twyman RM, Go S, Naqvi S, Farré G, Sabalza M et al (2010) Promoter diversity in multigene transformation. Plant Mol Biol 73:363–378
Phani V, Gowda MT, Dutta TK (2023) Grafting vegetable crops to manage plant-parasitic nematodes: a review. J Pest Sci. https://doi.org/10.1007/s10340-023-01658-w
Rosso MN, Jones JT, Abad P (2009) RNAi and functional genomics in plant parasitic nematodes. Annu Rev Phytopathol 47:207–232
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108
Severino FE, Brandalise M, Costa CS, Wilcken SRS, Maluf MP, Goncalves W et al (2012) CaPrx, a Coffea arabica gene encoding a putative class III peroxidase induced by root-knot nematode infection. Plant Sci 191–192:35–42
Sharma A, Sharma D, Verma SK (2019) Zinc binding proteome of a phytopathogen Xanthomonas translucens pv. undulosa. R Soc Open Sci 6:190369
Shirasu K, Lahaye T, Tan MW, Zhou F, Azevedo C, Schulze-Lefert P (1999) A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell 99:355–366
Shivakumara TN, Chaudhary S, Kamaraju D, Dutta TK, Papolu PK, Banakar P, Sreevathsa R, Singh B, Manjaiah KM, Rao U (2017) Host-induced silencing of two pharyngeal gland genes conferred transcriptional alteration of cell wall-modifying enzymes of Meloidogyne incognita vis-à-vis perturbed nematode infectivity in eggplant. Front Plant Sci 8:473
Siddique S, Endres S, Atkins JM, Szakasits D, Wieczorek K, Hofmann J et al (2009) Myo-inositol oxygenase genes are involved in the development of syncytia induced by Heterodera schachtii in Arabidopsis roots. New Phytol 184:457–472
Siddique S, Wieczorek K, Szakasits D, Kreil DP, Bohlmann H (2011) The promoter of a plant defensin gene directs specific expression in nematode induced syncytia in Arabidopsis roots. Plant Physiol Biochem 49:1100–1107
Sindhu AS, Maier TR, Mitchum MG, Hussey RS, Davis EL, Baum TJ (2009) Effective and specific in planta RNAi in cyst nematodes: expression interference of four parasitism genes reduces parasitic success. J Exp Bot 60:315–324
Southey JF (1986) Laboratory methods for work with plant and soil nematodes. Fisheries and Food Great Britain: Ministry of Agriculture, London, p 202
Sukno S, Shimerling O, Mccuiston J, Tsabary G, Shani Z, Shoseyov O et al (2006) Expression and regulation of the Arabidopsis thaliana Cel1 endo 1,4 beta glucanase gene during compatible plant-nematode interactions. J Nematol 38:354–361
Urwin PE, Lilley CJ, McPherson MJ, Atkinson HJ (1997) Resistance to both cyst and root-knot nematodes conferred by transgenic Arabidopsis expressing a modified plant cystatin. Plant J 12:455–461
Ventura V, Frisio DG (2021) The economics of RNAi-based innovation: from the innovation landscape to consumer acceptance. In: RNAi for plant improvement and protection. CABI, Wallingford, pp 159–166
Vercauteren I, van der Schueren E, Van Montagu M, Gheysen G (2001) Arabidopsis thaliana genes expressed in the early compatible interaction with root knot nematodes. Mol Plant-Microbe Interact 14:288–299
Vieira P, Gleason C (2019) Plant-parasitic nematode effectors-insights into their diversity and new tools for their identification. Curr Opin Plant Biol 50:37–43
Wheatley MS, Yang Y (2021) Versatile applications of the CRISPR/Cas toolkit in plant pathology and disease management. Phytopathology 111:1080–1090
Yadav BC, Veluthambi K, Subramaniam K (2006) Host-generated double stranded RNA induces RNAi in plant-parasitic nematodes and protects the host from infection. Mol Biochem Parasitol 148:219–222
Zschiesche W, Barth O, Daniel K, Böhme S, Rausche J, Humbeck K (2015) The zinc-binding nuclear protein HIPP 3 acts as an upstream regulator of the salicylate-dependent plant immunity pathway and of flowering time in Arabidopsis thaliana. New Phytol 207:1084–1096
Acknowledgements
We profusely thank the staffs of National Phytotron Facility, IARI for maintenance of our transgenic lines.
Funding
The present research was funded by the Indian Council of Agricultural Research (ICAR) via National Agricultural Innovative Project (NAIP/C4/C1092) and National Agricultural Science Fund (NFBSFARA/RNA-3022/2012-13).
Author information
Authors and Affiliations
Contributions
Conceptualization: YET, TKD, AS; methodology: YET, TKD; formal analysis: TKD; resources: PKJ, KS; writing—original draft: TKD; writing—review and editing: AS; Supervision: AS.
Corresponding authors
Ethics declarations
Conflict of interest
Authors declare that no competing interests is associated with this manuscript.
Additional information
Communicated by Marcelo Menossi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Thorat, Y.E., Dutta, T.K., Jain, P.K. et al. A nematode-inducible promoter can effectively drives RNAi construct to confer Meloidogyne incognita resistance in tomato. Plant Cell Rep 43, 3 (2024). https://doi.org/10.1007/s00299-023-03114-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00299-023-03114-6