
Abstract
DNA repair is essential to maintain genome integrity. In addition to various DNA repair pathways dealing with specific types of DNA lesions, DNA damage tolerance (DDT) promotes the bypass of DNA replication blocks encountered by the replication fork to prevent cell death. Budding yeast Rad5 plays an essential role in the DDT pathway and its structure indicates that Rad5 recognizes damaged DNA or stalled replication forks, suggesting that Rad5 plays an important role in the DDT pathway choice. It has been reported that Rad5 forms subnuclear foci in the presence of methyl methanesulfonate (MMS) during the S phase. By analyzing the formation of Rad5 foci after MMS treatment, we showed that some specific DNA structures rather than mono-ubiquitination of proliferating cell nuclear antigen are required for the recruitment of Rad5 to the damaged site. Moreover, inactivation of the base excision repair (BER) pathway greatly decreased the Rad5 focus formation, suggesting that Rad5 recognizes specific DNA structures generated by BER. We also identified a negative role of overexpressed translesion synthesis polymerase Polη in the formation of Rad5 foci. Based on these data, we propose a modified DDT pathway model in which Rad5 plays a role in activating the DDT pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Numerous DNA lesions are generated in cells per day (Boiteux and Jinks-Robertson 2013). In response to these lesions, cells carry out DNA damage repair as well as a tolerance mechanism to circumvent the lesions. During replication, the replication fork stalls when it encounters a replication-blocking lesion on the DNA template (Pages 2016). The stalled fork is a fragile structure, and the prolonged stalling of replication forks will lead to fork collapse (Adamczyk et al. 2016; Alexander and Orr-Weaver 2016). The DNA damage tolerance (DDT) pathway can restart this stalled replication fork through bypassing the lesion with specialized polymerases rather than actually repairing the damaged DNA (Andersen et al. 2008). In budding yeast, a critical step to activating the DDT pathway is triggered by the mono-ubiquitination of proliferating cell nuclear antigen (PCNA) at the K164 residue by a Rad6-Rad18 E2-E3 complex. Mono-ubiquitination of PCNA provides a signal to activate the translesion DNA synthesis (TLS) branch of the DDT pathway, while further poly-ubiquitination of PCNA by Mms2-Ubc13-Rad5 triggers the error-free lesion bypass (Hoege et al. 2002; Pastushok and Xiao 2004).
The TLS branch is also termed error-prone DDT because of its association with increased mutagenesis in yeast (Lemontt 1971; Prakash 1981). TLS polymerases Rev1, Polη (Y-family) and Polζ (B family) directly bypass replication-blocking lesions in a highly mutagenic manner due to lack of 3′–5′ proofreading activities (Prakash et al. 2005). Polη, encoded by RAD30 in S. cerevisiae, is able to insert adenine (AA) opposite the ultraviolet (UV)-induced cis-syn thymine (TT) dimers in an error-free manner compared to other TLS polymerases (Johnson et al. 1999a; Masutani et al. 1999; Prakash et al. 2005). When (6–4) photoproduct is induced on the template DNA, both yeast and human Polη bypass it by inserting a guanine (G) opposite the 3′-T (Horton et al. 2013; Johnson et al. 2001; Mukhopadhyay et al. 2004). In the Rev1- and Polζ-mediated TLS, Rev1, a deoxycytidyl transferase, catalyzes the insertion of a deoxycytidine monophosphate (dCMP) opposite the lesion site on the DNA (Haracska et al. 2002; Nelson et al. 1996), while Polζ (consists of Rev3 and Rev7) carries out an extension until a replicative polymerase takes over (Johnson et al. 1999a; Prakash and Prakash 2002). Rev1 also plays a noncatalytic role in TLS as an adaptor between PCNA and Polζ/Polη through its PCNA- and ubiquitin (Ub)-binding domains (Sale et al. 2012).
In the error-free DDT branch, the monoubiquitinated PCNA is polyubiquitinated by the Mms2-Ubc13-Rad5 complex, in which Rad5 is well known to function as an E3 that activates the error-free DDT pathway with Mms2 and Ubc13 (Hoege et al. 2002). How the polyubiquitinated PCNA promotes error-free lesion bypass remains unclear. Two mechanisms have been proposed, namely template switch through homologous recombination (Ball et al. 2009; Zhang and Lawrence 2005) and fork reversal (Blastyak et al. 2007) followed by synthesis, both using newly synthesized system chromatid as a template, and hence considered error-free (Xu et al. 2015). Moreover, a recent study suggested that homologous recombination to repair double-strand breaks (DSBs) is RAD5-independent (Fasullo and Sun 2017).
Recent studies have revealed a role of Rad5 in the TLS pathway through physical interaction with Rev1 (Kuang et al. 2013; Pages et al. 2008; Xu et al. 2016) through its very N terminus (Xu et al. 2016) and independent of the Mms2-Ubc13 function (Gangavarapu et al. 2006). Analyzing the protein structure, the C terminus of Rad5 contains a RING domain embedded in a helicase domain consisting of seven conserved SWI2/SNF2 motifs (Johnson et al. 1992; Lorick et al. 1999). A HIRAN domain (Iyer et al. 2006) and a leucine zipper are located on the Rad5 N terminus, which is predicted to bind DNA or proteins. Meanwhile, RAD5 historically has been identified as REV2 because its mutant can suppress the UV-induced mutagenesis rate (Lemontt 1971). Interestingly, this suppression is independent of its E3 or helicase function (Pages et al. 2008). Furthermore, this phenotype is restricted to the reversion of ochre (UAA) alleles, such as arg4-17 (Johnson et al. 1992). So far, the detailed function of Rad5 in TLS remains elusive.
In the presence of methyl methanesulfonate (MMS), Rad5 accumulates and forms subnuclear foci in the S phase (Ortiz-Bazan et al. 2014). Here, we analyze the Rad5 focus formation under different conditions and in different mutants. We show that a specific DNA lesion structure is required for the recruitment of Rad5 to the damaged site and that Polη impedes Rad5 focus formation. Moreover, mutations in the base excision repair (BER) pathway dramatically decrease the formation of Rad5 foci, indicating that specific DNA structures produced in BER are recognized by Rad5. Together, our data favor a model in which Rad5 plays an important role in mediating the pathway choice between TLS and error-free DDT by recognizing and binding to damaged DNA.
Materials and methods
Cell preparation and microscopy
Yeast strains were cultured in synthetic dropout medium (SD medium) overnight at 30° and subcultured in fresh SD medium for 4 h. The cells were treated with 10 mg/mL hydroxyurea (HU, Sigma H8627-10G), followed by incubation with or without 0.12% MMS (Sigma 129925-5G) for 1 h. Then cells were harvested by centrifuging at 3,000g for 1 min, and washed with 0.1 M phosphate buffer (PBS, pH 7.2). The pellet was resuspended in 20 μL 0.1 M PBS with 1.2 M sorbitol (pH 7.2) for microscopy observation. The cells in S phase were counted by their morphological features and analyzed with an Olympus IX81 confocal fluorescence microscope, using 488 nm for sfGFP and 561 nm for mCherry. The percentage of cells with foci was counted in three independent experiments, and at least 500 cells were counted in each experiment. Data were analyzed by t test.
Yeast two-hybrid assay
The plasmids used in yeast two-hybrid assay were based on pGBT9 (Gal4BD) and pGAD424 (Gal4AD) (Fields and Song 1989). REV1, PCNA, RAD5 and their truncations were cloned into Gal4AD and Gal4BD plasmids, and the plasmids were co-transformed into PJ69-4a. The Y2H assay was conducted as described in Xu et al. (Xu et al. 2016).
Yeast strains
All yeast strains used in this study are listed in Table S1. Strains were made from HK578 and BY4741 background except for PJ69-4a (James et al. 1996), and null mutations were created using a one-step gene deletion method (Rothstein 1983).
Strains and plasmids are available upon request. Detailed descriptions are included in Supplementary materials.
Results
MMS treatment leads to Rad5 relocalization in the HU-induced S phase
It has been reported that MMS induces Rad5 relocation in the S phase, but no foci have been observed when treating yeast cells with UV radiation and HU (Ortiz-Bazan et al. 2014). Similarly, we hardly detected any Rad5 foci in yeast cells even after a prolonged HU treatment. Therefore, we used HU to arrest yeast cells in the S phase. After treating HU-induced S phase cells with 0.12% MMS, we observed that 81.9% wild-type cells contained Rad5 foci in contrast to 3.7% Rad5 focus-positive cells in the absence of MMS treatment (Fig. 1).

Rad5 focus formation under different conditions and in different mutants. The cells in S phase were counted and analyzed by fluorescence microscopy. a The formation of Rad5 foci when cells were treated with different DNA-damaging agents. FY0341 cells were arrested in the S phase by 10 mg/mL HU for 1 h, followed by the incubation with different DNA-damaging agents for the indicated time. Arrows indicate the Rad5 foci. b Percentage of cells displaying Rad5-sfGFP foci in different DDT mutants. For MMS treatment, cells were arrested in S phase by HU treatment and exposed to 0.12% MMS for 1 h. WT represents the results from the strain FY0341. The histograms represent the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001; for each treatment, more than 500 cells were counted. Error bar represents standard deviation (SD); w/o without
We further tested whether other DNA-damaging agents resulted in the Rad5 focus formation. Zeocin treatment causes DNA DSBs (Ehrenfeld et al. 1987), and HO endonuclease also generates DSB at the mating type (MAT) locus on chromosome III of S. cerevisiae. We did not observe Rad5 foci in the cells treated with 0.06 µM zeocin or after overexpressing HO endonuclease (Fig. 1a), indicating that Rad5 foci are not induced by DNA DSBs. cis-Platinum is known to generate intra- or inter-strand, as well as DNA–protein, crosslinking (Ali-Osman et al. 1995; Poklar et al. 1996). Camptothecin (CPT) leads to the formation of DNA–protein adduct by inhibiting topoisomerase I and shows a crosslinking-like phenotype (Hsiang et al. 1985). As shown in Fig. 1a, neither cis-platinum nor CPT-treated cells displayed Rad5 foci. Since Rad5 foci are not induced by the genotoxic stress caused by UV irradiation, HU, DSBs, and DNA crosslinking agents, it implies that the Rad5 focus formation depends on the specific type(s) of DNA lesions. Accordingly, infer that the formation of Rad5 foci is due to Rad5 binding or processing specific DNA lesions.
The Rad5 focus formation does not depend on mono-ubiquitination of PCNA
Given that Rad5 functions in the DDT pathway, we examined whether inactivation of different genes in the DDT pathway affected the Rad5 focus formation. We created a series of mutant strains, including rad30Δ, rad18Δ, siz1Δ and pol30-K164R in the FY0341 background. In budding yeast, the DDT pathway is initiated by mono-ubiquitination of PCNA at the K164 residue by the Rad6-Rad18 complex. The pol30-K164R point mutation prevents ubiquitination at the PCNA-K164 residues. In the presence of MMS, we observed Rad5 foci in all these mutants (Supplementary Figure S1). As with the previous observation (Ortiz-Bazan et al. 2014), we found that the Rad5 focus formation did not depend on RAD18. Meanwhile, neither the deletion of RAD30 nor the pol30-K164R point mutation affected the efficiency of Rad5 focus formation (Fig. 1b). Taken together, we conclude that the focus formation of Rad5 in response to MMS is independent of PCNA mono-ubiquitination.
It has been reported that Rad5-13,14AA is essential for the interaction of Rad5 with Rev1 (Xu et al. 2016). Indeed cells harbouring the rad5-FN13,14AA point mutation show TLS deficient phenotype. Given that the recruitment of Rad5 is independent of mono-ubiquitination of PCNA, we investigated whether this point mutation in Rad5 affected the Rad5 focus formation. We created a rad5-FN13,14AA mutant strain in the FY0341 background. Interestingly, the rad5-FN13,14AA point mutation increased the Rad5 focus formation, as we observed Rad5 foci in 96% of rad5-FN13,14AA mutant cells (Fig. 1b). This observation implies that TLS pathway plays a role in the formation of Rad5 foci.
The HIRAN domain is essential for Rad5 relocalization
To further explore the mechanism of the Rad5 focus formation, we deleted the N-terminal 430 amino acids (aa) of Rad5 and the HIRAN domain (176-285 aa) of Rad5. First, we examined the interaction between Rad5 and Rev1. Using the yeast two-hybrid assay, we confirmed that the N-terminal 430 aa of Rad5 are sufficient to interact with Rev1, whereas the truncation without the N-terminal 430 aa lost the interaction with Rev1 (Fig. 2a). Deletion of the Rad5 HIRAN domain (∆176-285 aa) does not interfere with its interaction with Rev1, indicating that the HIRAN domain is not essential for the Rad5-Rev1 interaction. However, deletion of either the HIRAN domain alone or the N-terminal truncation (∆1-430 aa) abolished the interaction between Rad5 and PCNA. It suggests that the recruitment of Rad5 to DNA lesions may still need PCNA although Rad5 focus formation is independent of PCNA mono-ubiquitination. Taken together, these data indicate that the HIRAN domain might be necessary for Rad5 focus formation.

The formation of MMS-induced Rad5 foci relies on the HIRAN domain. Cells were treated with MMS and analyzed as described in Fig. 1. a The results of yeast two-hybrid assay. BD binding domain, AD activating domain. The reporter gene is HIS3, and basal expression of HIS3 is controlled by adding 1 mM 3-AT (3-Amino-1,2,4-triazole). b FY0341 cells were transformed with plasmids that overexpressed Rad5-mCherry or the indicated Rad5 truncated mutants. Arrows indicate the Rad5 foci. c A schematic diagram of functional domains at Rad5 N-terminal region. The black line, FN13,14. The blue box, HIRAN domain. The green box, leucine zipper motif. d Percentage of cells displaying Rad5-mCherry foci in the cells described in b. After deleting the Rad5 N terminus, the efficiency of Rad5 focus formation significantly is decreased. e Percentage of cells displaying Rad5-sfGFP foci in different BER mutants. Histograms represent the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001; n ≥ 500; error bar represents SD; OE overexpression, w/o without
Next, we cloned RAD5-mCherry cassette as well as serials of Rad5 truncations into the pSCW231 vector, a YEp-based multicopy plasmid with a strong PADH1 constitutive promoter (Sung et al. 1987) to examine which region in Rad5 is essential for the focus formation. Same as Rad5-mCherry expressed from the genome, Rad5-mCherry expressed from the plasmids also formed foci under the same condition (Fig. 2b). After exposure to MMS, 68.3% of wild-type cells bearing RAD5-mCherry overexpressing plasmids formed subnuclear foci (Fig. 2d). For cells overexpressing the N-terminally truncated mutant (Δ1-430 aa) or HIRAN domain-deleted mutant (Δ176-285 aa), we detected Rad5-mCherry foci in only 25.1 or 27.9% of cells, respectively (Fig. 2d). The statistic data of Rad5 foci efficiency for each mutant strain are shown in Fig. 2d. Accordingly, the formation of Rad5 foci relies on the HIRAN domain.
APN1 and APN2 in the BER pathway contribute to Rad5 focus formation
Previous research and our study indicate that Rad5 foci are formed at specific DNA damage sites caused by MMS or H2O2 treatment. Although MMS and H2O2 generate different types of DNA lesions, the BER pathway can repair the DNA lesions they caused (Boiteux and Radicella 1999; Lindahl and Wood 1999). To assess the contribution of the BER pathway to the Rad5 focus formation, we analyzed Rad5 foci in APN1, APN2, NTG1, NTG2, and MAG1 single mutants. 82% of wild-type FY0341 cells displayed Rad5 foci after MMS treatment (Fig. 1b), while only around 16% of apn1Δ or apn2Δ null mutant cells exhibited Rad5 foci (Fig. 2e). In contrast, deletion of MAG1 displayed a moderate effect (about 60%) on the Rad5 focus formation after MMS treatment (Fig. 2e). After deleting NTG1 and NTG2, which encode two apurinic/apyrimidinic (AP) lyases, around 53.2 and 60.6% of cells, respectively, displayed Rad5 foci in the presence of MMS (Fig. 2e). Considering that Apn1 and Apn2 are AP endonucleases (Johnson and Demple 1988), it is possible that Rad5 relocalization is caused by the HIRAN domain recognizing the intermediate products in the BER pathway.
Overexpression of RAD30 abolishes the formation of Rad5 foci
On the basis of the above data, we envisioned that Rad5 foci would be abolished when the DNA lesion is bypassed. To test this hypothesis, we transformed the FY0341 strain with RAD30-mCherry overexpressing plasmids. After MMS treatment, only 32.1% of cells with RAD30 overexpression displayed Rad5 foci (Fig. 3a). Meanwhile, the Polη-mCherry itself did not form foci in the nuclei (Supplementary Figure S3A). These observations indicate that overexpressed Polη has a negative contribution to Rad5 binding on the DNA damage sites. We further examined the efficiency of Rad5 focus formation in the rad30∆ strains. To our surprise, deletion of RAD30 did not affect the Rad5 focus formation (Fig. 1b), suggesting that the low level of Rad30 does not affect the accumulation of Rad5 in MMS treated cells. Considered together, these data imply that Polη plays a role in the binding of Rad5 to the DNA damage sites.

The impact of overexpression of RAD30-mCherry and its point mutants on Rad5 focus formation. Cells were treated with MMS and analyzed as described in Fig. 1. a The cells were transformed with plasmids that overexpressed RAD30-mCherry. b The cells were transformed with plasmids that overexpressed the indicated RAD30 point mutants. In the figure, the histograms represent the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001; n ≥ 500; error bars, SD; OE overexpression, w/o without
Furthermore, we examined whether overexpressed Polη abolishes the Rad5 focus formation in the background of other mutants. In both rev1Δ and rad5-FN13,14AA mutants, Rad5 foci were restored in the presence of excessive Polη (Fig. 3a). Since Rad5-FN13,14AA abolished the interaction with Rev1, it suggests that the Polη competes or removes Rad5 from the DNA damage site in a Rev1-dependent manner.
Disrupting the function of polη can decrease the influence on Rad5 focus formation
We sought to identify the potential key motifs in Polη required for the removal of Rad5 from the damaged DNA. The F35 residue of Polη functions to avoid ribonucleotide incorporation (Donigan et al. 2015). The D155A substitution abolishes the Polη polymerase activity and the damage-induced cohesion phenomenon (Enervald et al. 2013). The D570 residue is indispensable for binding ubiquitin (Parker et al. 2007), and F627, F628 residues are important for Rad30 interaction with Rev1 (Boehm et al. 2016). We overexpressed rad30-F35A, rad30-D155A, rad30-FF627,628AA, or rad30-D570A in yeast cells and analyzed the Rad5 focus formation after treating with MMS. All of these point mutations restored the Rad5-sfGFP focus formation rate to the wild-type level (Fig. 3b, and Supplementary Figure S3B). These observations further confirm that the holoenzyme is essential for high-level Polη to remove Rad5 from the damaged site.
Discussion
Rad5 is an important protein involved in budding yeast DDT pathway. Rad5 can form foci in the S phase when cells are treated with MMS, and the foci are indication of Rad5 binding to or processing specific DNA lesions. In this study, we investigated the Rad5 focus formation by different DNA-damaging agents and in different genetic backgrounds. We found that Rad5 focus formation is independent of mono-ubiquitination at the PCNA-K164 residue but requires the Rad5 HIRAN domain. Moreover, we observed that the percentage of cells with Rad5 foci significantly decreased when the BER pathway genes were deleted or RAD30 was overexpressed. These findings contribute to elucidate molecular mechanisms of the DDT pathway, and we addressed a novel activating role of Rad5 in DDT.
The classic DDT model predicts that RAD5 functions downstream of RAD18 as well as PCNA mono-ubiquitination (Hoege et al. 2002). The observation that Rad5 foci were not dependent on either RAD18 or PCNA-K164 supports a recently modified DDT model (Xu et al. 2016). On the basis of the fact that Rad5 does not have a putative PCNA-binding motif, such as PIP box (MacNeill 2016; Warbrick et al. 1998), KA box (Xu et al. 2001) or ABH2 PCNA-binding motif (Gilljam et al. 2009), the physical interaction between Rad5 and PCNA (Carlile et al. 2009; Hoege et al. 2002; Parker and Ulrich 2009) likely is mediated by other proteins (such as Rev1) or by a specific DNA structure rather than direct binding. Rad5 can bind various forms of DNA in vitro, such as Y-fork, three-way junction and four-way junction (Blastyak et al. 2007). Interestingly, all the DNA substrates used in the in vitro DNA binding assay possess free 3′-OH groups. The HIRAN domain of Rad5 has been predicted to recognize and bind some specific DNA structures generated by DNA damage (Iyer et al. 2006). The HIRAN domain of DNA translocase HLTF, a Rad5 homolog in human cells, functions to bind the 3′-OH of the single-strain DNA and duplex DNA to facilitate fork progression (Hishiki et al. 2015; Kile et al. 2015). However, the HIRAN domain of Rad5 homolog in Arabidopsis thaliana, AtRAD5A, shows high affinity for leading strand gap structure, and its DNA binding activity does not need a free 3′-OH group at the DNA ends (Kobbe et al. 2016). In this study, we showed that the HIRAN domain of Rad5 is essential for its focus formation. It is very likely that Rad5 is recruited to the damage site through the interaction between its HIRAN domain and the specific DNA structure caused by DNA damage. The specific DNA structure might be a free 3′-OH group.
The fact that apn1Δ or apn2Δ severely decreased the efficiency of the Rad5 focus formation indicates that the direct lesions caused by MMS or H2O2 might not be the signal for recruiting Rad5. Some intermediate products that arose in the BER pathway are important to recruit Rad5. In budding yeast, Apn1 and Apn2 cleave at the AP site to generate a 5′-deoxyribose phosphate (5′-dRP) and a 3′-OH group (Boiteux and Guillet 2004). Furthermore, the recruitment of Rad5 to the processed AP site is consistent with the indispensable role of Rad5 in the TLS bypassing an AP site (Pages et al. 2008). Therefore, the free 3′-OH structure produced during BER might be important to recruit Rad5 for TLS bypassing. Considering that proteins involved in BER pathway progress together with replication fork, we cannot exclude the possibility that Rad5 can also interact with AP endonucleases or other proteins involved in BER.
The difference of foci percentage between apn1Δ or apn2Δ mutants (16%) and untreated wild-type cells (4%) might result from the alternative pathway to process AP sites, which could be mediated by AP lyases Ntg1 and Ntg2 (Scharer and Jiricny 2001). Although the process of AP site by Ntg1 and Ntg2 yields a 3′-α,β-unsaturated aldehydic (3′-dRP) rather than a 3′-OH (Boiteux and Guillet 2004; Fromme et al. 2004), the product can be further processed into a 3′-OH group by Apn1 or Apn2 (Unk et al. 2002, 2000) or by Rad1-Rad10 (Boiteux and Guillet 2004; Guillet and Boiteux 2002). Mag1 generates an AP site after removing the damaged base, which functions upstream of Apn1/Apn2 and Ntg1/Ntg2 (Bjoras et al. 1995; Xiao et al. 2001). We found that deletion of MAG1 moderately affects the Rad5 focus formation. This could be due to the fact that some other glycosylases, such as Ogg1 (van der Kemp et al. 1996) and Ung1 (Crosby et al. 1981) are able to yield AP sites. Moreover, AP sites are also generated by spontaneous depurination or depyrimidination in a glycosylase-independent manner (Lindahl and Nyberg 1972).
Rad5 and Polη were first thought to mediate two competing pathways to repair UV-induced DNA damage, because the rad5Δ rad30Δ double mutant exhibits an increased UV-induced mutation rate than either single mutant (Johnson et al. 1999b; McDonald et al. 1997). Later, RAD5 and RAD30 were found to be epistatic on the TLS bypassing of the (6–4) TT photoproduct (Pages et al. 2008), suggesting that Polη handles certain type(s) of DNA damage in the same pathway as Rad5. Given that rad30Δ and rad5Δ showed the synergistic effect on the TLS bypassing of a cis-syn TT dimer (Pages et al. 2008), Polη and Rad5 also deal with some types of DNA damage in different pathways. Taken together, it is likely that the type of DNA damage determines whether Polη can bypass the DNA damage in a Rad5-dependent or independent manner. Here, we observed a phenomenon that overexpressed Polη can remove Rad5 from damaged sites, and this activity is dependent on the Rev1-binding motif of Rad5. Meanwhile, the overexpression of Polη point mutants did not affect the Rad5 focus formation. Our data imply that Rad5 will be removed from the DNA damage sites if Polη/Rev1 bypasses the damaged DNA to diminish the 3′-OH signal.
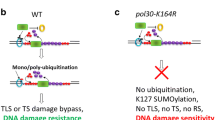
On the basis of previous observations and our data, we propose a modified DDT model, in which Rad5 recruitment also provides a signal to make a choice for different DDT pathways (Fig. 4). When DNA damage repaired by BER pathway leads to a specific DNA structure, such as a free 3′-OH group, Rad5 recognizes and binds to it through the HIRAN domain. Meanwhile, Rev1 also is recruited by the DNA lesion in a PCNA-K164 mono-ubiquitination-dependent manner. After being activated by Rad5 through an unknown mechanism, Rev1 recruits Polζ (Rev3/Rev7) to bypass the DNA damage (Xu et al. 2016). If the lesion is a favorite substrate for the translesion polymerase, the TLS polymerase will replace Rad5 from DNA and complete the synthesis. If the translesion polymerase cannot bypass the lesion, Rad5 will stay on the damaged site. When accumulating to some level or staying long enough, abundant Mms2-Ubc13 will be recruited to the damaged site, by which PCNA will be multi-ubiquitinated at the K164 residue, thus activating the recombination-mediated template switch pathway (Xu et al. 2015). The DDT pathway is conserved from yeast to human, thus our findings could help better understand the molecular mechanism for the maintenance of genome stability, and how this process goes awry in human diseases.

A modified model to illustrate the role of Rad5 in the DDT pathway choice. Asterisk, damaged base; triangle, specific DNA structure; black line, DNA strands; yellow circle, ubiquitin. Proteins involved in DDT are indicated. The damaged base is first recognized and repaired by the BER pathway. Specific DNA structures could be produced if the BER pathway is interrupted. When the replication fork encounters these structures, Rad5 is recruited and interacts with Rev1, Activated Rev1 further recruits translesion polymerases. If the lesion is bypassed by TLS polymerases, Rad5 longer binds to the site. Otherwise, it would stay longer or accumulate at this site. Probably, after passing a threshold, Rad5 can form a complex with Mms2-Ubc13 to polyubiquitinate PCNA and initiate the error-free DDT pathway
References
Adamczyk J, Deregowska A, Panek A, Golec E, Lewinska A, Wnuk M (2016) Affected chromosome homeostasis and genomic instability of clonal yeast cultures. Curr Genet 62:405–418. https://doi.org/10.1007/s00294-015-0537-3
Alexander JL, Orr-Weaver TL (2016) Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev 30:2241–2252. https://doi.org/10.1101/gad.288142.116
Ali-Osman F, Rairkar A, Young P (1995) Formation and repair of 1,3-bis-(2-chloroethyl)-1-nitrosourea and cisplatin induced total genomic DNA interstrand crosslinks in human glioma cells. Cancer Biochem Biophys 14:231–241
Andersen PL, Xu F, Xiao W (2008) Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res 18:162–173. https://doi.org/10.1038/cr.2007.114
Ball LG, Zhang K, Cobb JA, Boone C, Xiao W (2009) The yeast Shu complex couples error-free post-replication repair to homologous recombination. Mol Microbiol 73:89–102
Bjoras M, Klungland A, Johansen RF, Seeberg E (1995) Purification and properties of the alkylation repair DNA glycosylase encoded the MAG gene from Saccharomyces cerevisiae. Biochemistry 34:4577–4582
Blastyak A, Pinter L, Unk I, Prakash L, Prakash S, Haracska L (2007) Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell 28:167–175. https://doi.org/10.1016/j.molcel.2007.07.030
Boehm EM, Powers KT, Kondratick CM, Spies M, Houtman JC, Washington MT (2016) The proliferating cell nuclear antigen (PCNA)-interacting protein (PIP) motif of DNA polymerase eta mediates its interaction with the C-terminal domain of Rev1. J Biol Chem 291:8735–8744. https://doi.org/10.1074/jbc.M115.697938
Boiteux S, Guillet M (2004) Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 3:1–12
Boiteux S, Jinks-Robertson S (2013) DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics 193:1025–1064. https://doi.org/10.1534/genetics.112.145219
Boiteux S, Radicella JP (1999) Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress. Biochimie 81:59–67
Carlile CM, Pickart CM, Matunis MJ, Cohen RE (2009) Synthesis of free and proliferating cell nuclear antigen-bound polyubiquitin chains by the RING E3 ubiquitin ligase Rad5. J Biol Chem 284:29326–29334. https://doi.org/10.1074/jbc.M109.043885
Crosby B, Prakash L, Davis H, Hinkle DC (1981) Purification and characterization of a uracil-DNA glycosylase from the yeast Saccharomyces cerevisiae. Nucleic Acids Res 9:5797–5809
Donigan KA, Cerritelli SM, McDonald JP, Vaisman A, Crouch RJ, Woodgate R (2015) Unlocking the steric gate of DNA polymerase eta leads to increased genomic instability in Saccharomyces cerevisiae. DNA Repair 35:1–12. https://doi.org/10.1016/j.dnarep.2015.07.002
Ehrenfeld GM et al (1987) Copper-dependent cleavage of DNA by bleomycin. Biochemistry 26:931–942
Enervald E, Lindgren E, Katou Y, Shirahige K, Strom L (2013) Importance of Poleta for damage-induced cohesion reveals differential regulation of cohesion establishment at the break site and genome-wide. PLoS Genet 9:e1003158. https://doi.org/10.1371/journal.pgen.1003158
Fasullo MT, Sun M (2017) Both RAD5-dependent and independent pathways are involved in DNA damage-associated sister chromatid exchange in budding yeast. AIMS genetics 4:84–102. https://doi.org/10.3934/genet.2017.2.84
Fields S, Song O (1989) A novel genetic system to detect protein-protein interactions. Nature 340:245–246. https://doi.org/10.1038/340245a0
Fromme JC, Banerjee A, Verdine GL (2004) DNA glycosylase recognition and catalysis. Curr Opin Struct Biol 14:43–49. https://doi.org/10.1016/j.sbi.2004.01.003
Gangavarapu V, Haracska L, Unk I, Johnson RE, Prakash S, Prakash L (2006) Mms2-Ubc13-dependent and -independent roles of Rad5 ubiquitin ligase in postreplication repair and translesion DNA synthesis in Saccharomyces cerevisiae. Mol Cell Biol 26:7783–7790. https://doi.org/10.1128/MCB.01260-06
Gilljam KM et al (2009) Identification of a novel, widespread, and functionally important PCNA-binding motif. J Cell Biol 186:645–654. https://doi.org/10.1083/jcb.200903138
Guillet M, Boiteux S (2002) Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. Embo J 21:2833–2841. https://doi.org/10.1093/emboj/21.11.2833
Haracska L, Prakash S, Prakash L (2002) Yeast Rev1 protein is a G template-specific DNA polymerase. J Biol Chem 277:15546–15551. https://doi.org/10.1074/jbc.M112146200
Hishiki A, Hara K, Ikegaya Y, Yokoyama H, Shimizu T, Sato M, Hashimoto H (2015) Structure of a novel DNA-binding domain of Helicase-like transcription factor (HLTF) and its functional implication in DNA damage tolerance. J Biol Chem 290:13215–13223. https://doi.org/10.1074/jbc.M115.643643
Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and. SUMO Nature 419:135–141. https://doi.org/10.1038/nature00991
Horton JK et al (2013) Preventing oxidation of cellular XRCC1 affects PARP-mediated DNA damage responses. DNA Repair 12:774–785. https://doi.org/10.1016/j.dnarep.2013.06.004
Hsiang YH, Hertzberg R, Hecht S, Liu LF (1985) Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260:14873–14878
Iyer LM, Babu MM, Aravind L (2006) The HIRAN domain and recruitment of chromatin remodeling and repair activities to damaged. DNA Cell Cycle 5:775–782. https://doi.org/10.4161/cc.5.7.2629
James P, Halladay J, Craig EA (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436
Johnson AW, Demple B (1988) Yeast DNA 3′-repair diesterase is the major cellular apurinic apyrimidinic endonuclease—substrate-specificity and kinetics. J Biol Chem 263:18017–18022
Johnson RE, Henderson ST, Petes TD, Prakash S, Bankmann M, Prakash L (1992) Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol Cell Biol 12:3807–3818
Johnson RE, Kondratick CM, Prakash S, Prakash L (1999a) hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285:263–265
Johnson RE, Prakash S, Prakash L (1999b) Requirement of DNA polymerase activity of yeast Rad30 protein for its biological function. J Biol Chem 274:15975–15977
Johnson RE, Haracska L, Prakash S, Prakash L (2001) Role of DNA polymerase eta in the bypass of a (6 – 4) TT photoproduct. Mol Cell Biol 21:3558–3563. https://doi.org/10.1128/MCB.21.10.3558-3563.2001
Kile AC et al (2015) HLTF’s ancient HIRAN domain binds 3′ DNA ends to drive replication fork. Reversal Mol Cell 58:1090–1100. https://doi.org/10.1016/j.molcel.2015.05.013
Kobbe D et al (2016) AtRAD5A is a DNA translocase harboring a HIRAN domain which confers binding to branched DNA structures and is required for DNA repair in vivo. Plant J Cell Mol Biol. https://doi.org/10.1111/tpj.13283
Kuang L, Kou H, Xie Z, Zhou Y, Feng X, Wang L, Wang Z (2013) A non-catalytic function of Rev1 in translesion DNA synthesis and mutagenesis is mediated by its stable interaction with Rad. 5. DNA Repair 12:27–37. https://doi.org/10.1016/j.dnarep.2012.10.003
Lemontt JF (1971) Mutants of yeast defective in mutation induced by ultraviolet light. Genetics 68:21–33
Lindahl T, Nyberg B (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry 11:3610–3618
Lindahl T, Wood RD (1999) Quality control by DNA repair. Science 286:1897–1905
Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM (1999) RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA 96:11364–11369
MacNeill SA (2016) PCNA-binding proteins in the archaea: novel functionality beyond the conserved core. Curr Genet 62:527–532. https://doi.org/10.1007/s00294-016-0577-3
Masutani C et al (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399:700–704. https://doi.org/10.1038/21447
McDonald JP, Levine AS, Woodgate R (1997) The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism. Genetics 147:1557–1568
Mukhopadhyay S, Clark DR, Watson NB, Zacharias W, McGregor WG (2004) REV1 accumulates in DNA damage-induced nuclear foci in human cells and is implicated in mutagenesis by benzo[a]pyrenediolepoxide. Nucleic Acids Res 32:5820–5826. https://doi.org/10.1093/nar/gkh903
Nelson JR, Lawrence CW, Hinkle DC (1996) Deoxycytidyl transferase activity of yeast REV1 protein. Nature 382:729–731. https://doi.org/10.1038/382729a0
Ortiz-Bazan MA, Gallo-Fernandez M, Saugar I, Jimenez-Martin A, Vazquez MV, Tercero JA (2014) Rad5 plays a major role in the cellular response to DNA damage during chromosome replication. Cell Rep 9:460–468. https://doi.org/10.1016/j.celrep.2014.09.005
Pages V (2016) Single-strand gap repair involves both RecF and RecBCD pathways. Curr Genet 62:519–521. https://doi.org/10.1007/s00294-016-0575-5
Pages V, Bresson A, Acharya N, Prakash S, Fuchs RP, Prakash L (2008) Requirement of Rad5 for DNA polymerase zeta-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics 180:73–82. https://doi.org/10.1534/genetics.108.091066
Parker JL, Ulrich HD (2009) Mechanistic analysis of PCNA poly-ubiquitylation by the ubiquitin protein ligases Rad18 and Rad5. Embo J 28:3657–3666. https://doi.org/10.1038/emboj.2009.303
Parker JL, Bielen AB, Dikic I, Ulrich HD (2007) Contributions of ubiquitin- and PCNA-binding domains to the activity of Polymerase eta in Saccharomyces cerevisiae. Nucleic Acids Res 35:881–889. https://doi.org/10.1093/nar/gkl1102
Pastushok L, Xiao W (2004) DNA postreplication repair modulated by ubiquitination and sumoylation. Adv Protein Chem 69:279–306
Poklar N, Pilch DS, Lippard SJ, Redding EA, Dunham SU, Breslauer KJ (1996) Influence of cisplatin intrastrand crosslinking on the conformation, thermal stability, and energetics of a 20-mer DNA duplex. Proc Natl Acad Sci USA 93:7606–7611
Prakash L (1981) Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol General Gene MGG 184:471–478
Prakash S, Prakash L (2002) Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev 16:1872–1883. https://doi.org/10.1101/gad.1009802
Prakash S, Johnson RE, Prakash L (2005) Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74:317–353. https://doi.org/10.1146/annurev.biochem.74.082803.133250
Rothstein RJ (1983) One-step gene disruption in yeast. Methods Enzymol 101:202–211
Sale JE, Lehmann AR, Woodgate R (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol 13:141–152. https://doi.org/10.1038/nrm3289
Scharer OD, Jiricny J (2001) Recent progress in the biology, chemistry and structural biology of DNA glycosylases. BioEssays News Rev Mol Cell Dev Biol 23:270–281. https://doi.org/10.1002/1521-1878(200103)23:3<270::AID-BIES1037>3.0.CO;2-J
Sung P, Prakash L, Weber S, Prakash S (1987) The RAD3 gene of Saccharomyces cerevisiae encodes a DNA-dependent ATPase. Proc Natl Acad Sci USA 84:6045–6049
Unk I, Haracska L, Johnson RE, Prakash S, Prakash L (2000) Apurinic endonuclease activity of yeast Apn2 protein. J Biol Chem 275:22427–22434. https://doi.org/10.1074/jbc.M002845200
Unk I, Haracska L, Gomes XV, Burgers PM, Prakash L, Prakash S (2002) Stimulation of 3′–>5′ exonuclease and 3′-phosphodiesterase activities of yeast apn2 by proliferating cell nuclear antigen. Mol Cell Biol 22:6480–6486
van der Kemp PA, Thomas D, Barbey R, de Oliveira R, Boiteux S (1996) Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc Natl Acad Sci USA 93:5197–5202
Warbrick E, Heatherington W, Lane DP, Glover DM (1998) PCNA binding proteins in Drosophila melanogaster: the analysis of a conserved PCNA binding domain. Nucleic Acids Res 26:3925–3932
Xiao W, Chow BL, Hanna M, Doetsch PW (2001) Deletion of the MAG1 DNA glycosylase gene suppresses alkylation-induced killing and mutagenesis in yeast cells lacking AP endonucleases. Mutat Res/DNA Repair 487:137–147
Xu H, Zhang P, Liu L, Lee MY (2001) A novel PCNA-binding motif identified by the panning of a random peptide display library. Biochemistry 40:4512–4520
Xu X, Blackwell S, Lin A, Li F, Qin Z, Xiao W (2015) Error-free DNA-damage tolerance in Saccharomyces cerevisiae. Mutat Res Rev Mutat Res 764:43–50. https://doi.org/10.1016/j.mrrev.2015.02.001
Xu X et al (2016) Involvement of budding yeast Rad5 in translesion DNA synthesis through physical interaction with Rev1. Nucleic Acids Res 44:5231–5245. https://doi.org/10.1093/nar/gkw183
Zhang H, Lawrence CW (2005) The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci USA 102:15954–15959. https://doi.org/10.1073/pnas.0504586102
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant number 31371264); CAS Interdisciplinary Innovation Team; and the Newton Advanced Fellowship from the Royal Society (Grant number NA140085). Q. W and W. X were supported by the National Natural Science Foundation of China (Grant number 31670068).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest.
Additional information
Communicated by M. Kupiec.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fan, Q., Xu, X., Zhao, X. et al. Rad5 coordinates translesion DNA synthesis pathway by recognizing specific DNA structures in saccharomyces cerevisiae. Curr Genet 64, 889–899 (2018). https://doi.org/10.1007/s00294-018-0807-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-018-0807-y