
Abstract
Although the plastid genomes of diatoms maintain a conserved architecture and core gene set, considerable variation about this core theme exists and can be traced to several different processes. Gene duplication, pseudogenization, and loss, as well as intracellular transfer of genes to the nuclear genome, have all contributed to variation in gene content among diatom species. In addition, some noncoding sequences have highly restricted phylogenetic distributions that suggest a recent foreign origin. We sequenced the plastid genome of the marine diatom, Toxarium undulatum, and found that the genome contains three genes (chlB, chlL, and chlN) involved in light-independent chlorophyll a biosynthesis that were not previously known from diatoms. Phylogenetic and syntenic data suggest that these genes were differentially retained in this one lineage as they were repeatedly lost from most other diatoms. Unique among diatoms and other heterokont algae sequenced so far, the genome also contains a large group II intron within an otherwise intact psaA gene. Although the intron is most similar to one in the plastid-encoded psaA gene of some green algae, high sequence divergence between the diatom and green algal introns rules out recent shared ancestry. We conclude that the psaA intron was likely introduced into the plastid genome of T. undulatum, or some earlier ancestor, by horizontal transfer from an unknown donor. This genome further highlights the myriad processes driving variation in gene and intron content in the plastid genomes of diatoms, one of the world’s foremost primary producers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although the plastid genomes of diatoms encompass a relatively narrow spectrum of the distribution in size, gene content, and architecture of organelle genomes (Smith and Keeling 2015), they nevertheless exhibit substantial variation among species in gene and intergenic sequence content (Brembu et al. 2014; Ruck et al. 2014). This variation reflects numerous underlying processes that continue to drive genomic divergence among species. These processes include: (1) gene duplication, pseudogenization, and loss (Ruck et al. 2014); (2) ongoing intracellular transfer of genes to the nucleus (Lommer et al. 2010; Sabir et al. 2014), and; (3) acquisition of foreign DNA from sources inside, and perhaps even outside, the cell (Brembu et al. 2014; Ruck et al. 2014). Altogether, these processes appear to have affected roughly 10 % of the total protein-coding gene set and have occurred against the backdrop of a genome that, despite pervasive rearrangements, maintains the prototypical circular mapping, tripartite plastid genome architecture and a conserved, AT-rich nucleotide composition (Ruck et al. 2014).
Our ability to discern even broad-scale patterns of evolutionary change in the organelle and nuclear genomes of diatoms is, however, limited by the scarcity of genomic data for this group. Among the tens of thousands of extant diatom species (Mann and Vanormelingen 2013), just a few dozen have fully sequenced plastid genomes. Even fewer species have fully sequenced mitochondrial genomes, and nuclear genomes have been sequenced for only a small number of mostly model species (Armbrust et al. 2004; Bowler et al. 2008; Lommer et al. 2012). Sparse or biased taxonomic sampling can mislead biological interpretations of genomic data. For example, as more genomes are sampled, improved models can lead to the discovery of genes that were once thought missing (Simola et al. 2013), and genes that were once thought to have a foreign origin can be revealed as native (Salzberg et al. 2001). Dense sampling is an absolute prerequisite to understand the evolution of gene families with especially complex histories, such as the algal por gene, which has been serially duplicated, lost, and passed around among sometimes distant relatives by horizontal gene transfer (Hunsperger et al. 2015).
We sequenced the plastid genome of the tropical marine diatom, Toxarium undulatum Bailey, providing a first glimpse into plastid genome evolution in this part of the diatom phylogenetic tree. The presence of a complete set of genes encoding the light-independent protochlorophyllide oxidoreductase (LIPOR) pathway, previously thought missing in diatoms, expands our understanding of the functional photosynthetic repertoire of the ancestral diatom and further highlights the paradoxical retention in some taxa of a biochemical pathway that is clearly expendable. The genome also contains a putatively foreign group II intron not previously known from diatoms. Among sequenced diatom plastid genomes, these two features are found only in T. undulatum, but we show that these parallel patterns likely resulted from very different underlying processes.
Materials and methods
Diatom culturing, DNA extraction, and sequencing
A clonal culture of Toxarium undulatum (strain ECT3802) was established from a field collection of epiphytes associated with Gab Gab Reef, Apra Harbor, Guam, USA (13.44°N, 144.64°W) in October 2008. The culture was maintained in L1 medium (Guillard 1975) at 22 °C on a 12:12 h light:dark cycle. Live cells were concentrated by centrifugation, frozen, and then disrupted by mixing with glass beads using a Mini-Beadbeater-24 (BioSpec Products). Genomic DNA was extracted with a Qiagen DNeasy DNA Plant Mini Kit and sequenced on the Illumina MiSeq platform at the Institute for Genomics and Systems Biology at Argonne National Laboratory, generating 150-nt paired-end reads from libraries 300 nt in length.
Genome assembly and analysis
We assembled sequencing reads with Ray ver. 2.2.0 using default settings and a kmer length of 31 (Boisvert et al. 2010). We then used Geneious ver. 5.4 (Biomatters Ltd., Auckland, New Zealand) to identify gaps and finish the assembly. We annotated protein-coding genes with DOGMA (Wyman et al. 2004) and used ARAGORN (Laslett and Canback 2004) to identify predicted tRNAs and tmRNAs. We checked the boundaries of rRNA and ffs genes by comparisons to previously sequenced diatom genomes using the National Center for Biotechnology Information’s (NCBI) BLASTN software. The annotated genome sequence can be downloaded from GenBank using accession number KX619437.
To reconstruct the phylogenetic history of chl genes in T. undulatum, we collected chl gene sequences for representative heterokonts, red algae, green algae, and cyanobacteria from NCBI’s GenBank sequence repository. We then aligned conceptual amino acid translations with MAFFT (Katoh and Standley 2013) using default settings. In preliminary tree searches with the nucleotide alignments, we found that removal of visually poorly aligned regions did not affect the phylogenetic placement of T. undulatum, so all analyses used the full-length alignments. The individual chlB, chlL, and chlN gene trees were congruent with respect to the placement of T. undulatum (Fig. S1), so the three genes were concatenated to produce a single and more strongly supported phylogenetic hypothesis. Using IQtree v.1.4.1 (Nguyen et al. 2015), we identified cpREV + R5 (Adachi et al. 2000) as the best substitution model for the concatenated amino acid alignment. In this model, R represents the number of rate categories in the FreeRates model for among-site rate variation (Yang 1995). We used IQtree to perform 25 maximum likelihood optimizations with default settings, choosing the tree with the highest likelihood as the best one. Support for inferred relationships was obtained using bootstrap analysis with 500 pseudoreplicates. Multiple sequence alignments were deposited in an online data repository hosted by Zenodo (doi:10.5281/zenodo.58229).
Results and discussion
General features
The plastid genome of T. undulatum mapped as a single, circular molecule of length 141,681 nt (Table 1; Fig. 1), though it is unclear whether this topology exists in vivo (Bendich 2004). The genome has the tripartite architecture conserved across diatoms and many other plastid-bearing lineages (Smith and Keeling 2015), with small and large single copy regions separated by a pair of large inverted repeats (Fig. 1). Similar to the plastid genomes of most other eukaryotes, including diatoms (Ruck et al. 2014; Smith 2012), the plastid genome of T. undulatum is AT-rich (Table 1). Overall, the plastid genome of T. undulatum contains the vast majority of conserved open reading frames, protein-coding genes, and rRNA and tRNA genes that are near-universally conserved across diatoms. Several less conserved, mostly accessory genes, are missing from the genome, including acpP, bas1, ilvB, ilvH, and petJ. Absence of these genes reflects either deep losses within diatoms (ilvB, ilvH, and petJ) or more recent losses (acpP and bas1) (Ruck et al. 2014; Sabir et al. 2014). Pinpointing the exact timing of these losses will require more genomic sampling across diatoms.

Annotated map of the plastid genome of the diatom, Toxarium undulatum. Genes drawn on the inside of the circle are transcribed in the clockwise direction, whereas those on the outside of the circle are transcribed in the counterclockwise direction. The interior gray bar plot shows the average G + C content. The genome map was rendered with OGDRAW (Lohse et al. 2007)
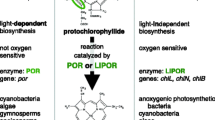
Light-independent protochlorophyllide oxidoreductase genes
Oxygenic photosynthesis in diatoms relies on chlorophyll a and the accessory pigment, chlorophyll c (Round et al. 1990). The last steps of chlorophyll a biosynthesis involve reduction of protochlorophyllide a to chlorophyllide a, the direct precursor molecule to chlorophyll a (Armstrong 1998). Although this conversion can be carried out through either light-dependent (POR) or light-independent (LIPOR) pathways, LIPOR genes have been lost repeatedly within virtually all major lineages of photosynthetic eukaryotes. The genes are missing from angiosperms and some gymnosperms (Ueda et al. 2014), Euglenophytes and many green algae, haptophytes, some cryptophytes and rhodophytes, and numerous algal heterokonts (Fong and Archibald 2008; Hunsperger et al. 2015). The presence of LIPOR genes in the plastid genomes of some heterokonts, including Triparma laevis—the sister lineage to diatoms—and their absence from the entire set of sequenced diatom plastid and nuclear genomes, suggested that these genes were lost in diatoms following their split from Parmales (Hunsperger et al. 2015; Tajima et al. 2016).
The plastid genome of T. undulatum contains three intact genes—chlB, chlL, and chlN—that together encode the complete LIPOR pathway (Fig. 1). This is the first report of these genes from among the few dozen completely sequenced diatom nuclear and plastid genomes. Given this highly restricted distribution, we used phylogenetic analysis to test two competing hypotheses that could account for their exclusive presence in T. undulatum: (1) the LIPOR genes were transferred into T. undulatum by a foreign donor, or (2) the genes were present in the plastid genome of the ancestral diatom and differentially retained in T. undulatum. We compiled LIPOR genes from a phylogenetically broad set of photosynthetic organisms, including cyanobacteria, land plants, and a diverse set of algae in the “red” plastid lineage (Fig. 2). The dataset included LIPOR genes from several algal heterokonts, including Tr. laevis, the sister lineage to diatoms. Phylogenetic analyses showed that each of the three genes individually was placed within heterokonts and sister to Tr. laevis (Fig. S1). A concatenated tree provided strong support for this relationship (Fig. 2), ruling out horizontal transfer as the source of these genes in T. undulatum and instead indicating that LIPOR genes were ancestrally present in diatoms. This hypothesis is also supported by conserved synteny. Although the T. undulatum and Tr. laevis genomes are highly rearranged compared to one another, the structure and arrangement of the LIPOR genes are similar in the two genomes. The genes are located in single-copy regions of the genome in both species, with a syntenic and identically oriented chlL–chlN–rps6 gene cluster distantly separated from a psaF–chlB cluster (Fig. 1).

Phylogenetic tree of amino acid sequences from three concatenated LIPOR genes (chlB, chlL, and chlN) present in the plastid genome of the diatom, Toxarium undulatum. This is the best of 25 optimizations using IQtree with default settings and a cpREV + R5 model of amino acid replacement. Numbers at nodes are standard bootstrap proportions from 500 pseudoreplicates
Repeated gene loss is a familiar theme in diatom plastid genomes, with some genes lost six or more separate times over the course of diatom evolution (Ruck et al. 2014). Another common theme is that many of the same genes repeatedly lost in diatoms are dispensable in other algal groups as well (e.g., bas1; Sánchez-Puerta et al. 2005). Based on our current understanding of phylogenetic relationships within diatoms (Theriot et al. 2015) and what we know from the relatively small sample of diatom plastid genomes, we estimate that LIPOR genes were lost at least five separate times across diatoms as they were maintained, fully intact and presumably functional, within T. undulatum (Fig. 3). Further sampling will show whether these genes are present throughout the genus Toxarium and in related genera, including Ardissonea, Climacosphenia, and allies (Medlin et al. 2008).

Different processes account for the unique presence, among diatoms, of LIPOR (chl) genes and an intron within the psaA gene in the plastid genome of Toxarium undulatum. The chl genes were lost at least five times (“–CHL” branch annotation) following the split of diatoms from Parmales, whereas the intron appears to have been recently acquired, presumably from a foreign donor. Phylogenetic relationships are based on Theriot et al. (2015), and genomic comparisons were based on plastid genomes from Ruck et al. (2014), Sabir et al. (2014), and Tajima et al. (2016)
Several compelling hypotheses have been proposed to account for the preferential reliance on the POR pathway for enzymatic reduction of protochlorophyllide in most taxa. The LIPOR complex likely originated in anoxygenic bacteria and at a time when atmospheric oxygen levels were much lower than they are today (Raymond et al. 2004; Reinbothe et al. 1996). The LIPOR protein complex contains an oxygen-sensitive iron sulfur cluster that exhibits reduced functionality in oxygen-rich conditions associated with high light or long photoperiods (Ueda et al. 2014; Yamazaki et al. 2006). The POR pathway, by contrast, evolved in the more photosynthetically active cyanobacteria and is insensitive to oxygen. The LIPOR pathway is, as a result, essentially incompatible with the high levels of oxygenic photosynthesis that characterize modern photosynthetic eukaryotes. In addition, many diatoms live in offshore regions of the ocean where iron is growth-limiting (Boyd et al. 2007). Several offshore species show specific adaptations to these environments, either through reduced iron demands (Peers and Price 2006; Strzepek and Harrison 2004) or luxury uptake and storage of iron (Marchetti et al. 2009). Repeated loss of the LIPOR pathway across diatoms may, therefore, reflect another type of adaptation to low iron in ancestral diatom taxa (Hunsperger et al. 2015).
The more vexing question is, however, why LIPOR genes are maintained alongside POR genes in some lineages (Fong and Archibald 2008; Hunsperger et al. 2015). Although they may confer some advantage to taxa regularly exposed to low light or with a frequent dependency on heterotrophic growth, neither of these seems to be particularly applicable to T. undulatum—a neritic, benthic diatom capable of active motility (Hasle and Syvertsen 1997; Kooistra et al. 2003). The three LIPOR genes in T. undulatum are highly conserved in sequence, with T. undulatum chlB and chlN showing >76 % amino acid identity with Tr. laevis, and chlL showing fully 92 % identity between T. undulatum and Tr. laevis—two taxa that split more than 150 Mya (Sorhannus 2007). In short, these genes appear to have been conserved and maintained in the T. undulatum lineage since the origin of diatoms and are by no means in the process of being lost, as LIPOR genes are in some lineages (Fong and Archibald 2008). Additional genomic sampling will show how far back in Toxariales these genes have been conserved, whether any other diatom lineages have also retained LIPOR genes, and whether retention or losses have ecological or other genomic correlates (e.g., the coincidental duplication of por genes; Hunsperger et al. 2015).
Novel group II intron in psaA
Among plastid genomes, those of diatoms are somewhat unusual in their propensity to collect and maintain noncoding DNA sequences, including deteriorating pseudogenes, plasmid-derived genes, and ORFs or other sequences of unknown origin (Ruck et al. 2014). Although largely unrecognizable, the restricted phylogenetic distributions of sequences in the latter category suggest that they may be recent acquisitions, either from the cell’s own (unsequenced) nuclear genome or some foreign source (Ruck et al. 2014). At least a few of the most recognizable “extra” sequences were, indeed, very likely acquired from an outside donor. For example, among the few dozen diatom species with a sequenced plastid genome, just one of them, Seminavis robusta, contains introns (Brembu et al. 2014). One of these is a group I intron within the large subunit rRNA (rnl) gene. The intron contains an intron-encoded protein (IEP) with a LAGLIDADG-type homing endonuclease, which is known to have facilitated the horizontal spread of similar introns among organelle genomes in other lineages (Belfort and Perlman 1995; Sánchez-Puerta et al. 2008; Turmel et al. 1995). The plastid genome of S. robusta also contains a group II intron located in the atpB gene (Brembu et al. 2014). This intron contains an IEP encoding a reverse transcriptase—a common feature of introns located in rRNA genes that also facilitates intron mobility and horizontal transfer (Johansen et al. 2007). Both introns in Seminavis are most similar to ones found in green algae, which led to the conclusion that they were acquired by horizontal transfer from green algal donors (Brembu et al. 2014). Given the available data, however, it cannot be ruled out that the transfer occurred in the other direction, from diatoms to chlorophytes, or that these two lineages never directly exchanged the intron at all (see discussion below). Introns with aberrant, highly restricted phylogenetic distributions have been found in other algal plastid genomes, too—for example, the nested group II/III “twintrons” in some cryptophytes that either were inherited vertically (Perrineau et al. 2015) or were acquired from a euglenoid-like donor (Khan and Archibald 2008).
The plastid psaA gene in T. undulatum is interrupted by a large (2844 nt in length) group II intron (Fig. 1), the first report of this intron in diatom plastid genomes. The intron is of type IIB, with the six predicted domains common to group II introns and the conserved 5′ (GUGYG) and 3′ (AY) end sequences (Zimmerly and Semper 2015). Presence of the catalytic AGC triad in domain V and a bulging A motif in domain VI, which are critical for splicing, together suggest that the intron is intact and autocatalytic (Gordon and Piccirilli 2001; Zimmerly and Semper 2015). Domain IV of the intron contains an IEP that is 507 amino acids in length (Fig. 1). A BLASTP search of the IEP against NCBI’s Conserved Domain Database (Marchler-Bauer et al. 2015) revealed the characteristic reverse transcriptase (RT), maturase (X), and DNA-binding (D) domains, but it lacked the H–N–H endonuclease domain found in many IEPs (Blocker et al. 2005). The reverse transcriptase domain includes all 13 putative active sites, all 8 putative dNTP binding sites, and the nucleic acid binding site (Qu et al. 2016). Finally, the psaA gene itself is fully intact, with no signs of degradation or coconversion of the flanking exon sequences (Belfort and Perlman 1995; Lambowitz and Belfort 1993), so available evidence suggests that the intron has not disrupted the functionality of this important photosystem gene.
To better understand the origin of this intron in T. undulatum, we used NCBI-BLASTN to search the entire 2.8 kb intron sequence against NCBI’s non-redundant nucleotide sequence database. The only strong matches were to the IEP, so we used NCBI-BLASTX to search the IEP against NCBI’s non-redundant protein sequence database. The top 10 hits matched IEPs within group II introns in plastid genes of green algae in the division Chlorophyta, with nine of them matching Chlorophyceae (e.g., the Chlamydomonas and Volvox lineage). Five of the top 10 hits matched IEPs located in the plastid psaA genes of chlorophycean green algae. The top matching green algal IEPs aligned along nearly the entire length of the gene but shared just 40–50 % amino acid identity, with very little matching sequence outside the IEP. The low similarity of these matches, combined with the relatively small number of matches outside of green algae, indicated that phylogenetic analysis would offer no additional information about the ancestry of this intron.
Although currently available data preclude identification of the putative donor, the highly mobile nature of group II introns (Belfort and Perlman 1995; Lambowitz and Belfort 1993) combined with the sporadic distribution of this particular intron—both within diatoms and across algae—are consistent with its introduction into T. undulatum or an earlier ancestor by horizontal transfer. Although most similar to group II introns within psaA genes of chlorophycean green algae, they are unlikely to be the proximal donor of this intron to T. undulatum. Phylogenetic studies of green algae have relied heavily on plastid genome data (Lemieux et al. 2014, 2015), so green algae are disproportionately represented in algal plastid genome databases. As a result, although the diatom and green algal psaA introns almost certainly have some shared history, the apparent close relationship found here and elsewhere (Brembu et al. 2014) may simply be a sampling artifact. Finally, the best horizontal transfer hypotheses have both strong phylogenetic support and either a plausible hypothesis for the transfer mechanism (Rice et al. 2013) or evidence of an intimate relationship between exchanging species (Mower et al. 2010; Sloan et al. 2014). To the best of our knowledge, no such relationship exists between diatoms and green algae. Several studies (Deschamps and Moreira 2012; Woehle et al. 2011) have cast serious doubt on the hypothesis that diatoms temporarily harbored an ancient “green” endosymbiont (Moustafa et al. 2009).
Additional sampling of plastid genomes is necessary to reconstruct the phylogenetic history of this intron, including what other algal lineages may harbor them, how recently they all diverged, and the precise pattern and directions of exchange among species. There is a growing appreciation for the intimate relationships between diatoms and both bacteria (Amin et al. 2012) and viruses (Tomaru and Nagasaki 2011), so it may be that intron transfers have been mediated by shared bacterial, viral, or plasmid vectors.
In summary, although currently available data suggest that, within diatoms, the psaA intron in T. undulatum is a recent arrival from a foreign donor, additional plastid genome data will provide the ultimate test of this hypothesis. If upheld, these data may shed valuable light on the mechanism of transfer, highlighting previously unknown associations of diatoms with other organisms. The possibility exists that further sampling may reveal the presence of this intron in other diatoms or heterokonts, strengthening support for vertical inheritance and widespread loss, similar to building evidence for the ancestral presence of twintrons in cryptophyte plastid genomes (Khan and Archibald 2008; Perrineau et al. 2015). Regardless, data from this study further underscore the dynamic nature of diatom plastid genomes. Ongoing work to sequence plastid genomes from a more phylogenetically diverse sample of diatoms will allow us to reconstruct the full pan-genome of diatom plastids and tease apart fine-scale patterns of gains and losses of aberrantly distributed genomic features.
References
Adachi J, Waddell JP, Martin W, Hasegawa M (2000) Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA. J Mol Evol 50:348–358
Amin SA, Parker MS, Armbrust EV (2012) Interactions between diatoms and bacteria. Microbiol Mol Biol Rev 76:667–684
Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, Zhou S, Allen AE, Apt KE, Bechner M, Brzezinski MA, Chaal BK, Chiovitti A, Davis AK, Demarest MS, Detter JC, Glavina T, Goodstein D, Hadi MZ, Hellsten U, Hildebrand M, Jenkins BD, Jurka J, Kapitonov VV, Kroger N, Lau WWY, Lane TW, Larimer FW, Lippmeier JC, Lucas S, Medina M, Montsant A, Obornik M, Parker MS, Palenik B, Pazour GJ, Richardson PM, Rynearson TA, Saito MA, Schwartz DC, Thamatrakoln K, Valentin K, Vardi A, Wilkerson FP, Rokhsar DS (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306:79–86
Armstrong GA (1998) Greening in the dark: light-independent chlorophyll biosynthesis from anoxygenic photosynthetic bacteria to gymnosperms. J Photochem Photobiol B 43:87–100
Belfort M, Perlman PS (1995) Mechanisms of intron mobility. J Biol Chem 270:30237–30240
Bendich AJ (2004) Circular chloroplast chromosomes: the grand illusion. Plant Cell 16:1661–1666
Blocker FJ, Mohr G, Conlan LH, Qi LI, Belfort M, Lambowitz AM (2005) Domain structure and three-dimensional model of a group II intron-encoded reverse transcriptase. RNA 11:14–28
Boisvert S, Laviolette F, Corbeil J (2010) Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol 17:1519–1533
Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, Maheswari U, Martens C, Maumus F, Otillar RP, Rayko E, Salamov A, Vandepoele K, Beszteri B, Gruber A, Heijde M, Katinka M, Mock T, Valentin K, Verret F, Berges JA, Brownlee C, Cadoret JP, Chiovitti A, Choi CJ, Coesel S, De Martino A, Detter JC, Durkin C, Falciatore A, Fournet J, Haruta M, Huysman MJ, Jenkins BD, Jiroutova K, Jorgensen RE, Joubert Y, Kaplan A, Kroger N, Kroth PG, La Roche J, Lindquist E, Lommer M, Martin-Jezequel V, Lopez PJ, Lucas S, Mangogna M, McGinnis K, Medlin LK, Montsant A, Oudot-Le Secq MP, Napoli C, Obornik M, Parker MS, Petit JL, Porcel BM, Poulsen N, Robison M, Rychlewski L, Rynearson TA, Schmutz J, Shapiro H, Siaut M, Stanley M, Sussman MR, Taylor AR, Vardi A, von Dassow P, Vyverman W, Willis A, Wyrwicz LS, Rokhsar DS, Weissenbach J, Armbrust EV, Green BR, Van de Peer Y, Grigoriev IV (2008) The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456:239–244
Boyd PW, Jickells T, Law CS, Blain S, Boyle EA, Buesseler KO, Coale KH, Cullen JJ, de Baar HJ, Follows M, Harvey M, Lancelot C, Levasseur M, Owens NP, Pollard R, Rivkin RB, Sarmiento J, Schoemann V, Smetacek V, Takeda S, Tsuda A, Turner S, Watson AJ (2007) Mesoscale iron enrichment experiments 1993-2005: synthesis and future directions. Science 315:612–617
Brembu T, Winge P, Tooming-Klunderud A, Nederbragt AJ, Jakobsen KS, Bones AM (2014) The chloroplast genome of the diatom Seminavis robusta: new features introduced through multiple mechanisms of horizontal gene transfer. Mar Genom 16:17–27
Deschamps P, Moreira D (2012) Reevaluating the green contribution to diatom genomes. Genome Biol Evol 4:683–688
Fong A, Archibald JM (2008) Evolutionary dynamics of light-independent protochlorophyllide oxidoreductase genes in the secondary plastids of cryptophyte algae. Eukaryot Cell 7:550–553
Gordon PM, Piccirilli JA (2001) Metal ion coordination by the AGC triad in domain 5 contributes to group II intron catalysis. Nat Struct Biol 8:893–898
Guillard RRL (1975) Culture of phytoplankton for feeding marine invertebrates. In: Smith WL, Chanley MH (eds) Culture of marine invertebrate animals. Plenum Press, New York, pp 26–60
Hasle GR, Syvertsen EE (1997) Marine diatoms. In: Tomas CR (ed) Identifying marine phytoplankton. Academic Press, San Diego, pp 5–386
Hunsperger HM, Randhawa T, Cattolico RA (2015) Extensive horizontal gene transfer, duplication, and loss of chlorophyll synthesis genes in the algae. BMC Evol Biol 15:1–19
Johansen SD, Haugen P, Nielsen H (2007) Expression of protein-coding genes embedded in ribosomal DNA. Biol Chem 388:679–686
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Khan H, Archibald JM (2008) Lateral transfer of introns in the cryptophyte plastid genome. Nucleic Acids Res 36:3043–3053
Kooistra WHCF, De Stefano M, Mann DG, Salma N, Medlin LK (2003) Phylogenetic position of Toxarium, a pennate-like lineage within centric diatoms (Bacillariophyceae). J Phycol 39:185–197
Lambowitz AM, Belfort M (1993) Introns as mobile genetic elements. Annu Rev Biochem 62:587–622
Laslett D, Canback B (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32:11–16
Lemieux C, Otis C, Turmel M (2014) Chloroplast phylogenomic analysis resolves deep-level relationships within the green algal class Trebouxiophyceae. BMC Evol Biol 14:1–15
Lemieux C, Vincent AT, Labarre A, Otis C, Turmel M (2015) Chloroplast phylogenomic analysis of chlorophyte green algae identifies a novel lineage sister to the Sphaeropleales (Chlorophyceae). BMC Evol Biol 15:1–13
Lohse M, Drechsel O, Bock R (2007) OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52:267–274
Lommer M, Roy AS, Schilhabel M, Schreiber S, Rosenstiel P, LaRoche J (2010) Recent transfer of an iron-regulated gene from the plastid to the nuclear genome in an oceanic diatom adapted to chronic iron limitation. BMC Genom 11:718. doi:10.1186/1471-2164-11-718
Lommer M, Specht M, Roy A-S, Kraemer L, Andreson R, Gutowska M, Wolf J, Bergner S, Schilhabel M, Klostermeier U, Beiko R, Rosenstiel P, Hippler M, LaRoche J (2012) Genome and low-iron response of an oceanic diatom adapted to chronic iron limitation. Genome Biol 13:R66
Mann DG, Vanormelingen P (2013) An inordinate fondness? The number, distributions, and origins of diatom species. J Eukaryot Microbiol 60:414–420
Marchetti A, Parker MS, Moccia LP, Lin EO, Arrieta AL, Ribalet F, Murphy MEP, Maldonado MT, Armbrust EV (2009) Ferritin is used for iron storage in bloom-forming marine pennate diatoms. Nature 457:467–470
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43:D222–D226
Medlin LK, Sato S, Mann DG, Kooistra WHCF (2008) Molecular evidence confirms sister relationship of Ardissonea, Climacosphenia, and Toxarium within the bipolar centric diatoms (Bacillariophyta, Mediophyceae), and cladistic analyses confirm that extremely elongated shape has arisen twice in the diatoms. J Phycol 44:1340–1348
Moustafa A, Beszteri B, Maier UG, Bowler C, Valentin K, Bhattacharya D (2009) Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 324:1724–1726
Mower J, Stefanovic S, Hao W, Gummow J, Jain K, Ahmed D, Palmer J (2010) Horizontal acquisition of multiple mitochondrial genes from a parasitic plant followed by gene conversion with host mitochondrial genes. BMC Biol 8:150
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Peers G, Price NM (2006) Copper-containing plastocyanin used for electron transport by an oceanic diatom. Nature 441:341–344
Perrineau M-M, Price DC, Mohr G, Bhattacharya D (2015) Recent mobility of plastid encoded group II introns and twintrons in five strains of the unicellular red alga Porphyridium. PeerJ 3:e1017
Qu G, Kaushal PS, Wang J, Shigematsu H, Piazza CL, Agrawal RK, Belfort M, Wang H-W (2016) Structure of a group II intron in complex with its reverse transcriptase. Nat Struct Mol Biol 23:549–557
Raymond J, Siefert JL, Staples CR, Blankenship RE (2004) The natural history of nitrogen fixation. Mol Biol Evol 21:541–554
Reinbothe S, Reinbothe C, Apel K, Lebedev N (1996) Evolution of chlorophyll biosynthesis—the challenge to survive photooxidation. Cell 86:703–705
Rice DW, Alverson AJ, Richardson AO, Young GJ, Sanchez-Puerta MV, Munzinger J, Barry K, Boore JL, Zhang Y, Knox EB (2013) Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342:1468–1473
Round FE, Crawford RM, Mann DG (1990) The diatoms: biology and morphology of the genera. Cambridge University Press, Cambridge
Ruck EC, Nakov T, Jansen RK, Theriot EC, Alverson AJ (2014) Serial gene losses and foreign DNA underlie size and sequence variation in the plastid genomes of diatoms. Genome Biol Evol 6:644–654
Sabir JSM, Yu M, Ashworth MP, Baeshen NA, Baeshen MN, Bahieldin A, Theriot EC, Jansen RK (2014) Conserved gene order and expanded inverted repeats characterize plastid genomes of Thalassiosirales. PLoS One 9:e107854
Salzberg SL, White O, Peterson J, Eisen JA (2001) Microbial genes in the human genome: lateral transfer or gene loss? Science 292:1903–1906
Sánchez-Puerta MV, Bachvaroff TR, Delwiche CF (2005) The complete plastid genome sequence of the haptophyte Emiliania huxleyi: a comparison to other plastid genomes. DNA Res 12:151–156
Sánchez-Puerta MV, Cho Y, Mower JP, Alverson AJ, Palmer JD (2008) Frequent, phylogenetically local horizontal transfer of the cox1 group I Intron in flowering plant mitochondria. Mol Biol Evol 25:1762–1777
Simola DF, Wissler L, Donahue G, Waterhouse RM, Helmkampf M, Roux J, Nygaard S, Glastad KM, Hagen DE, Viljakainen L, Reese JT, Hunt BG, Graur D, Elhaik E, Kriventseva EV, Wen J, Parker BJ, Cash E, Privman E, Childers CP, Muñoz-Torres MC, Boomsma JJ, Bornberg-Bauer E, Currie CR, Elsik CG, Suen G, Goodisman MAD, Keller L, Liebig J, Rawls A, Reinberg D, Smith CD, Smith CR, Tsutsui N, Wurm Y, Zdobnov EM, Berger SL, Gadau J (2013) Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality. Genome Res 23:1235–1247
Sloan DB, Nakabachi A, Richards S, Qu J, Murali SC, Gibbs RA, Moran NA (2014) Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol Biol Evol 31:857–871
Smith DR (2012) Updating our view of organelle genome nucleotide landscape. Front Genet 3:175
Smith DR, Keeling PJ (2015) Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. P Natl Acad Sci USA 112:10177–10184
Sorhannus U (2007) A nuclear-encoded small-subunit ribosomal RNA timescale for diatom evolution. Mar Micropaleontol 65:1–12
Strzepek RF, Harrison PJ (2004) Photosynthetic architecture differs in coastal and oceanic diatoms. Nature 431:689–692
Tajima N, Saitoh K, Sato S, Maruyama F, Ichinomiya M, Yoshikawa S, Kurokawa K, Ohta H, Tabata S, Kuwata A, Sato N (2016) Sequencing and analysis of the complete organellar genomes of Parmales, a closely related group to Bacillariophyta (diatoms). Curr Genet. doi:10.1007/s00294-016-0598-y
Theriot EC, Ashworth MP, Nakov T, Ruck E, Jansen RK (2015) Dissecting signal and noise in diatom chloroplast protein encoding genes with phylogenetic information profiling. Mol Phylogenet Evol 89:28–36
Tomaru Y, Nagasaki K (2011) Diatom viruses. In: Seckbach J, Kociolek P (eds) The diatom world. Springer, Netherlands, pp 211–225
Turmel M, Cote V, Otis C, Mercier JP, Gray MW, Lonergan KM, Lemieux C (1995) Evolutionary transfer of ORF-containing group I introns between different subcellular compartments (chloroplast and mitochondrion). Mol Biol Evol 12:533–545
Ueda M, Tanaka A, Sugimoto K, Shikanai T, Nishimura Y (2014) chlB requirement for chlorophyll biosynthesis under short photoperiod in Marchantia polymorpha L. Genome Biol Evol 6:620–628
Woehle C, Dagan T, Martin WF, Gould SB (2011) Red and problematic green phylogenetic signals among thousands of nuclear genes from the photosynthetic and apicomplexa-related Chromera velia. Genome Biol Evol 3:1220–1230
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255
Yamazaki S, Nomata J, Fujita Y (2006) Differential operation of dual protochlorophyllide reductases for chlorophyll biosynthesis in response to environmental oxygen levels in the cyanobacterium Leptolyngbya boryana. Plant Physiol 142:911–922
Yang Z (1995) A space-time process model for the evolution of DNA sequences. Genetics 139:993–1005
Zimmerly S, Semper C (2015) Evolution of group II introns. Mob DNA 6:1–19
Acknowledgments
We thank Colton Kessenich for help with the genome assembly; David Chafin, Jeff Pummill, and Pawel Wolinski for providing computational support through the Arkansas High Performance Computing Center (AHPCC); and Suresh Kumar and Jeffrey Lewis for critical comments on an earlier version of the manuscript. This material is based upon work supported by the National Science Foundation (NSF) under Grant No. DEB-1353131 and an award from the Arkansas Biosciences Institute. This research used computational resources available through the AHPCC, which is funded through multiple NSF grants and the Arkansas Economic Development Commission.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Electronic supplementary material
Below is the link to the electronic supplementary material.
294_2016_652_MOESM1_ESM.pdf
Figure S1. Individual gene phylogenies based on amino acid alignments for the chlB, chlL, and chlN genes. Numbers at nodes are standard bootstrap proportions from 500 pseudoreplicates (PDF 22 kb)
Rights and permissions
About this article

Cite this article
Ruck, E.C., Linard, S.R., Nakov, T. et al. Hoarding and horizontal transfer led to an expanded gene and intron repertoire in the plastid genome of the diatom, Toxarium undulatum (Bacillariophyta). Curr Genet 63, 499–507 (2017). https://doi.org/10.1007/s00294-016-0652-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0652-9