
Abstract
For the last two decades, the natural variation of the yeast Saccharomyces cerevisiae has been massively exploited with the aim of understanding ecological and evolutionary processes. As a result, many new genetic variants have been uncovered, providing a large catalogue of alleles underlying complex traits. These alleles represent a rich genetic resource with the potential to provide new strains that can cope with the growing demands of industrial fermentation processes. When surveyed in detail, several of these variants have proven useful in wine and beer industries by improving nitrogen utilisation, fermentation kinetics, ethanol production, sulphite resistance and aroma production. Here, I illustrate how allele-specific expression and polymorphisms within the coding region of GDB1 underlie fermentation kinetic differences in synthetic wine must. Nevertheless, the genetic basis of how GDB1 variants and other natural alleles interact in foreign genetic backgrounds remains unclear. Further studies in large sets of strains, recombinant hybrids and multiple parental pairs will broaden our knowledge of the molecular and genetic basis of trait adaptation for utilisation in applied and industrial processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Quantitative genetic studies and their industrial applications
The budding yeast Saccharomyces cerevisiae represents an outstanding model for evolutionary, molecular biology and genetics studies, mostly due to its ease of cultivation and laboratory manipulation, alongside its (prominent) role as a cornerstone of the genomics era (Liti 2015; Voordeckers et al. 2015). The importance of yeast became clear more than 80 years ago, with the beginning of genetics studies in brewing. Indeed, beer production aided the birth of yeast genetics by pioneering crosses between different strains to enhance the quality, flavour and stability of the final fermented product (Barnett 2007; Gibbons and Rinker 2015). Since then, yeast has become a key player in many industrial fermentation processes such as wine and sake as well as the production of fermented food like bread (Legras et al. 2007; Querol et al. 2003).
The close association between S. cerevisiae and humans implies that budding yeast corresponds to a domesticated species; however, re-sequencing studies demonstrated that it is not necessarily domesticated and many new wild isolates are continually being described (Bergstrom et al. 2014; Cromie et al. 2013; Liti et al. 2009; Skelly et al. 2013; Wang et al. 2012). In this context, field surveys in habitats remote from human activity have demonstrated that China harbours an important pool of different natural isolates with extensive genetic variation (Wang et al. 2012). This standing natural diversity of different S. cerevisiae strains provides a unique niche for the industry. Consequently, hundreds to thousands of strains are being used for molecular and quantitative genetic studies in different laboratories around the world, with the aim of: on one hand understanding yeast biology and on the other, providing the appropriate strains for utilisation in industry (Steensels et al. 2014). In an industrial setting, exploiting new genetic variants allows fine tuning of the final preferred product by, for example, increasing substrate utilisation for the production of desirable secondary metabolites or modifying metabolic fluxes in a particular genetic background (Marsit and Dequin 2015). Thus, deciphering the genetics underlying traits of industrial interest has enormous potential to identify new alleles that could serve to ameliorate fermentation processes, representing an opportunity to cope with the demands of increased productivity.
Throughout the last two decades several linkage mapping studies have been conducted in recombinant populations in order to elucidate the inheritance patterns of complex traits in yeast and identify Quantitative trait loci (QTLs) (Ehrenreich et al. 2009; Liti and Louis 2012). Most of these advances were performed utilising laboratory strains, such as S288c and their derivatives (Ehrenreich et al. 2009), providing little information about the allelic variants that explain the phenotypic diversity of the species in the wild (Liti 2015) and limiting their industrial use. With the advent of the genomic revolution and new sequencing technologies; the genome sequence of hundreds of strains is now available allowing the study of a greater genetic panel (Peter and Schacherer 2016). Thus, the latest quantitative genetic studies exploiting yeast allelic diversity expanded our knowledge of the ecology and evolutionary biology in this simple model organism by unveiling many new variants that could be useful in applied studies (Ambroset et al. 2011; Cubillos et al. 2013; Gutierrez et al. 2013; Hou et al. 2016; Jara et al. 2014; Parts et al. 2011; Salinas et al. 2012; Steyer et al. 2012; Tesniere et al. 2015; Wilkening et al. 2014).
The fermentation of beer and wine represents the main two beverages for which yeast has been utilised for centuries (Sicard and Legras 2011). The mapping of QTLs in S. cerevisiae during wine must fermentation has uncovered genetic variants for many traits that have likely been exposed to selection, such as: nitrogen consumption (Brice et al. 2014; Gutierrez et al. 2013; Jara et al. 2014), ethanol tolerance and production (Duitama et al. 2014; Snoek et al. 2016; Swinnen et al. 2012; Tilloy et al. 2014), fermentation kinetics (Kessi-Perez et al. 2016), sulphite resistance (Zimmer et al. 2014) and aroma production (Steyer et al. 2012). For example, utilising bulk segregant analysis coupled with whole-genome sequencing, four genetic variants which can improve nitrogen uptake in wine strains were discovered (Brice et al. 2014). Similarly, consumers and the wine industry are demanding lower alcohol levels and for this, directed evolved populations grown in the laboratory under limiting conditions have provided new artificial genetic variants to cope with this request (Tilloy et al. 2014). Furthermore, linkage analysis in a cross between CBS6412 and ER7A (an industrial strain) revealed SSK1, an osmosensor member of the HOG pathway, as responsible for ethanol yield differences between these two strains. The CBS6412 allele affected growth, volumetric productivity and had a low glycerol/high ethanol production ratio (Hubmann et al. 2013). One of the advantages of this type of study is that two independent genetic variants with completely opposite phenotypes may be equally useful for the industry. For example, an allele which is responsible for the production of greater ethanol levels may not be desirable in the wine industry, but it would be highly prized in the bioethanol business (Pais et al. 2013). In this way, deciphering genetic variants can have multiple applications in different industrial processes with high economic impact.
What drives natural phenotypic variation?
Originally, the majority of the genetic studies focused their efforts in finding differences between strains in coding regions. This bias is based on the premise that most allelic variants within ORFs will significantly affect the protein structure and therefore non-synonymous mutations represent key targets in the search for causal polymorphisms (Ehrenreich et al. 2009; Liti and Louis 2012). These variants were thought for years to be the principal force behind natural phenotypic variation. However, it has recently been argued that these types of mutations are usually deleterious within natural populations and most protein coding sequences are conserved even between different species (Wray 2007). Alternatively, coding regions could be finely modulated through their gene expression. In the past decades, experimental evidence has been gathered, predominantly in higher eukaryotes, demonstrating that natural gene expression variation represents a key factor shaping an individual’s phenotype, where polymorphisms within, e.g., transcription factor binding sites, can lead to phenotypic differences between strains (Chidi et al. 2016; Cubillos et al. 2012; Fraser et al. 2012; Salinas et al. 2016; Wittkopp and Kalay 2012). Moreover, in recent years gene expression regulation has been at the forefront of genetic and evolution studies in multiple species. By finely tuning mutations in both portions of the genome (coding and regulatory regions), organisms can exhibit distinct phenotypes and adapt to stressful conditions (Salinas et al. 2016). By modifying gene expression patterns individuals can exhibit an extraordinary regulatory elasticity, allowing them to withstand unfavourable environmental conditions. A well-known approach to quantify the effect of natural variants acting in cis (this is near the encoded transcript) is to study allele-specific expression (ASE) through massive RNA sequencing (McManus et al. 2010; Salinas et al. 2016). ASE is the difference in expression levels between two parental alleles within a hybrid and reflects the outcome of polymorphisms located upstream of the ORF. Therefore, ASE is a highly useful tool for understanding the impact of genetic variation within regulatory regions (Fig. 1).

Allele-specific expression pipeline. F1 hybrids generated from two individuals genetically diverged (Strain A in red and Strain B in blue) can be utilised to estimate allele-specific expression (ASE) and quantify expression divergence between genetic backgrounds. To accomplish this, the F1 hybrid is subjected to RNA-seq and then specific reads belonging to Strain A or Strain B are quantified for each parental background. Significant deviations from a 50/50 distribution for each allele are labelled as ASE between strains
ASE in natural yeast isolates
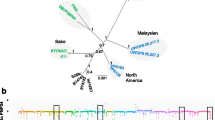
Previously, we were able to demonstrate that ASE is highly widespread between natural S. cerevisiae isolates by quantifying the effect of cis-variants in a grid of six F1 hybrids derived from the cross of four divergent strains (Salinas et al. 2016). Furthermore, several efforts have demonstrated that allelic expression differences can directly impact a phenotype (Fay et al. 2004; Gerke et al. 2009; Salinas et al. 2016). By estimating the consumption of aspartic acid and glutamic acid in wine fermentation must in two strains of different geographic origin, we were able to show that polymorphisms in both portion of the gene ASN1 (coding and regulatory), an asparagine synthetase that catalyses the synthesis of asparagine from aspartic acid (Salinas et al. 2016), were partly responsible for nitrogen assimilation differences between these two genetic backgrounds. Interestingly, ASN1 was not the only case where we could directly correlate allele-specific expression and phenotype. Among the thousands of alleles differentially expressed, we also found GDB1, a glycogen debranching enzyme required for glycogen degradation and relevant in the fermentation process (Apweiler et al. 2012). In our ASE strategy, we found that a Wine/European isolate (DBVPG6765, named as WE) exhibited greater expression levels compared to any other surveyed strain (Fig. 2a). To determine the influence of dissimilar GDB1 allelic variants in the fermentation process and its potential industrial application, we performed a reciprocal hemizygosity assay between WE and a North American (NA) strain, since they showed the greatest allelic expression differences, and characterised them for fermentation kinetics in synthetic wine must. We observed significant differences in the total CO2 output between reciprocal hemizygotes, with the NA-gdb1Δ/WE-GDB1 hybrid having a greater rate of CO2 production than the NA-GDB1/WE-gdb1Δ hemizygote, in agreement with greater expression levels in the WE background (Fig. 2b). These strains differ by several polymorphisms in both, the regulatory and coding regions within GDB1. In order to determine whether the phenotypic differences were due to either category of polymorphic changes, we performed an allele swap assay where we swapped, on one hand the promoters of the two strains and on the other, the coding portion (Salinas et al. 2016) (Fig. 2c). Based on this approach, we reconstructed all the possible reciprocal hemizygote combinations in the parental backgrounds by varying either the ORF or the regulatory region. After 21 days of fermentation, we observed in the NA background that the CO2 production increased by 14 % when the GDB1 WE promoter was introduced (Fig. 2d(i)), in agreement with greater expression levels of the GDB1 WE. Surprisingly, when we introduced the NA promoter into the WE background, the NA promoter allele did not change the fermentation kinetics, suggesting a background-dependent effect on the ORF being expressed (Fig. 2d(ii)). Subsequently, when we performed the corresponding experiment replacing the ORF region, we observed an 18 % lower CO2 output when we introduced the GDB1 NA allele into the WE background (Fig. 2d(iv)). These results demonstrate that, just like in ASN1, polymorphisms located in regulatory and coding regions in GDB1 explain the phenotypic differences observed in reciprocal hemizygotes and ultimately, between NA and WE strains. Based on these results, GDB1 alleles (and many others so far described in the literature) could represent potential genetic variants for applied yeast studies.

Phenotypic validation of ASE in GDB1. a Diagram representing the three F1 hybrids and ASE levels for GDB1 between the Wine/European (WE) isolate crossed against the Y12 (Sake, SA), DBVPG6044 (West African, WA) and YPS128 (North American, NA) isolates. b CO2 output (gl/L) for GDB1 reciprocal hemizygotes. NA-GDB1/WE-gdb1Δ (green) denotes hemizygotes carrying the NA allele, while NA-gdb1Δ/WE-GDB1 (orange) denotes hemizygotes carrying the WE allele. c Selected promoter and ORF swap strategies are shown. The WE/NA promoters and ORFs were swapped in the opposite genetic backgrounds, generating four different combinations. d CO2 output (gl/L) for GDB1 reciprocal hemizygotes carrying swapped promoters (graphs i and ii) or ORF (graphs iii and iv). p denotes the promoter and o denotes the ORF that the reciprocal hemizygote carries in each case. The relative percentage of CO2 output difference is indicated in the two cases where significant differences were found between hemizygotes carrying different combinations of promoter and ORF
Perspectives
Quantitative genetic studies have provided a wide set of natural allelic variants which can be used to tackle the needs of the fermentation industry. In the near future, low sequencing costs will expand the repertoire of sequenced strains, revealing an even larger number of genetic variants to explore. However, the phenotypic contribution of these alleles may vary when placed in different genetic backgrounds and therefore large screens to estimate gene–gene (G × G) interactions should be explored before they can be extrapolated to other strains. In this context, establishing not only how these allelic variants interact with other genes from different backgrounds, but also how they interact with the environment (G × E) is a milestone that has not been reached. Screens in offspring derived from dozens of parental pairs (Hou et al. 2016) or the utilisation of recombinant hybrids (Hallin et al. 2016) grown under an array of environments will provide the means for dissecting and understanding complex genetic interactions. Part of these screens can be complemented by determining the effects of polymorphisms upon gene expression or protein structure. Thus far, although many studies have described differences in expression levels, the molecular mechanisms underlying transcript abundance variation are still not clear and represent a current challenge in modern genetics. Thus, deciphering G × G and G × E interactions will help to understand how allelic variants respond to genetic and environmental interactions and generate better models for their application in the industry.
References
Ambroset C, Petit M, Brion C, Sanchez I, Delobel P, Guerin C, Chiapello H, Nicolas P, Bigey F, Dequin S, Blondin B (2011) Deciphering the molecular basis of wine yeast fermentation traits using a combined genetic and genomic approach. G3 (Bethesda) 1:263–281. doi:10.1534/g3.111.000422
Apweiler E, Sameith K, Margaritis T, Brabers N, van de Pasch L, Bakker LV, van Leenen D, Holstege FC, Kemmeren P (2012) Yeast glucose pathways converge on the transcriptional regulation of trehalose biosynthesis. BMC Genom 13:239. doi:10.1186/1471-2164-13-239
Barnett JA (2007) A history of research on yeasts 10: foundations of yeast genetics. Yeast 24:799–845. doi:10.1002/yea.1513
Bergstrom A, Simpson JT, Salinas F, Barre B, Parts L, Zia A, Nguyen Ba AN, Moses AM, Louis EJ, Mustonen V, Warringer J, Durbin R, Liti G (2014) A high-definition view of functional genetic variation from natural yeast genomes. Mol Biol Evol 31:872–888. doi:10.1093/molbev/msu037
Brice C, Sanchez I, Bigey F, Legras JL, Blondin B (2014) A genetic approach of wine yeast fermentation capacity in nitrogen-starvation reveals the key role of nitrogen signaling. BMC Genom 15:495. doi:10.1186/1471-2164-15-495
Chidi BS, Rossouw D, Bauer FF (2016) Identifying and assessing the impact of wine acid-related genes in yeast. Curr Genet 62:149–164. doi:10.1007/s00294-015-0498-6
Cromie GA, Hyma KE, Ludlow CL, Garmendia-Torres C, Gilbert TL, May P, Huang AA, Dudley AM, Fay JC (2013) Genomic sequence diversity and population structure of Saccharomyces cerevisiae assessed by RAD-seq. G3 (Bethesda) 3:2163–2171. doi:10.1534/g3.113.007492->
Cubillos FA, Coustham V, Loudet O (2012) Lessons from eQTL mapping studies: non-coding regions and their role behind natural phenotypic variation in plants. Curr Opin Plant Biol 15:192–198. doi:10.1016/j.pbi.2012.01.005
Cubillos FA, Parts L, Salinas F, Bergstrom A, Scovacricchi E, Zia A, Illingworth CJ, Mustonen V, Ibstedt S, Warringer J, Louis EJ, Durbin R, Liti G (2013) High-resolution mapping of complex traits with a four-parent advanced intercross yeast population. Genetics 195:1141–1155. doi:10.1534/genetics.113.155515
Duitama J, Sanchez-Rodriguez A, Goovaerts A, Pulido-Tamayo S, Hubmann G, Foulquie-Moreno MR, Thevelein JM, Verstrepen KJ, Marchal K (2014) Improved linkage analysis of Quantitative Trait Loci using bulk segregants unveils a novel determinant of high ethanol tolerance in yeast. BMC Genom 15:207. doi:10.1186/1471-2164-15-207
Ehrenreich IM, Gerke JP, Kruglyak L (2009) Genetic dissection of complex traits in yeast: insights from studies of gene expression and other phenotypes in the BYxRM cross. Cold Spring Harb Symp Quant Biol 74:145–153. doi:10.1101/sqb.2009.74.013
Fay JC, McCullough HL, Sniegowski PD, Eisen MB (2004) Population genetic variation in gene expression is associated with phenotypic variation in Saccharomyces cerevisiae. Genome Biol 5:R26. doi:10.1186/gb-2004-5-4-r26
Fraser HB, Levy S, Chavan A, Shah HB, Perez JC, Zhou Y, Siegal ML, Sinha H (2012) Polygenic cis-regulatory adaptation in the evolution of yeast pathogenicity. Genome Res 22:1930–1939. doi:10.1101/gr.134080.111
Gerke J, Lorenz K, Cohen B (2009) Genetic interactions between transcription factors cause natural variation in yeast. Science 323:498–501. doi:10.1126/science.1166426
Gibbons JG, Rinker DC (2015) The genomics of microbial domestication in the fermented food environment. Curr Opin Genet Dev 35:1–8. doi:10.1016/j.gde.2015.07.003
Gutierrez A, Beltran G, Warringer J, Guillamon JM (2013) Genetic basis of variations in nitrogen source utilization in four wine commercial yeast strains. PLoS ONE 8:e67166. doi:10.1371/journal.pone.0067166
Hallin J, Martens K, Young A, Zackrisson M, Salinas F, Parts L, Warringer J, Lit G (2016) Powerful decomposition of complex traits in a diploid model using Phased Outbred Lines. bioRxiv http://dx.doi.org/10.1101/042176
Hou J, Sigwalt A, Pflieger D, Peter J, Montigny Jd, Dunham M, Schacherer J (2016) The hidden complexity of Mendelian traits across yeast natural populations. bioRxiv http://dx.doi.org/10.1101/039693
Hubmann G, Mathe L, Foulquie-Moreno MR, Duitama J, Nevoigt E, Thevelein JM (2013) Identification of multiple interacting alleles conferring low glycerol and high ethanol yield in Saccharomyces cerevisiae ethanolic fermentation. Biotechnol Biofuels 6:87. doi:10.1186/1754-6834-6-87
Jara M, Cubillos FA, Garcia V, Salinas F, Aguilera O, Liti G, Martinez C (2014) Mapping genetic variants underlying differences in the central nitrogen metabolism in fermenter yeasts. PLoS ONE 9:e86533. doi:10.1371/journal.pone.0086533
Kessi-Perez EI, Araos S, Garcia V, Salinas F, Abarca V, Larrondo LF, Martinez C, Cubillos FA (2016) RIM15 antagonistic pleiotropy is responsible for differences in fermentation and stress response kinetics in budding yeast. FEMS Yeast Res. doi:10.1093/femsyr/fow021
Legras JL, Merdinoglu D, Cornuet JM, Karst F (2007) Bread, beer and wine: Saccharomyces cerevisiae diversity reflects human history. Mol Ecol 16:2091–2102. doi:10.1111/j.1365-294X.2007.03266.x
Liti G (2015) The fascinating and secret wild life of the budding yeast S. cerevisiae. Elife. doi:10.7554/eLife.05835
Liti G, Louis EJ (2012) Advances in quantitative trait analysis in yeast. PLoS Genet 8:e1002912. doi:10.1371/journal.pgen.1002912
Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, Tsai IJ, Bergman CM, Bensasson D, O’Kelly MJ, van Oudenaarden A, Barton DB, Bailes E, Nguyen AN, Jones M, Quail MA, Goodhead I, Sims S, Smith F, Blomberg A, Durbin R, Louis EJ (2009) Population genomics of domestic and wild yeasts. Nature 458:337–341. doi:10.1038/nature07743
Marsit S, Dequin S (2015) Diversity and adaptive evolution of Saccharomyces wine yeast: a review. FEMS Yeast Res. doi:10.1093/femsyr/fov067
McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ (2010) Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20:816–825. doi:10.1101/gr.102491.109
Pais TM, Foulquie-Moreno MR, Hubmann G, Duitama J, Swinnen S, Goovaerts A, Yang Y, Dumortier F, Thevelein JM (2013) Comparative polygenic analysis of maximal ethanol accumulation capacity and tolerance to high ethanol levels of cell proliferation in yeast. PLoS Genet 9:e1003548. doi:10.1371/journal.pgen.1003548
Parts L, Cubillos FA, Warringer J, Jain K, Salinas F, Bumpstead SJ, Molin M, Zia A, Simpson JT, Quail MA, Moses A, Louis EJ, Durbin R, Liti G (2011) Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res 21:1131–1138. doi:10.1101/gr.116731.110
Peter J, Schacherer J (2016) Population genomics of yeasts: towards a comprehensive view across a broad evolutionary scale. Yeast 33:73–81. doi:10.1002/yea.3142
Querol A, Fernandez-Espinar MT, del Olmo M, Barrio E (2003) Adaptive evolution of wine yeast. Int J Food Microbiol 86:3–10
Salinas F, Cubillos FA, Soto D, Garcia V, Bergstrom A, Warringer J, Ganga MA, Louis EJ, Liti G, Martinez C (2012) The genetic basis of natural variation in oenological traits in Saccharomyces cerevisiae. PLoS ONE 7:e49640. doi:10.1371/journal.pone.0049640
Salinas F, de Boer CG, Abarca V, Garcia V, Cuevas M, Araos S, Larrondo LF, Martinez C, Cubillos FA (2016) Natural variation in non-coding regions underlying phenotypic diversity in budding yeast. Sci Rep 6:21849. doi:10.1038/srep21849
Sicard D, Legras JL (2011) Bread, beer and wine: yeast domestication in the Saccharomyces sensu stricto complex. C R Biol 334:229–236. doi:10.1016/j.crvi.2010.12.016
Skelly DA, Merrihew GE, Riffle M, Connelly CF, Kerr EO, Johansson M, Jaschob D, Graczyk B, Shulman NJ, Wakefield J, Cooper SJ, Fields S, Noble WS, Muller EG, Davis TN, Dunham MJ, Maccoss MJ, Akey JM (2013) Integrative phenomics reveals insight into the structure of phenotypic diversity in budding yeast. Genome Res 23:1496–1504. doi:10.1101/gr.155762.113
Snoek T, Verstrepen KJ, Voordeckers K (2016) How do yeast cells become tolerant to high ethanol concentrations? Curr Genet. doi:10.1007/s00294-015-0561-3
Steensels J, Snoek T, Meersman E, Picca Nicolino M, Voordeckers K, Verstrepen KJ (2014) Improving industrial yeast strains: exploiting natural and artificial diversity. FEMS Microbiol Rev 38:947–995. doi:10.1111/1574-6976.12073
Steyer D, Ambroset C, Brion C, Claudel P, Delobel P, Sanchez I, Erny C, Blondin B, Karst F, Legras JL (2012) QTL mapping of the production of wine aroma compounds by yeast. BMC Genom 13:573. doi:10.1186/1471-2164-13-573
Swinnen S, Schaerlaekens K, Pais T, Claesen J, Hubmann G, Yang Y, Demeke M, Foulquie-Moreno MR, Goovaerts A, Souvereyns K, Clement L, Dumortier F, Thevelein JM (2012) Identification of novel causative genes determining the complex trait of high ethanol tolerance in yeast using pooled-segregant whole-genome sequence analysis. Genome Res 22:975–984. doi:10.1101/gr.131698.111
Tesniere C, Brice C, Blondin B (2015) Responses of Saccharomyces cerevisiae to nitrogen starvation in wine alcoholic fermentation. Appl Microbiol Biotechnol 99:7025–7034. doi:10.1007/s00253-015-6810-z
Tilloy V, Ortiz-Julien A, Dequin S (2014) Reduction of ethanol yield and improvement of glycerol formation by adaptive evolution of the wine yeast Saccharomyces cerevisiae under hyperosmotic conditions. Appl Environ Microbiol 80:2623–2632. doi:10.1128/AEM.03710-13
Voordeckers K, Kominek J, Das A, Espinosa-Cantu A, De Maeyer D, Arslan A, Van Pee M, van der Zande E, Meert W, Yang Y, Zhu B, Marchal K, DeLuna A, Van Noort V, Jelier R, Verstrepen KJ (2015) Adaptation to High Ethanol Reveals Complex Evolutionary Pathways. PLoS Genet 11:e1005635. doi:10.1371/journal.pgen.1005635
Wang QM, Liu WQ, Liti G, Wang SA, Bai FY (2012) Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol Ecol. doi:10.1111/j.1365-294X.2012.05732.x
Wilkening S, Lin G, Fritsch ES, Tekkedil MM, Anders S, Kuehn R, Nguyen M, Aiyar RS, Proctor M, Sakhanenko NA, Galas DJ, Gagneur J, Deutschbauer A, Steinmetz LM (2014) An evaluation of high-throughput approaches to QTL mapping in Saccharomyces cerevisiae. Genetics 196:853–865. doi:10.1534/genetics.113.160291
Wittkopp PJ, Kalay G (2012) Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet 13:59–69. doi:10.1038/nrg3095
Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8:206–216. doi:10.1038/nrg2063
Zimmer A, Durand C, Loira N, Durrens P, Sherman DJ, Marullo P (2014) QTL dissection of Lag phase in wine fermentation reveals a new translocation responsible for Saccharomyces cerevisiae adaptation to sulfite. PLoS ONE 9:e86298. doi:10.1371/journal.pone.0086298
Acknowledgments
I would like to thank Gianni Liti, Francisco Salinas and Verónica García for constructive comments on the manuscript. Francisco Cubillos is supported by Comisión Nacional de Investigación Científica y Tecnológica CONICYT FONDECYT (Grant 11140097) and MN-FISB (Grant NC120043).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Cubillos, F.A. Exploiting budding yeast natural variation for industrial processes. Curr Genet 62, 745–751 (2016). https://doi.org/10.1007/s00294-016-0602-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0602-6