
Abstract
The dendrimer has a high degree of geometric symmetry, a precise and controllable molecular size, a large number of surface-active functional groups, a rich cavity inside the molecule, and a controlled molecular chain growth. The unique structural properties of the above-mentioned macromolecules have made it a research hot spot in many fields. Molecular simulation technology, as a new scientific research method, plays an important role in the basic theory and applied research of dendrimers. This paper reviews the basic progress of molecular simulation technology in the field of dendrimers in recent years, including the application of dendrimers in medicine, DNA, pharmaceutical carriers, proteins, amino acids, and so on.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The important member in the macromolecular system is dendrimers. Dendrimer is firstly reported in 1978 by Vögtel et al. by applying Michael addition and reduction approaches [1]. They succeeded to synthesize a branched tripropylamine-based macromolecule by utilizing a primary amine and acrylonitrile to give a dinitrile, which called it a cascade molecule. Following this achievement, Tomalia et al. synthesized branched polyamide-amine (PAMAM) for the first time in 1985 [2]. Accordingly, the word of dendrimer as a specific member of macromolecule has become popular in scientific researches since the 1990s. In this regard, Tomalia and Fréchet wrote an interesting review article about the historical perspective concerning the discovery of dendrimers [3]. The features of a dendrimer are an exact molecular structure, a precise molecular weight, and monodispersity with repeated and regular branch [4]. Contrary to linear polymers, dendrimers illustrate a unique class of synthetic polymers, highly rigid and strongly branched molecules which can be synthesized from a branch point or central segment [5].
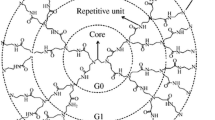
In general, a dendrimer consists of three distinct segments: a core, scaffold, and surface structure. The core is placed in the center of the molecule and attached with a given number of branches which are called dendrons. Each dendron is composed of the scaffold (number of branching points) and surface groups. The number of branches characterizes the generation and the scale of a dendrimer, i.e., the number of branch points, the functionality, and the length of the spacer. It should be noted that the physical and chemical properties of the dendrimer are determined by the nature of functional groups, which extend to the surroundings [6].
Some of the important structural features of dendrimers are: abundant surface functional groups, various types of functionalized, precise molecular arrangements, precise nanoscale structure, highly geometric symmetry, and homologous series of cavity size. Therefore, they have many performance characteristics such as: solubility, hydrodynamic performance, unique viscosity behavior, and versatility [7]. Various potential applications of dendrimers, including biological [8], biomedical [9], detection therapeutic, diagnostic, and detection [10] for cancer treatment [11], pharmaceutical, nanocarriers, and drug delivery [12], tissue engineering [13], brain delivery and cancer therapy [14], sensing [15], catalysis [16], molecular electronics [17], photonics [18], nanomedicine [19], magnetic resonance imaging [20], gene delivery [21], optoelectronic applications [22], dendrimer liquid membranes for gas separation [23], and so on [24, 25], have been proposed because of their unique nanostructures and excellent physical properties.
Synthesis of dendrimers
There are two major routes to synthesize dendrimers consisting divergent and convergent. The divergent methodology was first introduced independently by Newkome et al. [26] and Tomalia et al. [2]. Divergent method is summarized in four steps as below: Firstly, a reaction starts with a core, and this core must have some features such as have reactive groups and a small functional molecule. Secondly, this core reacts with some blocks with some characterizations such as having a well-designed building and having some functional groups which are able to transform into a new reactive point to form the first-generation dendrimer (G1). This procedure is well known as dendritic growth. Thirdly, the G1 dendrimer will expose the reactive points on its surface, and G2 could be made after the second-stage dendritic growth. Finally, by repeating activation and growth process, higher-generation dendrimers could be achieved [27].
On the other hand, the synthesis dendrimers from divergent method suffer from some important issues such as time-consuming and steric-shielding effects. It should be noted that both of these limitations have a strong effect for synthesis of a big dendrimer. Besides the mentioned problems, if the final functional groups react with the interior of the dendrimer, it is unable to react with the building blocks. This procedure is known as dense-core theory and is common in the flexible dendrimers. After each generation growth, surface space for each active point is reduced, leading to a defect in the structure and unreacted active when making higher-generation dendrimers. This procedure is called a dense-shell concept and is common in the rigid dendrimers [28]. It should be noted that according to the flexibility of the backbones of dendrimers, they are divided into flexible and rigid dendrimers.
On the other hand, the convergence methodology was reported by Hawker and Fréchet in 1990 while they synthesized the poly (aryl ether) dendrimers [29]. This applies a reverse growth process as compared with the divergent one. Convergence approach starts from the building block and reacts with a focal-activated building block to form a dendron inward toward to the core to give birth to the dendrimer [30].
Dendrimers are designed into a given category base on the diverse functional groups, types of functionalized, and architecture of dendrimers. The important categories of dendrimers are: (1) carbon- or oxygen-based dendrimers such as polyether, polyester, and glycodendrimers [31], (2) chiral dendrimers including chirality base on the core and chirality base on the branching unit [32], (3) metallodendrimer such as poly (propylene imine) pyridyl imine palladium [33] and poly (bis (imino) pyridyl) iron(II) [34], (4) peptide dendrimers which consists of a peptidyl branching core or covalently attached surface functional points such as multiple antigen peptide (MAP) [35], (5) phosphorus dendrimers [36], (6) porphyrin dendrimers [37], (7) silicon dendrimers including silane, carbosilane, siloxane, and carbosiloxane [38], (8) triazine dendrimers [39], (9) hybrid dendrimers [40], (10) PAMAM dendrimers [41], (11) polyamidoamine organosilicon (PAMANOS) dendrimers [42], (12) poly propylene imine (PPI) dendrimers [43], and (13) polylysine (PLL) dendrimers [44].
Characterization of dendrimers
A wide range of analytical techniques has been proposed for characterization of dendrimers according to the various applications of dendrimers. Much scientific research is focused on developing and improving on techniques for characterization of dendrimers. These techniques define the feature, property, and structure of dendrimers, such as optical activity, structural properties, thermodynamic properties, chemical composition, molecular mass, surface structure, size, shape, morphology, and homogeneity of dendrimers. Table 1 illustrates the summary of the technique for characterization of dendrimers.
PAMAM dendrimer
The first dendritic structure synthesized concerning to the divergent route which received widespread attention was Tomalia’s polyamide-amine (PAMAM) dendrimer.
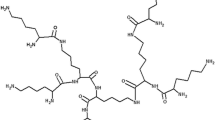
Today, PAMAM dendrimer is commercially available and has been studied extensively. PAMAM dendrimer is the first commercial and synthetic dendrimer member based on ethylene diamine core and amide repeating branched structures [67, 68]. Ethylene diamine (EDA)-based poly amide dendrimers have been extensively investigated in biomedical applications and composite base materials since synthesis [69]. The molecular structure of PAMAM is shown in Fig. 1.

PAMAM dendrimer. The core, G0, and G1 are shown, reprinted with permission from Ref. [116] copyright 2019 American Chemical Society
Molecular dynamics (MD) simulation
Simultaneously with the technology development, the applications and functions of molecular simulation techniques are becoming more and more demanding. In the environmental protection, chemical and chemical industry and energy conservation are constant themes, and the improvement and promotion of new functional products will be one of the major development trends in the future. The development of computers has introduced the calculation methods and theoretical techniques of chemical, physical, and materials science, which has promoted the progression of new products. With the rapid development of molecular simulation technology, the world’s major companies in order to meet the research needs of different fields have developed a variety of molecular simulation calculation software, such as TINKER, Gromacs, Materials Studio, and LAMMPS. MD simulation method is a widely used computer simulation method [70,71,72,73,74]. In a nutshell, molecular simulation is a systematic computer simulation of real experimental molecules. Since the computer can clearly display the microstructure of the molecule and calculate the performance of the target product, some experiments, in which it is difficult or impossible to get the data, can be done using molecular simulation. Based on the experiment, a set of calculation algorithms and calculation models through some basic principles are established, on the basis of which a reasonable molecular structure and molecular behavior are calculated. Molecular simulation methods mainly have four theoretical methods, including quantum mechanical method, molecular mechanics method, molecular dynamics method, and Monte Carlo method, in which quantum mechanics can describe the change of electronic structure, and molecular mechanics can describe the changes in the ground state atomic structure. These two methods, strictly speaking, describe the molecular structure of absolute zero. MD can be used to describe the physical structure of the average structure and molecular structure at various temperatures. With MD simulations, the particles are moved based on Newton’s equations of motion and the forces the particles exert on each other. In this case, the particles follow realistic trajectories which are important for the study of dynamic properties and systems that are out of equilibrium. The simulations can be done in an NVE, NVT, or NPT ensemble. Here the letters indicate which quantities are kept constant during the simulation. N is the number of particles, V the volume, E the energy, T the temperature, and P the pressure. The time steps the system take cannot be too big; otherwise, the trajectory of the particles is no longer realistic. For the NVE ensemble, the steps need to be even shorter to ensure that energy is conserved. The fact that particles follow a realistic trajectory can also be a disadvantage. When the system gets stuck in a local minimum, it may take a long time before it crosses the barrier. If we are only interested in the equilibrium properties of the system and not in the dynamics/time evolution, it is better to use a Monte Carlo method. The Monte Carlo method of the molecule can describe the average structure of various temperatures by the introduction of the Boltzmann factor. In terms of obtaining a statistical average structure of a certain state, the Monte Carlo method of the molecule is often more effective than the molecular dynamics method. MD methods have irreplaceable advantages when studying dynamic processes on short timescales.
Application of PAMAM evaluated using MD simulation
The first MD investigations of dendrimer base macromolecules were achieved by Goddard and coworkers [75]. They applied many ligand molecules to encapsulate inside the dendrimer and succeeded to coat fifth-generation poly propylene imine dendrimers with Bengal Rose. Ivanov and Jacobson applied molecular modeling (MM) to purify the molecular model proposed by the PAMAM protein agonist (CGS21680) bound to the A2A adenosine receptor dimerization in the guest molecule inside the dendrimer [76]. Efficient encapsulation was noticed in the interior of the backfolded molecule in comparison with their extended isomeric counterparts [77].
Application of PAMAM in medicine
MD simulation can obtain the structure of the complex and the driving effect behind it, but it is difficult to study the whole process of drug encapsulation and release. MD studies the PAMAM-based dendrimers and drug interactions mainly used in the full atomic MD. On this premise, Alderete et al. applied MD to investigate the complexation of mefenamic acid (MA) with low-generation (PAMAM-G2 and PAMAM-G3) PAMAM dendrimers [78]. They found that by increasing the dendrimer generation, the internal drug encapsulation is enhanced. They suggested that the PAMAM with the positively charged surface is the most relevant factor for drug association. Their MD results are in good agreement with experimental findings.
pH environment has a large effect on the efficacy of the drug in the human body, and therefore, MD is able to analyze the effects of drugs with different pH values, which can significantly improve the drug development rate. On this premise, Caballero and coworkers investigated the interaction between the nicotinic acid (NA) as a drug and PAMAM-G3 dendrimer at different pH by applying MD simulations [79]. They found that at pH = 3 the internal amine groups are protonated and the PAMAM cavities become less hydrophobic; therefore, the PAMAM–drug interactions become similar to solvent–drug interactions. They showed that VdW interactions between the methylene groups of the PAMAM-G3 dendrimer and drug stabilized the drug inside the PAMAM-G3 dendrimer at pH = 6 (Fig. 2).

Simulation snapshots of drug inside PAMAM-G3 at pH = 6, a conformation A, b conformation B, c conformation C, d radial distribution function (RDF) of the dendrimer around drug for conformations A, B, and C, e RDF of the water molecules around drug for conformations A, B, and C, reprinted from Ref. [79] Copyright (2019), with permission from Elsevier
Figure 2 illustrates the conformation of drug and the relation of them with a surface amine of PAMAM-G3 dendrimer (Fig. 2a–c). RDFs of dendrimer–drug and water–drug are shown in Fig. 2d and e, respectively. Drug is more exposed to the water in conformation A rather than conformation B, and drug is closer to methylene groups of PAMAM-G3 dendrimer in conformation C. Their simulation results showed that PAMAM-G3 dendrimer is more favorable for drug entrapment when pH = 6, and their complexes are very stable. Giri et al. investigated the impact of core chemistry, terminal group, and generation of dendrimer in binding of human serum albumin (HSA) to PAMAM dendrimers by measuring the HSA binding constants (Kb) of PAMAM dendrimers [80]. Their MD simulation results illustrated that Kb of HAS to PAMAM depends on their chemical composition and size of their terminal groups. Figure 3 shows the impact of the dendrimer terminal group on the HAS Kb of PAMAM-G4 dendrimer.

Effects of the dendrimer terminal group on the HAS Kb of PAMAM-G4 dendrimer, reprinted with permission from Ref. [80] copyright 2019 American Chemical Society
Figure 3 illustrates that the lowest Kb values are observed for the PAMAM-G4 dendrimer with neutral terminal groups due to weak hydrogen bonding interactions between the protein amino acid residues and terminal groups of PAMAM-G4 dendrimer. Figure 4 highlights the impact of dendrimer core chemistry on the HAS Kb of PAMAM-G4 dendrimer. From Fig. 3, it can be deduced that the HAS Kb of PAMAM-G4 dendrimer is not significantly related to core chemistry. Their results of the Kb value reveal some critical effects and interactions between the HSA protein and PAMAM dendrimer. Their MD results are in good agreement with their experimental findings. By using MD simulations, Maiti et al. tested the release pattern of two soluble drugs including l-alanine and (Ala) salicylic acid (Sal) and two insoluble drugs including primidone (Prim) and phenylbutazone (Pbz) [81]. These four ligands were placed inside the ethylenediamine (EDA) core of PAMAM-G5 dendrimer. Their potential of mean force (PMF) results showed that insoluble drugs (Prim and Pbz) have higher energy barriers than soluble drugs (Ala and Sal) (see Fig. 5). However, their biological activity depends on the surface charge properties of dendrimers. These data help to optimize and design the dendrimer-based drug delivery system.

Effects of dendrimer core chemistry on the HAS Kb of PAMAM-G4 dendrimer, reprinted with permission from Ref. [80] copyright 2019 American Chemical Society

PMF variation as a function of the drug–dendrimer, reprinted with permission from Ref. [81] copyright 2019 American Chemical Society
Tanis and Karatasos used atomistic MD simulation and applied AMBER force field to investigate the complexation of ibuprofen and PAMAM-G3 dendrimer in aqueous solution under various pH conditions [82]. They indicated that the PAMAM-G3 dendrimer–ibuprofen complex is unstable at low pH due to the lack of hydrogen bonding. No stable drug/dendrimer complex was detected at low pH, and the electrostatic interaction between ibuprofen and PAMAM-G3 dendrimer allows them to form stable complexes as shown in Fig. 6.

Average distance between the drug and the PAMAM-G3 dendrimer centers of mass, reprinted with permission from Ref. [82] copyright 2019 American Chemical Society
Also, Liu et al. used the Dreiding force field to find that surface grafting of PEG which promoted the PAMAM dendrimer to accommodate more drug molecules [83]. They found that at high pH, the PMF energy barrier of PAMAM dendrimers with anticancer drug molecules including CE6, SN38, DOX, and MTX is much lower than that of physiological pH, so the high pH environment is suitable for drug embedding because the drug–dendrimer complex is formed.
Application of PAMAM in DNA
By individualized analysis of tumor DNA, chemotherapy patients may prolong survival by a factor of six. Doctors have determined that the precise treatment of cancer is increasingly dependent on the genetic test results and guidance of tumors. PAMAM different algebras have different entanglement effects on single-stranded DNA. On this premise, Maiti and Bagchi studied sequence-dependent complexation between single-strand DNA (ssDNA) and various generation EDA-cored PAMAM dendrimers by using MD simulations and calculating free energy [84]. They revealed that the G2 and G3 did not have enough surface charge to neutralize ssDNA because part of the ssDNA far from PAMAM spread out in solution as shown in Fig. 7.

a Structure of ssDNA–dendrimer complex during various stages of the wrapping process at the interval of few ns. b Variation of the number of contact points between DNA and dendrimer, reprinted with permission from Ref. [84] copyright 2019 American Chemical Society
In another close study, the complexation between various generations of PAMAM dendrimers (G3–G5) and double-stranded DNA (dsDNA) have been studied by Nandy and Maiti [85]. They illustrated that dsDNA can be completely entangled on PAMAM-G5 dendrimers. Therefore, it is generally believed that the charge between the positively charged dendrimer and the negatively charged genetic material plays a key role in the structure of the complex. From the snapshots in Fig. 8, it is revealed that the dendrimer continues to search for a suitable binding position on DNA at the beginning and the dendrimer slides along the DNA backbone for both G3 and G4. They found that binding energies of the complexation follow the trend G5 > G4 > G3.

a Structure of the DNA-PAMAM-G4 dendrimer complex during various stages of complex formation. b the same for the DNA-PAMAM-G3 dendrimer complex, reprinted with permission from Ref. [85] copyright 2019 American Chemical Society
The stability of dsDNA entanglement on PAMAM is also one of the important indicators. In this regard, Yu and Larson used Monte Carlo simulation system to investigate the effects of PAMAM algebra, surface amidation, and solution salt concentration on the stability of PAMAM dendrimer and dsDNA complexes [86]. They showed that high salt concentration is not conducive to dsDNA and increased PAMAM dendrimer algebra in complex compress dsDNA more tightly. Also, Márquez-Miranda et al. applied MD simulations to study the effects of different surface chemical groups of PAMAM dendrimers on nucleic acid molecules [87, 88]. They demonstrated that the PAMAM can form a stable complex with ssDNA, when the PAMAM terminal group is an amine group and the PAMAM cannot form a stable complex with ssDNA when the terminal group is a hydroxyl group because ssDNA has only a small amount of contact with PAMAM, and they cannot pass through the cell membrane.
Not only the size and surface chemistry of dendrimers focus of attention, but also flexibility and stiffness of PAMAM dendrimers are another critical factor in the formation of dendrimers. On this premise, Pavan and coworkers used MD to investigate the effect of the stiffness of dendrimers on the structure of dendrimer–gene complexes [89]. The MD simulation results showed that the stiffness of PAMAM dendrimers plays a crucial role in the binding state. It is mainly regulated by combining the competition between enthalpy and entropy.
The curves of RDF in Fig. 9 demonstrate the atomic density with respect to time, and the high peaks correspond to areas of low atomic mobility and high density of atoms. They found that flexible molecules tend to form a spherical composite structure, while rigid molecules are rearranged such that their terminal groups make more contact with the oligonucleotide. Ainalem and Nylander wrote an interesting review article and discussed about the PAMAM algebra, ionic strength, and other factors which affect the morphology of PAMAM and DNA complexes [90].

RDF of G2-5 (a) and F2-1 (b), reprinted with permission from Ref. [89] copyright 2019 American Chemical Society
PAMAM for pharmaceutical carriers and biomedical applications
The development of novel PAMAM drugs and gene delivery with the greatest therapeutic potential and minimal side effects is a huge challenge for nanomedicine. As a delivery vector, the PAMAM must exceed many of the obstacles encountered before the biological agent is delivered to the target within the cell. As an important supplement to experimental methods, computer simulation has a good advantage for studying intermolecular interactions [91,92,93]. As transporters, when PAMAM-based dendrimers approach cells, they first interact with the cell membrane. Therefore, understanding the interaction between dendrimers and biofilms is important for designing efficient dendrimer-based carriers. Maiti et al. simulated the structure of the first to 11th-generation PAMAM dendrimers [94]. They found very little strain in these structures up to G6; however, for G10 there is considerable strain throughout the entire structure, which increases dramatically for G11. They suggested that the steric interactions of the surface groups prevent growth of full generations beyond G10. For example, in the case of PAMAM dendrimers with ethylenediamine as the initial nucleus, G1 to G3 cannot form a dense spatial internal structure, and each branch sparsely forms an ellipsoid [95]. Until G4 and G5, this macromolecule has a relatively complete spherical outline and internal space. Ma et al. recently studied the role of PAMAM and negatively charged asymmetric membranes, revealing the physical mechanism of dendrimers as carriers in gene transfection to cause gene–carrier complexes to escape from endocytosis [96, 97] and proposed utilization of pH-responsive and possible pathways for gene-targeted transport based on complex charge reversal [98]. Whether the stiffness of the dendrimer as a carrier can reach the index is key to successful delivery, so it must be considered whether the stiffness is optimal. Lyulin et al. used the coarse-grained MD simulation method to simulate the interaction between dendrimers and linear polyelectrolyte [99]. They observed the formation of compact dendrimer polyelectrolyte complexes, while strong electrostatic interactions induced dendrimer size reduction. Moreover, Lyulin et al. studied the structure and dynamics of dendritic macromolecules in dilute solutes by explicitly excluding volume and hydrodynamic interaction Brownian dynamics simulation, and compared the results with the mean field theory [100, 101]. In addition, the influence of the stiffness of polyelectrolyte on the dendrimer polyelectrolyte complex was also studied by coarse-grained MD simulation method [102]. It was found that with the increase in polyelectrolyte stiffness, the polyelectrolyte structure composited with PAMAM changed interestingly from curling. If the U or V shape becomes bar shape, there may be an optimal stiffness for the transport and release of biologically active guest molecules.
The PAMAM dendrimers need to penetrate into the drug body to act on the target cell, so there is a certain requirement for the target product embedding degree. Wang et al. used dissipative particle dynamics to find that increasing the PAMAM dendrimer algebra would enhance its permeability to bilayer membranes [103]. Yan et al. systematically studied the interactions between charged dendrimers and phospholipid bilayers and their complex structures by using dissipative particle dynamics [104]. They found that the effect of increasing the hydrophilic component and phospholipid head on the surface of the phospholipid bilayer led to the spreading of the dendrimer on the surface of the phospholipid bilayer, while the effect of increasing the hydrophobic component on the inside of the dendrimer on the phospholipid tail group led to the deeper embedding of the dendrimer into the phospholipid bilayer. Figure 10 shows the snapshots of the complexes comprised the charged G5 dendrimer with the lipid bilayer membrane.

Complexes between the charged G5 dendrimer and the lipid bilayer membrane. Panels d–f are the cross-sectional views of panels a–c respectively, reprinted with permission from Ref. [104] copyright 2019 American Chemical Society
In addition to considering the environmental pH as a drug, it is also necessary to consider the pH value of the carrier as a carrier. On this premise, Terao and Nakayama studied the structure of charged dendrimers at different pH values, as well as multiple generations (G5, G6, and G7) by random MD simulations [105]. Gurtovenko et al. used MD to simulate the calculated amount of charged dendrimers under explicit counterions and solvent molecules under neutral pH conditions [106]. They found that the addition of explicit counterions to the simulation has a large effect on the structure and kinetics of the charged dendrimer. Guo et al. used dissipative particle dynamic methods to study the structure–performance relationship of a series of pH-responsive polymer transport systems [107, 108]. Luo and Jiang combined MD and dissipative particle dynamics methods to study the loading and release of pH-responsive amphiphilic copolymer poly(-amino ester)-polyethylene glycol (PAE-PEG) anticancer drug camptothecin [109]. In addition, the terminal primary amino group of the PAMAM molecule is distributed throughout the molecule and can be close to the core inside the molecule, rather than being located entirely on the surface of the molecule. This indicates that the terminal group of the dendrimer has sufficient flexibility to be folded back into the interior of the molecule. It is consistent with the coarse-grained MD simulation performed by Zhong et al. using the Martini force field [91] and the all-atom MD results by Mait et al. using the Dreiding force field [110].
Application of dendrimers in proteins and amino acids
A large number of terminal functional groups and tight and precisely controlled molecular structure are the unique properties of dendrimers, which make them a good use in the field of proteins and amino acids. Su ling Chen et al. used coarse-grained molecular dynamics to simulate the interaction of G4 PAMAM dendrimers with KALP peptides in different pH solutions [111]. They found that KALP peptide had little effect on the size, shape, and density distribution of dendrimers in two pH environments, and there was a certain space inside the dendrimer to accommodate KALP polypeptide molecules. The calculation of free energy shows that the two molecules are not easy to form a stable composite structure in acidic and neutral environments. Also, Schneider et al. demonstrated the MD simulation of G0 dendritic macromolecules with alpha-chymotrypsinogen A (aCgn) and surface-modified guanidine (Gdm) and studied the effects of salt ions such as Cl−, SO42−, and H2PO4− [112]. They proposed a priority coefficient of action, the thermodynamics of free energy in the migration of proteins from water to additives, and insight into how dendrimer salts affect protein–protein interactions. It can also be used to measure the tendency of protein surface additives. The multi-surface group binds the dendrimer to the protein more strongly than the single functional group. Poly-l-lysine (PLL) dendrimers are amino acid macromolecules that act as drug delivery agents. Their branched structure allows them to be functionalized by different groups to encapsulate drugs into their structures. Rahimi et al. designed a process particle size model of PLL dendrimers and determined its parameters for simulating three generations of PLL dendrimers [113]. The results show that as the amount of production increases, dendrimers change. It is more spherical. At pH = 7, the PLL dendrimer has more holes, allowing more water molecules to be encapsulated inside. The formation of the spherical structure of the PLL dendrimer was confirmed by calculating the moment of inertia and the aspect ratio. Robert et al. [114] studied the structural changes of PLL dendrimers from the first-generation (G1) to the fifth-generation (G6) by means of all-atomic MD simulation and pointed out that the complexes of G1 and G6 dendrimers were spherical and regular, with highly recessed surfaces and dense nuclear structures. Neelov et al. [115] studied the properties of different PLL dendrimers using atomic MD simulations and reported that their properties are not dependent on temperature, but their internal group mobility is dependent on their formation. Kavyani et al. [116] used the GC-MD method to show that the length and nature of the PAMAM dendrimer core have an effect on the size and encapsulation capacity of dendrimers. Figure 11 highlights that at pH 7 the dendrimer terminals are closer to the core than at pH 5 and also proves that the PAMAM with the DAH core has the lowest RDF values, so the DAH core can create more cavities in the dendrimer structure.

RDF of G4s dendrimer terminal beads at both pHs 5 and 7 with various cores. The dotted rectangular in (a) is extended in (b), reprinted with permission from Ref. [116] copyright 2019 American Chemical Society
In another work, Lee and Larson used GC-MD simulation to study the effects of PLL and PAMAM dendrimers on the DMPC bilayer membrane [117]. They obtained the bonding interaction parameters of coarse crystal PAMAM dendrimers with histidine and arginine terminal groups at pH 5 and 7 [118]. They pointed out that as the amount of histidine in the terminal group of the dendrimer increases, the size of the formed complex becomes larger.
Dendrimers in other applications
MD simulation provides a general simulation method. There are efficient methods for simulating various natural processes at the molecular level [119,120,121,122,123]. Dendrimers have great application prospects in new nanocomposites. Therefore, understanding the interaction between dendrimers and surfaces is of great significance. Wolski and Wolski used the all-atom MD simulation method to study the behavior of PAMAM dendrimers adsorbed on the polarization model of gold surface [124]. The study found that with increase in pH value, the structure of dendrimers became more compact. Also, other applications of PAMAM dendrimers are fingerprint detection [125], biomedical applications [126], methanol oxidation [127], optical sensing [128], and so on [129].
Conclusions and perspectives
In recent years, due to the rapid development of fluid mechanics, quantum mechanics, quantum science, and other disciplines, providing solid theoretical techniques for experimental design, computational molecular simulation has received more and more attention, which will become the mainstream development trend in the future. Molecular modeling can help researchers get a lot of information that is difficult or impossible to obtain during the experiment. As the application of molecular simulation technology continues to expand, the simulation of dendrimers will be deeper. At present, the molecular simulation of dendrimers around the world is mainly focused on medicine, but dendrimers have a wide range of applications in the fields of surfactants, photographic materials, nanomaterials, and catalysis. Research on these areas has focused on theory rather than on practical applications in a specific area. Therefore, the future development trend of molecular simulation technology will be toward the practical application of broadening the dendrimer field.
References
Buhleier E, Wehner W, Vögtle F (1978) ‘Cascade’- and ‘Nonskid-chain-like’ syntheses of molecular cavity topologies. Chem Inf 9 25:228
Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S et al (1985) A new class of polymers: starburst-dendritic macromolecules. Polym J 17:117
Tomalia DA, Fréchet JM (2002) Discovery of dendrimers and dendritic polymers: a brief historical perspective. J Polym Sci Part A Polym Chem 40:2719–2728
Abbasi E, Aval SF, Akbarzadeh A, Milani M, Nasrabadi HT, Joo SW et al (2014) Dendrimers: synthesis, applications, and properties. Nanoscale Res Lett 9:247
Lee CC, MacKay JA, Fréchet JM, Szoka FC (2005) Designing dendrimers for biological applications. Nat Biotechnol 23:1517
Vögtle F, Richardt G, Werner N (2009) Dendrimer chemistry: concepts, syntheses, properties, applications. Wiley, Hoboken
Tang Z (2017) Research progress on synthesis and characteristic about dendrimers. In: IOP conference series: earth and environmental science. IOP Publishing, vol 100, p 012024
Cloninger MJ (2002) Biological applications of dendrimers. Curr Opin Chem Biol 6:742–748
Mintzer MA, Grinstaff MW (2011) Biomedical applications of dendrimers: a tutorial. Chem Soc Rev 40:173–190
Noriega-Luna B, Godínez LA, Rodríguez FJ, Rodríguez A, Larrea G, Sosa-Ferreyra C et al (2014) Applications of dendrimers in drug delivery agents, diagnosis, therapy, and detection. J Nanomater 2014:39
Wolinsky JB, Grinstaff MW (2008) Therapeutic and diagnostic applications of dendrimers for cancer treatment. Adv Drug Deliv Rev 60:1037–1055
Cheng Y, Wang J, Rao T, He X, Xu T (2008) Pharmaceutical applications of dendrimers: promising nanocarriers for drug delivery. Front Biosci 13:1447–1471
Joshi N, Grinstaff M (2008) Applications of dendrimers in tissue engineering. Curr Top Med Chem 8:1225–1236
Somani S, Dufès C (2014) Applications of dendrimers for brain delivery and cancer therapy. Nanomedicine 9:2403–2414
Astruc D, Ornelas C, Ruiz J (2008) Metallocenyl dendrimers and their applications in molecular electronics, sensing, and catalysis. Acc Chem Res 41:841–856
Niu Y, Crooks RM (2003) Dendrimer-encapsulated metal nanoparticles and their applications to catalysis. C R Chim 6:1049–1059
Astruc D, Ornelas C, Aranzaes JR (2008) Ferrocenyl-terminated dendrimers: design for applications in molecular electronics, molecular recognition and catalysis. J Inorg Organomet Polym Mater 18:4–17
Bergamini G, Marchi E, Ceroni P (2011) Metal ion complexes of cyclam-cored dendrimers for molecular photonics. Coord Chem Rev 255:2458–2468
Menjoge AR, Kannan RM, Tomalia DA (2010) Dendrimer-based drug and imaging conjugates: design considerations for nanomedical applications. Drug Discov Today 15:171–185
Langereis S, Dirksen A, Hackeng TM, Van Genderen MH, Meijer E (2007) Dendrimers and magnetic resonance imaging. New J Chem 31:1152–1160
Dufes C, Uchegbu IF, Schätzlein AG (2005) Dendrimers in gene delivery. Adv Drug Deliv Rev 57:2177–2202
Yokoyama S, Otomo A, Nakahama T, Okuno Y, Mashiko S (2003) Dendrimers for optoelectronic applications. In: Schalley CA, Vögtle F (eds) Dendrimers V. Topics in current chemistry, vol 228. Springer, Berlin, Heidelberg, pp 205–226
Kovvali AS, Sirkar K (2001) Dendrimer liquid membranes: CO2 separation from gas mixtures. Ind Eng Chem Res 40:2502–2511
Szymański P, Markowicz M, Mikiciuk-Olasik E (2011) Nanotechnology in pharmaceutical and biomedical applications: dendrimers. Nano 6:509–539
Caminade A-M, Ouali A, Laurent R, Turrin C-O, Majoral J-P (2016) Coordination chemistry with phosphorus dendrimers. Applications as catalysts, for materials, and in biology. Coord Chem Rev 308:478–497
Newkome GR, Yao Z, Baker GR, Gupta VK (1985) Micelles. Part 1. Cascade molecules: a new approach to micelles. A [27]-arborol. J Org Chem 50:2003–2004
Wiesler U-M, Berresheim A, Morgenroth F, Lieser G, Müllen K (2001) Divergent synthesis of polyphenylene dendrimers: the role of core and branching reagents upon size and shape. Macromolecules 34:187–199
Boris D, Rubinstein M (1996) A self-consistent mean field model of a starburst dendrimer: dense core vs dense shell. Macromolecules 29:7251–7260
Hawker CJ, Frechet JM (1990) Preparation of polymers with controlled molecular architecture. A new convergent approach to dendritic macromolecules. J Am Chem Soc 112:7638–7647
Grayson SM, Frechet JM (2001) Convergent dendrons and dendrimers: from synthesis to applications. Chem Rev 101:3819–3868
Twibanire JDAK, Grindley TB (2014) Polyester dendrimers: smart carriers for drug delivery. Polymers 6:179–213
Roy R, Shiao TC (2015) Glyconanosynthons as powerful scaffolds and building blocks for the rapid construction of multifaceted, dense and chiral dendrimers. Chem Soc Rev 44:3924–3941
Smith G, Chen R, Mapolie S (2003) The synthesis and catalytic activity of a first-generation poly (propylene imine) pyridylimine palladium metallodendrimer. J Organomet Chem 673:111–115
Zheng Z-J, Chen J, Li Y-S (2004) The synthesis and catalytic activity of poly (bis (imino) pyridyl) iron (II) metallodendrimer. J Organomet Chem 689:3040–3045
Luo K, Li C, Li L, She W, Wang G, Gu Z (2012) Arginine functionalized peptide dendrimers as potential gene delivery vehicles. Biomaterials 33:4917–4927
Caminade A-M, Ouali A, Laurent R, Turrin C-O, Majoral J-P (2015) The dendritic effect illustrated with phosphorus dendrimers. Chem Soc Rev 44:3890–3899
Fukuzumi S, Saito K, Ohkubo K, Khoury T, Kashiwagi Y, Absalom MA et al (2011) Multiple photosynthetic reaction centres composed of supramolecular assemblies of zinc porphyrin dendrimers with a fullerene acceptor. Chem Commun 47:7980–7982
Campos B, Algarra M, Alonso B, Casado C, Jiménez-Jiménez J, Rodríguez-Castellón E et al (2015) Fluorescent sensor for Cr(VI) based in functionalized silicon quantum dots with dendrimers. Talanta 144:862–867
Lim J, Simanek EE (2012) Triazine dendrimers as drug delivery systems: from synthesis to therapy. Adv Drug Deliv Rev 64:826–835
Xu X, Jian Y, Li Y, Zhang X, Tu Z, Gu Z (2014) Bio-inspired supramolecular hybrid dendrimers self-assembled from low-generation peptide dendrons for highly efficient gene delivery and biological tracking. ACS Nano 8:9255–9264
Najlah M, Freeman S, Khoder M, Attwood D, D’Emanuele A (2017) In vitro evaluation of third generation PAMAM dendrimer conjugates. Molecules 22:1661
Dvornic PR (2006) PAMAMOS: the first commercial silicon-containing dendrimers and their applications. J Polym Sci Part A Polym Chem 44:2755–2773
Bhargava M, Bhargava S, Bhargava V (2017) P3. 02c-002 Mannosylated poly (propylene imine) dendrimer mediated lung delivery of anticancer bioactive: topic: targeted therapy. J Thorac Oncol 12:S1272
Lataifeh A, Kraatz H-B (2019) Self-assembly of silver nanoparticles-low generation peptide dendrimer conjugates into poly-l-lysine. Mater Lett 254:353–356
Pande S, Crooks RM (2011) Analysis of poly (amidoamine) dendrimer structure by UV–Vis spectroscopy. Langmuir 27:9609–9613
Castagnola M, Zuppi C, Rossetti DV, Vincenzoni F, Lupi A, Vitali A et al (2002) Characterization of dendrimer properties by capillary electrophoresis and their use as pseudostationary phases. Electrophoresis 23:1769–1778
Soininen AJ, Kasëmi E, Schlüter AD, Ikkala O, Ruokolainen J, Mezzenga R (2010) Self-assembly and induced circular dichroism in dendritic supramolecules with cholesteric pendant groups. J Am Chem Soc 132:10882–10890
Appelhans D, Oertel U, Mazzeo R, Komber H, Hoffmann J, Weidner S et al (2009) Dense-shell glycodendrimers: UV/Vis and electron paramagnetic resonance study of metal ion complexation. Proc R Soc A Math Phys Eng Sci 466:1489–1513
Chiu MH, Prenner EJ (2011) Differential scanning calorimetry: an invaluable tool for a detailed thermodynamic characterization of macromolecules and their interactions. J Pharm Bioallied Sci 3:39
Pan Z, Xu M, Cheung EY, Harris KD, Constable EC, Housecroft CE (2006) Understanding the structural properties of a dendrimeric material directly from powder X-ray diffraction data. J Phys Chem B 110:11620–11623
Gautam SP, Gupta AK, Agrawal S, Sureka S (2012) Spectroscopic characterization of dendrimers. Int J Pharm Pharm Sci 4:77–80
Porcar L, Liu Y, Verduzco R, Hong K, Butler PD, Magid LJ et al (2008) Structural investigation of PAMAM dendrimers in aqueous solutions using small-angle neutron scattering: effect of generation. J Phys Chem B 112:14772–14778
Mullen DG, Desai A, van Dongen MA, Barash M, Baker JR Jr, Banaszak Holl MM (2012) Best practices for purification and characterization of PAMAM dendrimer. Macromolecules 45:5316–5320
Giordanengo R, Mazarin M, Wu J, Peng L, Charles L (2007) Propagation of structural deviations of poly (amidoamine) fan-shape dendrimers (generations 0–3) characterized by MALDI and electrospray mass spectrometry. Int J Mass Spectrom 266:62–75
Biricova V, Laznickova A (2009) Dendrimers: analytical characterization and applications. Bioorg Chem 37:185–192
Zhou L, Russell DH, Zhao M, Crooks RM (2001) Characterization of poly (amidoamine) dendrimers and their complexes with Cu2+ by matrix-assisted laser desorption ionization mass spectrometry. Macromolecules 34:3567–3573
Najlah M, Freeman S, Attwood D, D’Emanuele A (2006) Synthesis, characterization and stability of dendrimer prodrugs. Int J Pharm 308:175–182
Xu TH, Lu R, Qiu XP, Liu XL, Xue PC, Tan CH et al (2006) Synthesis and characterization of carbazole-based dendrimers with porphyrin cores. Eur J Org Chem 2006:4014–4020
Carr PL, Davies GR, Feast WJ, Stainton NM, Ward IM (1996) Dielectric and mechanical characterization of aryl ester dendrimer/PET blends. Polymer 37:2395–2401
Tintaru A, Ungaro R, Liu X, Chen C, Giordano L, Peng L et al (2015) Structural characterization of new defective molecules in poly (amidoamide) dendrimers by combining mass spectrometry and nuclear magnetic resonance. Anal Chim Acta 853:451–459
Sharma A, Gautam SP, Gupta AK (2011) Surface modified dendrimers: synthesis and characterization for cancer targeted drug delivery. Bioorg Med Chem 19:3341–3346
Popescu M-C, Filip D, Vasile C, Cruz C, Rueff J, Marcos M et al (2006) Characterization by Fourier transform infrared spectroscopy (FT-IR) and 2D IR correlation spectroscopy of PAMAM dendrimer. J Phys Chem B 110:14198–14211
Li J, Piehler L, Qin D, Baker J, Tomalia D, Meier D (2000) Visualization and characterization of poly (amidoamine) dendrimers by atomic force microscopy. Langmuir 16:5613–5616
Shi X, Sun K, Balogh LP, Baker JR Jr (2006) Synthesis, characterization, and manipulation of dendrimer-stabilized iron sulfide nanoparticles. Nanotechnology 17:4554
Xia C, Fan X, Locklin J, Advincula RC, Gies A, Nonidez W (2004) Characterization, supramolecular assembly, and nanostructures of thiophene dendrimers. J Am Chem Soc 126:8735–8743
Baytekin B, Werner N, Luppertz F, Engeser M, Brüggemann J, Bitter S et al (2006) How useful is mass spectrometry for the characterization of dendrimers?:“Fake defects” in the ESI and MALDI mass spectra of dendritic compounds. Int J Mass Spectrom 249:138–148
Tosh DK, Yoo LS, Chinn M, Hong K, Kilbey SM, Barrett MO et al (2010) Polyamidoamine (PAMAM) dendrimer conjugates of “clickable” agonists of the A3 adenosine receptor and coactivation of the P2Y14 receptor by a tethered nucleotide. Bioconjug Chem 21:372–384
Åkesson A, Lind TK, Barker R, Hughes A, Cárdenas M (2012) Unraveling dendrimer translocation across cell membrane mimics. Langmuir 28:13025–13033
Tomalia DA (2005) Birth of a new macromolecular architecture: dendrimers as quantized building blocks for nanoscale synthetic polymer chemistry. Prog Polym Sci 30:294–324
Foroutan M, Fatemi SM, Darvishi M (2018) Formation and stability of water clusters at the molybdenum disulfide interface: a molecular dynamics simulation investigation. J Phys Condens Matter 30:415001
Foroutan M, Darvishi M, Fatemi SM, Babazadeh KH (2018) Water chain formation on rutile TiO2 (110) nanocrystal: a molecular dynamics simulation approach. J Mol Liq 250:344–352
Foroutan M, Fatemi SM, Esmaeilian F (2017) A review of the structure and dynamics of nanoconfined water and ionic liquids via molecular dynamics simulation. Eur Phys J E 40:19
Foroutan M, Darvishi M, Fatemi SM (2017) Structural and dynamical characterization of water on the Au (100) and graphene surfaces: a molecular dynamics simulation approach. Phys Rev E 96:033312
Fatemi SM, Foroutan M (2015) Study of dispersion of carbon nanotubes by Triton X-100 surfactant using molecular dynamics simulation. J Iran Chem Soc 12:1905–1913
Miklis P, Çaǧin T, Goddard WA (1997) Dynamics of Bengal Rose encapsulated in the Meijer dendrimer box. J Am Chem Soc 119:7458–7462
Ivanov AA, Jacobson KA (2008) Molecular modeling of a PAMAM-CGS21680 dendrimer bound to an A2A adenosine receptor homodimer. Bioorg Med Chem Lett 18:4312–4315
Chasse TL, Sachdeva R, Li Q, Li Z, Petrie RJ, Gorman CB (2003) Structural effects on encapsulation as probed in redox-active core dendrimer isomers. J Am Chem Soc 125:8250–8254
Barra PA, Barraza L, Jiménez VA, Gavín JA, Alderete JB (2014) Complexation of mefenamic acid by low-generation PAMAM dendrimers: insight from nmr spectroscopy studies and molecular dynamics simulations. Macromol Chem Phys 215:372–383
Caballero J, Poblete H, Navarro C, Alzate-Morales JH (2013) Association of nicotinic acid with a poly (amidoamine) dendrimer studied by molecular dynamics simulations. J Mol Graph Model 39:71–78
Giri J, Diallo MS, Simpson AJ, Liu Y, Goddard WA III, Kumar R et al (2011) Interactions of poly (amidoamine) dendrimers with human serum albumin: binding constants and mechanisms. ACS Nano 5:3456–3468
Maingi V, Kumar MVS, Maiti PK (2012) PAMAM dendrimer–drug interactions: effect of pH on the binding and release pattern. J Phys Chem B 116:4370–4376
Tanis I, Karatasos K (2009) Association of a weakly acidic anti-inflammatory drug (ibuprofen) with a poly (amidoamine) dendrimer as studied by molecular dynamics simulations. J Phys Chem B 113:10984–10993
Zhang F-D, Liu Y, Xu J-C, Li S-J, Wang X-N, Sun Y et al (2015) Binding and conformation of dendrimer-based drug delivery systems: a molecular dynamics study. Adv Manuf 3:221–231
Maiti PK, Bagchi B (2006) Structure and dynamics of DNA—dendrimer complexation: role of counterions, water, and base pair sequence. Nano Lett 6:2478–2485
Nandy B, Maiti PK (2010) DNA compaction by a dendrimer. J Phys Chem B 115:217–230
Yu S, Larson RG (2014) Monte-Carlo simulations of PAMAM dendrimer–DNA interactions. Soft Matter 10:5325–5336
Márquez-Miranda V, Camarada MB, Araya-Durán I, Varas-Concha I, Almonacid DE, González-Nilo FD (2015) Biomimetics: from bioinformatics to rational design of dendrimers as gene carriers. PLoS ONE 10:e0138392
Márquez-Miranda V, Peñaloza JP, Araya-Durán I, Reyes R, Vidaurre S, Romero V et al (2016) Effect of terminal groups of dendrimers in the complexation with antisense oligonucleotides and cell uptake. Nanoscale Res Lett 11:66
Pavan GM, Mintzer MA, Simanek EE, Merkel OM, Kissel T, Danani A (2010) Computational insights into the interactions between DNA and siRNA with “rigid” and “flexible” triazine dendrimers. Macromolecules 11:721–730
Ainalem M-L, Nylander T (2011) DNA condensation using cationic dendrimers—morphology and supramolecular structure of formed aggregates. Soft Matter 7:4577–4594
Zhong T, Ai P, Zhou J (2011) Structures and properties of PAMAM dendrimer: a multi-scale simulation study. Fluid Phase Equilib 302:43–47
Yang L, da Rocha SR (2014) PEGylated, NH2-terminated PAMAM dendrimers: a microscopic view from atomistic computer simulations. Mol Pharm 11:1459–1470
Lin X, Bai T, Zuo YY, Gu N (2014) Promote potential applications of nanoparticles as respiratory drug carrier: insights from molecular dynamics simulations. Nanoscale 6:2759–2767
Maiti PK, Çaǧın T, Wang G, Goddard WA (2004) Structure of PAMAM dendrimers: generations 1 through 11. Macromolecules 37:6236–6254
Charles S, Vasanthan N, Kwon D, Sekosan G, Ghosh S (2012) Surface modification of poly (amidoamine)(PAMAM) dendrimer as antimicrobial agents. Tetrahedron Lett 53:6670–6675
Ding H-M, Tian W-D, Ma Y-Q (2012) Designing nanoparticle translocation through membranes by computer simulations. ACS Nano 6:1230–1238
Tu CK, Chen K, Tian WD, Ma YQ (2013) Computational investigations of a peptide-modified dendrimer interacting with lipid membranes. Macromol Rapid Commun 34:1237–1242
Ma Y-Q (2012) pH-responsive dendrimers interacting with lipid membranes. Soft Matter 8:2627–2632
Lyulin SV, Vattulainen I, Gurtovenko AA (2008) Complexes comprised of charged dendrimers, linear polyelectrolytes, and counterions: insight through coarse-grained molecular dynamics simulations. Macromolecules 41:4961–4968
Lyulin SV, Evers L, van der Schoot P, Darinskii AA, Lyulin AV, Michels M (2004) Effect of solvent quality and electrostatic interactions on size and structure of dendrimers. Brownian dynamics simulation and mean-field theory. Macromolecules 37:3049–3063
Lyulin SV, Darinskii AA, Lyulin AV, Michels M (2004) Computer simulation of the dynamics of neutral and charged dendrimers. Macromolecules 37:4676–4685
Tian W-D, Ma Y-Q (2010) Complexation of a linear polyelectrolyte with a charged dendrimer: polyelectrolyte stiffness effects. Macromolecules 43:1575–1582
Wang Y-L, Lu Z-Y, Laaksonen A (2012) Specific binding structures of dendrimers on lipid bilayer membranes. Phys Chem Chem Phys 14:8348–8359
Yan L-T, Yu X (2009) Charged dendrimers on lipid bilayer membranes: insight through dissipative particle dynamics simulations. Macromolecules 42:6277–6283
Terao T, Nakayama T (2004) Molecular dynamics study of dendrimers: structure and effective interaction. Macromolecules 37:4686–4694
Gurtovenko AA, Lyulin SV, Karttunen M, Vattulainen I (2006) Molecular dynamics study of charged dendrimers in salt-free solution: effect of counterions. J Chem Phys 124:094904
Guo XD, Zhang LJ, Wu ZM, Qian Y (2010) Dissipative particle dynamics studies on microstructure of pH-sensitive micelles for sustained drug delivery. Macromolecules 43:7839–7844
Guo XD, Zhang LJ, Qian Y (2012) Systematic multiscale method for studying the structure–performance relationship of drug-delivery systems. Ind Eng Chem Res 51:4719–4730
Luo Z, Jiang J (2012) pH-sensitive drug loading/releasing in amphiphilic copolymer PAE–PEG: integrating molecular dynamics and dissipative particle dynamics simulations. J Control Release 162:185–193
Maiti PK, Goddard WA (2006) Solvent quality changes the structure of G8 PAMAM dendrimer, a disagreement with some experimental interpretations. J Phys Chem B 110:25628–25632
Chen S, Pan M, Tian W (2014) Computational study of the interaction between PAMAM dendrimer and KALP peptide. Acta Polym Sin 8:1062–1069
Schneider CP, Shukla D, Trout BL (2011) Effects of solute–solute interactions on protein stability studied using various counterions and dendrimers. PLoS ONE 6:e27665
Rahimi A, Amjad-Iranagh S, Modarress H (2016) Molecular dynamics simulation of coarse-grained poly (l-lysine) dendrimers. J Mol Model 22:59
Roberts BP, Scanlon MJ, Krippner GY, Chalmers DK (2009) Molecular dynamics of poly (l-lysine) dendrimers with naphthalene disulfonate caps. Macromolecules 42:2775–2783
Neelov I, Markelov D, Falkovich S, Ilyash MY, Okrugin B, Darinskii A (2013) Mathematical simulation of lysine dendrimers: temperature dependences. Polym Sci Ser C 55:154–161
Kavyani S, Amjad-Iranagh S, Modarress H (2014) Aqueous poly (amidoamine) dendrimer G3 and G4 generations with several interior cores at pHs 5 and 7: a molecular dynamics simulation study. J Phys Chem B 118:3257–3266
Lee H, Larson RG (2008) Lipid bilayer curvature and pore formation induced by charged linear polymers and dendrimers: the effect of molecular shape. J Phys Chem B 112:12279–12285
Lee H, Choi JS, Larson RG (2011) Molecular dynamics studies of the size and internal structure of the PAMAM dendrimer grafted with arginine and histidine. Macromolecules 44:8681–8686
Fatemi SM, Foroutan M (2016) Recent developments concerning the dispersion of carbon nanotubes in surfactant/polymer systems by MD simulation. J Nanostruct Chem 6:29–40
Fatemi SM, Foroutan M (2014) Study of the dynamic behavior of boron nitride nanotube (BNNT) and triton surfactant complexes using molecular dynamics simulations. Adv Sci Eng Med 6:583–590
Fatemi SM, Foroutan M (2015) Recent findings about ionic liquids mixtures obtained by molecular dynamics simulation. J Nanostruct Chem 5:243–253
Fatemi S, Foroutan M (2016) Review on carbon nanotubes and carbon nanotube bundles for gas/ion separation and water purification studied by molecular dynamics simulation. Int J Environ Sci Technol 13:457–470
Fatemi SM, Foroutan M (2014) Study of dispersion of boron nitride nanotubes by triton X-100 surfactant using molecular dynamics simulations. J Theor Comput Chem 13:1450063
Wolski P, Panczyk T (2019) Conformational properties of PAMAM dendrimers adsorbed on the gold surface studied by molecular dynamics simulation. J Phys Chem C 123:22603–22613
Jin Y-J, Luo Y-J, Li G-P, Li J, Wang Y-F, Yang R-Q et al (2008) Application of photoluminescent CdS/PAMAM nanocomposites in fingerprint detection. Forensic Sci Int 179:34–38
Yang H, Kao WJ (2006) Dendrimers for pharmaceutical and biomedical applications. J Biomater Sci Polym Ed 17:3–19
Maiyalagan T (2009) Pt–Ru nanoparticles supported PAMAM dendrimer functionalized carbon nanofiber composite catalysts and their application to methanol oxidation. J Solid State Electrochem 13:1561–1566
Soršak E, Valh JV, Urek ŠK, Lobnik A (2015) Application of PAMAM dendrimers in optical sensing. Analyst 140:976–989
Domański D, Klajnert B, Bryszewska M (2004) Influence of PAMAM dendrimers on human red blood cells. Bioelectrochemistry 63:189–191
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fatemi, S.M., Fatemi, S.J. & Abbasi, Z. PAMAM dendrimer-based macromolecules and their potential applications: recent advances in theoretical studies. Polym. Bull. 77, 6671–6691 (2020). https://doi.org/10.1007/s00289-019-03076-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-019-03076-4