
Abstract
Dendrimers are viewed as hyperbranched, three-dimensional, monodisperse globular macromolecules with branches emanating from each monomeric unit. For improving the solvability of hydrophobic drugs and raising their bioactivity accompanied by a persistent release action, dendrimeric nanoparticles are customarily employed as latent drug delivery devices. PAMAM dendrimers have been broadly investigated as new approaches for restrained drug delivery; nevertheless, the computational analysis of the dendrimer-drug complex is an intricate phenomenon ascribable to the conformational flexibility of dendrimers and the distinct characteristics of the interactions existing within the dendrimer-drug system. Traditional procedures for analyzing drug interaction have been intended mainly for protein-derived substrates and, thus, there is a necessity to create novel conventions to handle special views of dendrimers. In the present research investigation, cavities in generation-2 and generation-3 Polyamidoamine (PAMAM) dendrimers have been developed, followed by the employment of fully atomistic molecular dynamics (MD) simulations to analyze the interactions of dendrimer with multitudinous model drugs, encompassing Tricaprin, Cinnamide, and Chloramphenicol palmitate (CAP-P). The binding energies, along with the energies associated with highest occupied molecular orbital, lowest unoccupied molecular orbital, and energy gap values have been assessed for various dendrimeric (PAMAM)-drug complexes and it was ascertained that it was energetically feasible for the drug moieties to bind with the dendrimeric system. Among the multitudinal model drugs examined, it was found that CAP-P exhibited the greatest binding energy (− 13.09 kcal/mol), and lowest energy gap values of 6.32 eV toward G2 dendrimer, thereby signifying that PAMAM G2 was a suitable vehicle to carry CAP-P drug possessing greater reactivity, with reduced system stability.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Instead of emphasizing the production of novel drugs, the current trend in the pharmaceutical industry has focused on the amelioration of various characteristics of drugs that are presently utilized in diverse therapies, consequently preventing the investment of additional funding, resources, and time in the R&D of new chemical essences [1]. Significant advancements in this domain are essential to mitigate the side effects associated with drugs. Various nanobearers like nanospheres [2, 3], CNTs [4], polymersomes [5,6,7], and micelles [8] tend to impart a solution to this concern through aimed and restrained discharge without altering the chemical framework of drugs [9,10,11,12,13,14,15,16,17,18,19]. Considering their unusual morphological characteristics and biocompatibility with other macromolecular entities, “dendrimers” are contemplated to be emerging flexible nanoscopic structures bestowing several merits over the conventional linear chain polymers [20, 21], thereby leading to an exquisite polymeric system for catalysis [22], disease diagnosis [23], gene [24,25,26], and drug delivery [27], 28.
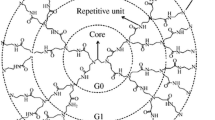
Dendrimers are spherical, three-dimensional, hyperbranched frameworks encompassing core, repetitive branches, and peripheral functional groups [21, 24, 27,28,29], as illustrated in Fig. 1. To improve the delivery of specific drugs, they possess hydrophobic or hydrophilic internal gaps and alterable peripheral molecules, thereby resulting in enhanced bioavailability and reduced cytotoxicity [30, 31]. There are two efficient means of forming dendrimer-drug complexes namely (1) the utilization of linkers via covalent interactions existing between the surface functional groups and (2) physical encapsulation of drug molecules within the interior hydrophobic gaps or periphery of the dendrimer using ionic, electrostatic interactions, hydrophobic or hydrogen bonding interactions [32, 33]. Poly(amidoamine) (PAMAM) was one of the predominantly investigated dendrimers for curtailing the impediments associated with drug delivery, as promulgated by Tomalia et al. [34, 35]. PAMAM dendrimer possesses some inherent advantages over other dendrimeric systems in the drug delivery domain inclusive of protecting normal cells from cytotoxic agents, lowering the dose-dependent side effects, and overcoming the drug resistance of infected cells [36].

Structure of a dendrimer
Novel perceptions of the dendrimer-drug complex are denoted by both theoretical and computational approaches, which could not be accomplished through employing experimentations alone [37,38,39,40,41]. Various analyses have been conducted on the transportation of several model drugs (and other organic molecules) with dendrimers which certainly proves the esoteric concern of research circles in this direction [42,43,44,45,46]. For the design and enhancement of dendrimer-based systems, computational studies play an essential role [39]. MD simulations were conducted by Goddard and colleagues to investigate the capsulation of Bengal Rose moieties within the Meijer dendrimer box constructed by the incorporation of tert-butyloxycarbonyl-l-Phe cap moieties to sixty-four preliminary amines of a fifth-generation Poly(propyleneimine) dendrimer [47]. MD simulations [48,49,50] of naphthyridine-based dendrimers to analyze binding energies were investigated by Posocco and co-workers using Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) [51]. A recent investigation was based on the calculation of interaction energies using the Metropolis Monte Carlo algorithm and semi-empirical quantum mechanical methods [52]. To find the mechanism behind the solubility of weak acid drugs in dendrimers, Lewis and Ganesan implemented a self-consistent field theory model [53].
Two main overtures of drug delivery schemes are (1) either to bind covalently to form dendrimer-drug conjugates or (2) to form complexes by interacting non-covalently. Even though it is not suitable if dendrimers discharge drugs too early before arriving at the aimed cells, drug molecules can be discharged more easily in dendrimer-drug complexes compared to conjugates. To achieve that goal, it is crucial to comprehend the underlying mechanisms governing interactions within the dendrimer-drug systems. In this regard, the present computational research emphasizes the analysis of the binding energy of these complexes via fully atomistic classical molecular dynamics (MD) simulations employing the PAMAM-drug complexes with three model drugs, namely, Tricaprin, Cinnamide, and Chloramphenicol palmitate (CAP-P). These three model drugs are used widely for different therapies (Table 1). In this investigation, we analyzed the binding strength of these drugs in the PAMAM dendrimer of different generations through the study of their binding energy (Eb) values. Furthermore, this research also accentuates the enumeration of the corresponding EHOMO (energy associated with highest occupied molecular orbital), ELUMO (energy accompanying the lowest unoccupied molecular orbital), and Eg (energy gap) values of various dendrimer-drug complexes.
Computational Analysis
Molecular Dynamics Simulation Procedure
To examine the binding strength of dendrimers, we created fully atomistic models of non-covalently bound drug-dendrimer complexes, designated as DN@PAMAM for the dendrimeric structures of different generations. DN represents drug molecules, where N = number of drug molecules that are incorporated within PAMAM dendrimer (Here N = 2, 3). Three types of model drug moieties were examined in the present research work namely, Tricaprin (TCAPIN10) (C66H124O12), Cinnamide (C36H36N4O4), and Chloramphenicol palmitate (CAP-P) (C108H168N8O24Cl8), encompassing G2 (C62H144N42O12) and G3 (C142H320N90O28) PAMAM-based dendrimers.
The binding of PAMAM with different drugs was examined by utilizing molecular dynamic simulation through Biovia Materials Studio Software (Fig. 2). The simulation method is composed of molecular mechanics and dynamics computations. The chemical structures of the drugs were imported from the Materials Studio Software library (Fig. 3). An ab initio force field, condensed phase optimized molecular potentials for atomistic simulation studies (COMPASS), was utilized for the computational simulation. Drugs were subjected to geometry optimization followed by an energy minimization step. Amorphous cells were constructed as per the relative densities of the chosen PAMAM dendrimer of specific generations. The dendrimeric macromolecule was then enclosed with the drug molecules through a layer builder. The layer formed was subjected to geometry optimization (5000 iterations) and dynamics (500 iterations) calculations, and the MD simulation was run.

Structure of A G2-PAMAM and B G3-PAMAM sketched using Biovia Materials Studio Software

Chemical structures of A Tricaprin, B Cinnamide, and C CAP-P imported from the library of Biovia Materials Studio software
We studied binding energy for the quantitative analysis of the binding strength of the dendrimer-drug complex. The binding strength of DN@PAMAM was enumerated using the following mathematical expression [Eq. (1)] [54]:
where Eb is the average binding energy associated with DN@PAMAM system; EP–D is the overall energy of DN@PAMAM complex constituting dendrimer and drug molecules; EP is the overall energy concomitant with PAMAM dendrimer, devoid of any drug molecules; and ED is the comprehensive energy of sequestered drug moiety, without dendrimer system.
The frontier molecular orbitals referred to as the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) exhibited a prominent role in optical, electrical characteristics, and quantum chemistry. HOMO refers to the outmost greater energy orbital encompassing electrons and therefore, it functions as an electron donor and exemplifies the propensity of the molecules against electrophilic attack. On the other hand, LOMO is associated with the lowest energy orbital serving as an electron acceptor, while characterizing susceptivity of molecules toward nucleophilic attack. The energy gap existing between the HOMO and LUMO orbitals assists in delineating kinetic stability and chemical reactivity of molecular systems [55]. In the present research work, Biovia Materials Studio Software was also utilized to enumerate the partial density of states and ascertain the involvement of PAMAM dendrimer and various drug molecules toward the HOMO–LUMO band gap energy changes (Eg), contingent with the following mathematical expression [Eq. (2)] [56]:
where \(E_{{{\text{HOMO}}}}\) is the energy associated with the highest occupied molecular orbital; \(E_{{{\text{LUMO}}}}\) is the energy inextricably linked to the lowest unoccupied molecular orbital; and \(E_{{\text{g}}}\) is the HOMO–LUMO energy gap value.
Results and Discussion
Binding energy (Eb), outlined in Eq. (1), was utilized to evaluate the binding strength of the DN-PAMAM system encompassing the interaction energies of drug molecules, PAMAM dendrimer, and dendrimer-drug moieties, with the aid of Forcite Modules employing Biovia Materials Studio software encompassing the COMPASS force field. Negative binding energy values were ascertained for the dendrimer-drug systems for both generations of dendrimers, signifying interactions between the drug molecules and dendrimer system in a manner stabilizing the complex. This can be attributed to the reduction in the overall system energy due to dendrimer-drug complex formation, indicating favorable interactions within the system thereby imparting stability to the generated system. Negative interaction energy values often associated with electronic and shape interactions of system indicate effectual molecular recognition, which is essential for the dendrimer to efficaciously encapsulate or deliver the drug.
Better binding is shown by the complex with a smaller Eb with a larger absolute value. The calculated Eb of the investigated model drug moieties reduces as per the following order: CAP-P (− 13.09 kcal/mol) > Tricaprin (− 3.46 kcal/mol) > Cinnamide (− 2.83 kcal/mol) in the D1@G2 complex and Cinnamide (− 5.68 kcal/mol) > Tricaprin (− 4.04 kcal/mol) > CAP-P (− 2.81 kcal/mol) in the D1@G3 complex (Table 2). We concluded that incorporating the drug molecules into the PAMAM dendrimeric system was energetically favorable owing to the attainment of negative binding energy values suggesting an interaction between the former and the latter for all the systems being investigated. Among the obtained binding energy values, CAP-P demonstrated maximum binding energy toward G2-PAMAM (Eb: − 13.09 kcal/mol), thereby substantiating that G2 dendrimer served as a good carrier for the drug moiety. Furthermore, it was noted that systems with more negative interaction energy values demonstrated stronger binding affinities within the dendrimer-drug system. This insinuates that the developed complexes possessed thermodynamic favourability, which is imperative in drug delivery systems where strong binding interactions can ameliorate the efficaciousness of selective drug molecules. It should be noted that a straightforward correspondence exists between the concepts of surface stability, and surface energy, manifesting that surfaces possessing lower overall surface energy values exhibit greater stability, and vice-versa [57]. In the present research work, CAP-P drug possessing the maximal binding energy value of − 13.09 kcal/mol toward G2-PAMAM dendrimer system was most stable, while the same drug molecule demonstrated the lowest binding energy value of − 2.83 kcal/mol toward G3-PAMAM dendrimer, thereby signifying lesser stability of the simulated drug-dendrimer complexes.
Furthermore, the corresponding ELUMO, EHOMO, and Eg values computed for the various dendrimer-drug complexes using the partial density of states theory are enlisted in Table 2. From the table, it was ascertained that the G2-PAMAM dendrimer system demonstrated the lowest energy gap (Eg) value of 6.32 eV toward CAP-P drug, with corresponding ELUMO and EHOMO values of − 2.35 and − 8.67 eV, respectively. On the other hand, the G3-PAMAM dendrimer manifested the greatest energy gap (Eg) value of 7.62 eV toward CAP-P drug molecules with corresponding ELUMO and EHOMO values of − 4.67 and − 12.29 eV, respectively. The test results obtained signify that the dendrimer-drug complex exhibiting lowest energy gap value possessed least stability with higher reactivity, compared to the systems possessing larger energy gap values [55]. This could be attributed to greater reactivity promoting facile electron transfer reactions that are crucial for interactions like binding of drug molecules with dendrimer systems. A lower energy gap value can result in robust binding interactions when the interactions between the dendrimer-drug systems are associated with transfer of electrons or substantial orbital overlap.
Conclusion
Dendrimers are considered to be a versatile nano-carrier for effective drug delivery due to their accurate molecular weight, polyvalent nature, biocompatibility, and greater water solubility. Both theoretical and computational studies are required for the investigation of these nano-carriers. This work is based on the molecular dynamics analysis of dendrimer-drug delivery complexes emphasizing the binding strength as a function of different types of drug molecules. The investigated dendrimer-drug delivery systems include G2 and G3 PAMAM dendrimers loaded with three model drugs: Tricaprin, Cinnamide, and CAP-P, respectively. Better binding is shown by the complex with a negative Eb (binding energy) with a larger absolute value as noticed from the Eg (energy gap) value of − 13.09 kcal/mol toward the G2-PAMAM dendrimer-CAP-P drug complex. Furthermore, this system also exhibited the lowest Eg value of 6.32 eV toward CAP-P drug signifying greater reactivity, with reduced stability of the dendrimer-drug system. Thus, it can be contemplated that incorporating drug molecules within PAMAM dendrimer was an energetically favorable phenomenon ascribable to the negative binding energy values obtained. Our research investigations manifest facile interaction of drug moieties with PAMAM dendrimer presuming that the design of PAMAM dendrimer is crucial to ameliorate its drug loading efficiency. The data deduced from this research work imparts valuable proof to evolve highly efficient dendrimer-drug delivery complexes.
Availability of Data and Materials
All data generated or analyzed during this study are included in the submitted manuscript and will be made available on request.
Code Availability
Not applicable.
References
V. Wagner, A. Dullaart, A.-K. Bock, A. Zweck, The emerging nanomedicine landscape. Nat. Biotechnol. 24, 1211–1217 (2006). https://doi.org/10.1038/nbt1006-1211
S.T. Bromley, E. Flikkema, New materials from fully coordinated SiO2 nanoclusters. Comput. Mater. Sci. 35, 382–386 (2006). https://doi.org/10.1016/j.commatsci.2004.08.018
A.D. Costache, L. Sheihet, K. Zaveri et al., Polymer−drug interactions in tyrosine-derived triblock copolymer nanospheres: a computational modeling approach. Mol. Pharm. 6, 1620–1627 (2009). https://doi.org/10.1021/mp900114w
B. Ghalandari, M. Monajjemi, F. Mollaamin, Theoretical investigation of carbon nanotube binding to DNA in view of drug delivery. J. Comput. Theor. Nanosci. 8, 1212–1219 (2011). https://doi.org/10.1166/jctn.2011.1801
D.E. Discher, V. Ortiz, G. Srinivas et al., Emerging applications of polymersomes in delivery: from molecular dynamics to shrinkage of tumors. Prog. Polym. Sci. 32, 838–857 (2007). https://doi.org/10.1016/j.progpolymsci.2007.05.011
S.M. Loverde, D.A. Pantano, D.A. Christian et al., Curvature, rigidity, and pattern formation in functional polymer micelles and vesicles—from dynamic visualization to molecular simulation. Curr. Opin. Solid State Mater. Sci. 15, 277–284 (2011). https://doi.org/10.1016/j.cossms.2011.06.003
Z. Pang, H. Gao, Y. Yu et al., Brain delivery and cellular internalization mechanisms for transferrin conjugated biodegradable polymersomes. Int. J. Pharm. 415, 284–292 (2011). https://doi.org/10.1016/j.ijpharm.2011.05.063
L. Zhao, F. Xu, H. Chen et al., Polyethylene glycol-polyethylenimine-tetrachloroplatinum (IV): a novel conjugate with good abilities of antitumor and gene delivery. J. Appl. Polym. Sci. 123, 1509–1517 (2012). https://doi.org/10.1002/app.34117
A.-Z. Chen, M.-Y. Chen, S.-B. Wang et al., Poly(L-histidine)-chitosan/alginate complex microcapsule as a novel drug delivery agent. J. Appl. Polym. Sci. 124, 3728–3736 (2012). https://doi.org/10.1002/app.35371
R. Yadav, K. Balasubramanian, Polyacrylonitrile/Syzygium aromaticum hierarchical hydrophilic nanocomposite as a carrier for antibacterial drug delivery systems. RSC Adv. 5, 3291–3298 (2015). https://doi.org/10.1039/C4RA12755B
V. Verma, K. Balasubramanian, Experimental and theoretical investigations of Lantana camara oil diffusion from polyacrylonitrile membrane for pulsatile drug delivery system. Mater. Sci. Eng. C 41, 292–300 (2014). https://doi.org/10.1016/j.msec.2014.04.061
R.S. Ambekar, B. Kandasubramanian, A polydopamine-based platform for anti-cancer drug delivery. Biomater. Sci. 7, 1776–1793 (2019). https://doi.org/10.1039/c8bm01642a
P.P. Mane, R.S.Ambekar, B. Kandasubramanian. Electrospun nanofiber-based cancer sensors: a review. Int. J. Pharmaceut. (2020). https://doi.org/10.1016/j.ijpharm.2020.119364
N. Mayilswamy, N. Jaya Prakash, B. Kandasubramanian, Design and fabrication of biodegradable electrospun nanofibers loaded with biocidal agents. Int. J. Polym. Mater. Polym. Biomater. (2022). https://doi.org/10.1080/00914037.2021.2021905
N. Palaniappan, J. Alphonsa, I.S. Cole, et al. Rapid investigation expiry drug green corrosion inhibitor on mild steel in NaCl medium. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. (2019). https://doi.org/10.1016/j.mseb.2019.114423
N. Jaya Prakash, D. Shanmugarajan, B. Kandasubramanian et al., Biodegradable silk-curcumin composite for sustained drug release and visual wound monitoring. Mater. Today Chem. 27, 101289 (2023). https://doi.org/10.1016/j.mtchem.2022.101289
R. Yadav, K. Balasubramanian. Bioabsorbable engineered nanobiomaterials for antibacterial therapy, in Engineering of Nanobiomaterials (Elsevier, 2016), pp. 77–117
V. More, S.V. Antanitta, R. Khonde, B. Kandasubramanian. Cellulose and derivatives serving as natural, versatile and biocompatible polymers in biomedical applications. Int. J. Polym. Mater. Polym. Biomater. (2024). https://doi.org/10.1080/00914037.2024.2376247
S.M. George, B. Kandasubramanian, Advancements in MXene-Polymer composites for various biomedical applications. Ceram. Int. 46, 8522–8535 (2020). https://doi.org/10.1016/j.ceramint.2019.12.257
S. Svenson, Dendrimers as versatile platform in drug delivery applications. Eur. J. Pharm. Biopharm. 71, 445–462 (2009). https://doi.org/10.1016/j.ejpb.2008.09.023
D.A. Tomalia, A.M. Naylor, W.A. Goddard, Starburst dendrimers: molecular-level control of size, shape, surface chemistry, topology, and flexibility from atoms to macroscopic matter. Angew. Chem. Int. Ed. Engl. 29, 138–175 (1990). https://doi.org/10.1002/anie.199001381
Y. Li, H. He, X. Jia et al., A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials 33, 3899–3908 (2012). https://doi.org/10.1016/j.biomaterials.2012.02.004
A. Patri, J. Kukowskalatallo, J. Bakerjr, Targeted drug delivery with dendrimers: comparison of the release kinetics of covalently conjugated drug and non-covalent drug inclusion complex. Adv. Drug Deliv. Rev. 57, 2203–2214 (2005). https://doi.org/10.1016/j.addr.2005.09.014
P.K. Maiti, T. Çaǧın, S.-T. Lin, W.A. Goddard, Effect of solvent and pH on the structure of PAMAM dendrimers. Macromolecules 38, 979–991 (2005). https://doi.org/10.1021/ma049168l
R.K. Tekade, P.V. Kumar, N.K. Jain, Dendrimers in oncology: an expanding horizon. Chem. Rev. 109, 49–87 (2009). https://doi.org/10.1021/cr068212n
M.F. Neerman, H.-T. Chen, A.R. Parrish, E.E. Simanek, Reduction of drug toxicity using dendrimers based on melamine. Mol. Pharm. 1, 390–393 (2004). https://doi.org/10.1021/mp049957p
D.A. Tomalia, Birth of a new macromolecular architecture: dendrimers as quantized building blocks for nanoscale synthetic polymer chemistry. Prog. Polym. Sci. 30, 294–324 (2005). https://doi.org/10.1016/j.progpolymsci.2005.01.007
P.K. Maiti, T. Çaǧın, G. Wang, W.A. Goddard, Structure of PAMAM dendrimers: generations 1 through 11. Macromolecules 37, 6236–6254 (2004). https://doi.org/10.1021/ma035629b
R.S. Ambekar, M. Choudhary, B. Kandasubramanian, Recent advances in dendrimer-based nanoplatform for cancer treatment: a review. Eur. Polym. J. 126, 109546 (2020). https://doi.org/10.1016/j.eurpolymj.2020.109546
M.A. Mintzer, E.L. Dane, G.A. O’Toole, M.W. Grinstaff, Exploiting dendrimer multivalency to combat emerging and re-emerging infectious diseases. Mol. Pharm. 9, 342–354 (2012). https://doi.org/10.1021/mp2005033
C. Giovino, I. Ayensu, J. Tetteh, J.S. Boateng, Development and characterisation of chitosan films impregnated with insulin loaded PEG-b-PLA nanoparticles (NPs): a potential approach for buccal delivery of macromolecules. Int. J. Pharm. 428, 143–151 (2012). https://doi.org/10.1016/j.ijpharm.2012.02.035
A. Demanuele, D. Attwood, Dendrimer–drug interactions. Adv. Drug Deliv. Rev. 57, 2147–2162 (2005). https://doi.org/10.1016/j.addr.2005.09.012
U. Boas, P.M.H. Heegaard, Dendrimers in drug research. Chem. Soc. Rev. 33, 43 (2004). https://doi.org/10.1039/b309043b
D.A. Tomalia, H. Baker, J. Dewald et al., A new class of polymers: starburst-dendritic macromolecules. Polym. J. 17, 117–132 (1985). https://doi.org/10.1295/polymj.17.117
N.K. Jain, U. Gupta, Application of dendrimer–drug complexation in the enhancement of drug solubility and bioavailability. Expert Opin. Drug Metab. Toxicol. 4, 1035–1052 (2008). https://doi.org/10.1517/17425255.4.8.1035
R. Esfand, D.A. Tomalia, Poly(amidoamine) (PAMAM) dendrimers: from biomimicry to drug delivery and biomedical applications. Drug Discov. Today 6, 427–436 (2001). https://doi.org/10.1016/S1359-6446(01)01757-3
Y. Li, T. Hou, Computational simulation of drug delivery at molecular level. Curr. Med. Chem. 17, 4482–4491 (2010). https://doi.org/10.2174/092986710794182935
S.H. Kim, M.H. Lamm, Multiscale modeling for host-guest chemistry of dendrimers in solution. Polymers 4, 463–485 (2012). https://doi.org/10.3390/polym4010463
W. Tian, Y. Ma, Theoretical and computational studies of dendrimers as delivery vectors. Chem. Soc. Rev. 42, 705–727 (2013). https://doi.org/10.1039/C2CS35306G
A.-M. Caminade, S. Fruchon, C.-O. Turrin et al., The key role of the scaffold on the efficiency of dendrimer nanodrugs. Nat. Commun. 6, 7722 (2015). https://doi.org/10.1038/ncomms8722
S. Kanchi, G. Suresh, U.D. Priyakumar et al., Molecular dynamics study of the structure, flexibility, and hydrophilicity of PETIM dendrimers: a comparison with PAMAM dendrimers. J. Phys. Chem. B 119, 12990–13001 (2015). https://doi.org/10.1021/acs.jpcb.5b07124
D. Kannaiyan, T. Imae, pH-dependent encapsulation of pyrene in PPI-Core:PAMAM-shell dendrimers. Langmuir 25, 5282–5285 (2009). https://doi.org/10.1021/la8039847
E. Boisselier, L. Liang, M. Dalko-Csiba et al., Interactions and encapsulation of vitamins C, B 3, and B 6 with dendrimers in water. Chem. Eur. J. 16, 6056–6068 (2010). https://doi.org/10.1002/chem.200902995
Y. Cheng, Z. Shao et al., Comparison of generation 3 polyamidoamine dendrimer and generation 4 polypropylenimine dendrimer on drug loading, complex structure, release behavior, and cytotoxicity. Int. J. Nanomed. (2011). https://doi.org/10.2147/IJN.S27028
Y. Haba, A. Harada, T. Takagishi, K. Kono, Rendering poly(amidoamine) or poly(propylenimine) dendrimers temperature sensitive. J. Am. Chem. Soc. 126, 12760–12761 (2004). https://doi.org/10.1021/ja047755g
D.L. Richter-Egger, A. Tesfai, S.A. Tucker, Spectroscopic investigations of poly(propyleneimine)dendrimers using the solvatochromic probe phenol blue and comparisons to poly(amidoamine) dendrimers. Anal. Chem. 73, 5743–5751 (2001). https://doi.org/10.1021/ac0155355
P. Miklis, T. Çaǧin, W.A. Goddard, Dynamics of bengal rose encapsulated in the meijer dendrimer box. J. Am. Chem. Soc. 119, 7458–7462 (1997). https://doi.org/10.1021/ja964230i
P. Govindaraj, B. Kandasubramanian, K.M. Kodam, Molecular interactions and antimicrobial activity of curcumin (Curcuma longa) loaded polyacrylonitrile films. Mater. Chem. Phys. 147, 934–941 (2014). https://doi.org/10.1016/j.matchemphys.2014.06.040
A. Kore, N. Mayilswamy, B. Kandasubramanian (2023) Molecular dynamics study of polymeric drug systems, Stavropol, Russia, p. 100032
R. Magisetty, A. Shukla, B. Kandasubramanian, Molecular dynamic simulation constructed interaction parameter investigation between poly(1,6-heptadiyne) and NiFe2O4 in nanocomposite. Mater. Today: Proc. 24, 1720–1728 (2020). https://doi.org/10.1016/j.matpr.2020.03.595
P. Posocco, M. Ferrone, M. Fermeglia, S. Pricl, Binding at the core. Computational study of structural and ligand binding properties of naphthyridine-based dendrimers. Macromolecules 40, 2257–2266 (2007). https://doi.org/10.1021/ma062610a
F. Avila-Salas, C. Sandoval, J. Caballero et al., Study of interaction energies between the PAMAM dendrimer and nonsteroidal anti-inflammatory drug using a distributed computational strategy and experimental analysis by ESI-MS/MS. J. Phys. Chem. B 116, 2031–2039 (2012). https://doi.org/10.1021/jp2069122
T. Lewis, V. Ganesan, Mean-field modeling of the encapsulation of weakly acidic molecules in polyelectrolyte dendrimers. J. Phys. Chem. B 116, 8269–8281 (2012). https://doi.org/10.1021/jp3033066
M. Masoumi, M. Jahanshahi, M.G. Ahangari, G.N. Darzi, Electronic, mechanical and thermal properties of SiO2 nanotube interacting with poly lactic-co-glycolic acid: density functional theory and molecular dynamics studies. Appl. Surf. Sci. 546, 148894 (2021). https://doi.org/10.1016/j.apsusc.2020.148894
A. Eşme, S.G. Sağdınç, Spectroscopic (FT–IR, FT–Raman, UV–Vis) analysis, conformational, HOMO-LUMO, NBO and NLO calculations on monomeric and dimeric structures of 4-pyridazinecarboxylic acid by HF and DFT methods. J. Mol. Struct. 1147, 322–334 (2017). https://doi.org/10.1016/j.molstruc.2017.06.110
R. Al-Shdefat, M.M. Kadhim, A.B. Mahdi et al., Theoretical evaluation of poly(amidoamine) dendrimers with different peripheral groups as a purinethol drug delivery system in aqueous medium. Colloids Surf. B 216, 112534 (2022). https://doi.org/10.1016/j.colsurfb.2022.112534
N.M. Mahani, A molecular dynamics study on polycaprolactone-metal oxide interactions. Mat Res 23, e20200188 (2020). https://doi.org/10.1590/1980-5373-mr-2020-0188
Acknowledgements
The authors acknowledge Dr. BHVS Narayana Murthy, Vice Chancellor of Defense Institute of Advanced Technology, Pune and Mr. K A Rajesh, Joint-Director & HoD, Central Institute of Petrochemical Engineering and Technology, Kochi, for their valuable support. The first author acknowledges Miss. Shruti Gupta and Miss. Payal Varma for their unwavering technical support throughout the writing.
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
Lisha V S contributed to Material preparation, data collection analysis, and writing of the article. Neelaambhigai Mayilswamy contributed to material preparation and writing. Balasubramanian Kandasubramanian made a substantial contribution to conceptualization and discussion and reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare no conflict of interest.
Consent to Participate
Not applicable.
Consent for Publication
The authors consent to publish the article on acceptance.
Human and Animals Rights Declaration
The submitted article complies with the ethical guidelines of the journal and does not contain the results of studies involving human and/or animals.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lisha, V.S., Mayilswamy, N. & Kandasubramanian, B. Molecular Dynamics Simulation of PAMAM Dendrimer-Drug Delivery Systems. Biomedical Materials & Devices (2024). https://doi.org/10.1007/s44174-024-00229-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44174-024-00229-6