
Abstract
Fungal communities are associated with healthy peanut crops and good crop production, through the regulation of pod rot disease. Rotted peanut pods and their surrounding soil samples were collected from locations in northern China. Fungal species were identified by next-generation sequencing, using the conserved sequences of their internal transcribed spacer regions. Results showed that rotted pod samples were rich in the phyla Ascomycota and Basidiomycota, and soil samples also contained these, plus Chytridiomycota and Zygomycota. There were regional variations in the species of fungi related to peanut pod rot and its surrounding soil, between locations. Fungal species of Cryptococcus and Fusarium were less abundant in soil samples than in rotted pod samples, and were the main pathogenic fungi identified in our study. Soil total carbon, nitrogen, and potassium had a strong influence on the fungal community, and total phosphorous and calcium ions, together with soil pH, had a modest influence. Only Mycosphaerella and Gibberella were not significantly affected by these factors. These findings may be of some help to control pod rot disease and reduce the production loss of peanut crops.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The peanut (Arachis hypogaea) is originally from South America but is now an important oil and food crop in many countries, especially in China, which contributes more than a quarter of global production [1]. Peanut pod rot, which leads to kernel damage, has become an important fungal disease worldwide, affecting the production and quality of peanuts. It is difficult, above the ground, to determine whether the peanut is affected by pod rot disease, and the specific fungi which cause the disease are difficult to distinguish [2]. Therefore, it is difficult to control the disease using traditional approaches such as chemical or cultivation methods [3].
Several species of fungi, and calcium ions (Ca2+), have been verified as important to the rotting of peanut pods and in the surrounding soils [4]. The uptake of Ca2+ by peanut pods is essential for the development of cell walls and seeds [5]. In field surveys, Pythium myriotylum, Rhizoctonia solani, and Fusarium solani have been frequently isolated from rotted peanut pods [6]. The primary cause of peanut pod rot in different districts also differs; P. myriotylum was the main cause of peanut pod rot in Cosigüina, Nicaragua, and F. solani in Egypt [7].
It is clear that changes in chemical, environmental factors regulate the microbial community via the ecosystem [8]. Soil quality, together with the soil-based microbial community, are constrained by pH, which can become the main factor controlling soil productivity and sustainability [9]. The microbial community plays an important role in mediating organic cycles in the soil and, in return, the limiting nutrients, nitrogen, phosphorus, and potassium, also regulate the microbial community [10]. How the major nutrient elements and the pH of soil act to modify the microbial community remains unclear.
A peanut production survey conducted in China showed several instances of peanut pod rot in Hebei Province, which led to production losses [11]. In order to reduce production loss, experimental trials are needed to provide theoretical support. Hence, our objective here is (1) to investigate the composition and structure of the fungal community causing peanut pod rot in northern China; (2) to compare the differences in fungal community between rotted pods and their surrounding soils; (3) to identify the main pathogenic fungal species causing peanut pod rot, and (4) to explore the correlations between soil chemical properties and the pathogenic fungal community.
Materials and Methods
Materials
Peanut (Arachis hypogaea L.) material Jihua 5 was the control variety of peanut used in the regional test in Hebei province. It has multi-resistance and wide adaptation, and is derived from a cross between Pu93-11 and Zheng86036-26-1. It was obtained from the Hebei Academy of Agriculture and Forestry Sciences. Conventional field planting was used, with protected areas around the study regions, with normal cultivation management, intertillage weeding, and no fungicide application.
Sampling
Five samples of rotted peanut pods and corresponding soil samples were collected, at the later stages of peanut growth, from four locations in the old Yellow River drainage area and the Sha River drainage area, including Baoding (BD), Tangshan (TS), Xinle (XL), and Handan (HD). Soil was classified as yellow loam in BD, TS, HD, and sandy loam in XL, with medium fertility, flat terrain, and good water retention. The affected pods and corresponding soil samples (10–15 cm deep around the infected peanut plant) were taken by a five-point method in each region, and repeated five times. All samples were stored in liquid nitrogen for transfer to the laboratory. The affected pods were cleaned with sterilized water, dried, and stored at −70 °C. Soil samples were forced through a 2 mm sieve, and then mixed thoroughly. Each soil sample was divided into two parts, one of which was used for high-throughput sequencing and stored at −70 °C. The other part was used for the determination of physical and chemical properties. The pod samples were labeled as BP, TP, XP, and HP, and the corresponding soil samples were marked as BS, TS, XS, and HS.
Sequencing of Fungal ITS Regions
Next-generation sequencing library preparation and Illumina MiSeq sequencing, together with DNA extraction using the CTAB method, were conducted at GENEWIZ, Inc. (Suzhou, China). DNA samples were quantified using a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). DNA (50–100 ng) was used to generate amplicons, using a panel of primers designed by GENEWIZ (GENEWIZ, Inc., South Plainfield, NJ, USA). Oligonucleotide primers were designed to anneal to the relatively conserved sequences spanning the internal transcribed spacer (ITS) regions of fungi. The ITS2 region was amplified using forward primer ITS1 (5′-GTGAATCATCGARTC-3′) and a reverse primer ITS4 (5′-TCCTCCGCTTATTGAT-3′), which were used as the universal fungus-specific primers [12]. In addition to ITS target-specific sequences, the primers ITS1 and ITS4 also contained adapter sequences, allowing uniform amplification of the library with high complexity, ready for downstream NGS sequencing on the Illumina Miseq platform. DNA libraries were validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and quantified using a Qubit 2.0 Fluorometer. DNA libraries were multiplexed and loaded on an Illumina MiSeq instrument according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was performed using a 2×300/250 paired-end (PE) configuration. Image analysis and base calling were conducted by the MiSeq Control Software (MCS) embedded in the MiSeq instrument.
Environmental Parameter Measurements
Total carbon, nitrogen, phosphorous, potassium, and calcium ions, together with soil pH in sampling locations, were measured according to the national standard of P.R.C. Total carbon was determined by potassium dichromate oxidation-outer heating method [13], total nitrogen by the automatic kjeldahl apparatus method [14], total phosphorus by molybdenum-antimony anticolorimetry method [15], and total potassium by extraction-flame photometer method [16]. Exchangeable calcium was measured by the volumetric method [17] and pH value by the acidimeter method [18].
Data Analysis
The QIIME data analysis package was used for statistical analysis of operational taxonomic units (OTUs) at 97% sequence identity [19]. The RDP classifier (Ribosomal database program) was used to assign a taxonomic category to all OTUs at a confidence threshold of 0.8 [20]. Quality filtering of joined sequences was performed, and sequences that did not fulfill the following criteria were discarded: sequence length < 200 bp, no ambiguous bases, and mean quality score ≥ 20. The UCHIME algorithm was used to classify the sequences, based on the RDP Gold database [20], and then the chimeric sequences were removed. Sequences were rarefied prior to calculation of alpha and beta diversity statistics [21]. A beta diversity distance-based, nonmetric, multidimensional scaling (NMDS) method was used to analyze and classify all samples. Cluster analysis was conducted using UPGMA (Unweighted pair group method with arithmetic mean) based on the Bray–Curtis similarity distance. Spearman correlation coefficients and P values were calculated, from a test of significance between species abundance and environmental factors. Correlation heatmaps were then drawn, according to the results, to visually show the relationship between environmental factors and community composition. An ordination analysis, redundancy analysis (RDA), was conducted to characterize the relationship between environmental factors and the microbial community. The symbols represent the location, and the arrow lines with capital letters represent environmental factors. R version 3.5.3 with the Vegan package was used to process the data and draw figures.
Results
Overview of Sequence Assignment
A total of 4,966,440 sequence reads and 1,488,951,007 bp were obtained from 40 samples, with an average Q30 of 78.54, GC of 58.36, and 0.18 N (ppm). Each sample had 56,667 effective reads, accounting for 91.39%, with an average sequence length of 324.37. The reliability of our outputs was tested at a similarity level of 97%, using rarefaction curves to predict the maximum OTU numbers. As sequencing depth increased, the number of OTUs increased. When the curve became flat, the number of observed OTUs did not rise with an increase in the amount of data extracted. The rarefaction curve of each sample reached saturation, which shows that sufficient sequencing outputs, for the rotted pod and soil samples, were used in this work (Figure S1).
Fungal Community Composition
Fungal community compositions at different taxonomic levels for rotted peanut pods and their surrounding soil samples were counted and summarized. At the phylum level, the number of fungal community taxons extracted from the rotted peanut pods and its surrounding soils were the same. The soil samples exhibited greater diversity than the pod samples at the levels of class, order, family, genus, and species. The number of fungal species extracted from pods ranged from 34 to 53, and from soils ranged from 127 to 179 (Table 1).
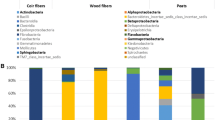
Most of the fungal community derived from rotted pod samples belonged to the phyla Ascomycota and Basidiomycota, and the soil samples contained both of these plus Chytridiomycota and Zygomycota. Sixteen fungal classes were observed. Sordariomycetes, Eurotiomycetes, Dothideomycetes, Agaricomycetes, Tremellomycetes, and Leotiomycetes were common in both rotted pods and soil samples, and the remaining ten classes, Orbiliomycetes, Chytridiomycetes, Glomeromycetes, Saccharomycetes, Pezizomycetes, Ustilaginomycetes, Microbotryomycetes, Pucciniomycetes, Exobasidiomycetes, and Agaricostilbomycetes were mainly found in soil samples (Fig. 1). A relative abundance heatmap, representing the composition of the fungal community, showed a difference in species composition (Figure S2). Almost all fungal species observed in soil samples were more abundant than those of rotted pod samples, except for Fusarium and Cryptococcus.

Relative abundances of top 30 of biological species at the phylum level. The abscissa is the name of the pod and soil sample. The ordinate is the relative abundance of different species (relative abundance). The bottom-up color in the bar chart corresponds to the species name of the following legend. Other presented the taxonomic name except for the first 30 species of relative abundance
Fungal Community Diversity
The microbial taxonomic diversity of the rotted pod samples was distinct from that of the soil samples (Table 2). Good coverage was evidenced by the same value of observed species index in both soil and pod samples. All soil samples had a higher Ace index and Chao1 index than their corresponding rotted peanut pods. The soil samples had more calculable OTUs than rotted pod samples, and the community richness of soil samples was higher than that of rotted peanut pod samples. Each soil sample had a higher Shannon index than its corresponding pod samples, and the community diversity of soil samples was higher than that of rotted pod samples. The Simpson index identified that the soil samples had a smaller ecological dominance and a higher evenness than rotted pod samples. The rank abundance curves of all soil and rotted pod samples were used to visualize relative abundance, and soil samples exhibited lower species evenness and higher diversity and richness than rotted pod samples (Figure S3).
The similarity of fungal community composition was clarified by Euclidean similarity distance-based cluster analysis and Bray–Curtis similarity distance-based nonmetric multidimensional scaling (Fig. 2). When the stress was less than 0.2, NMDS accurately reflected the differentiation of samples. An NMDS stress value lower than 0.104 indicated excellent ordination representation. The rotted pod samples from XL showed a bigger difference between samples, and we suggest that sample BD3 might be contaminated. The fungal community extracted from soil samples only differed between locations, not within a single location. However, the fungal community extracted from rotted pod samples differed between samples in the same location, and between locations. The fungal diversity of rotted peanut pods was affected by the fungal community of its corresponding soil, but was not determined by it.

NMDS (a) and clustering (b) analysis of fungal communities. a Each point represents a sample, and the distance between points represents the degree of difference. Samples from the same group were marked in the same color. b Each branch represents a sample, and different color branches represent different groups
Fungal Community Difference
Similarities were analyzed to verify the overall similarity and significance. The difference between groups was extremely significant and was greater than that within groups (Table 3). Based on the community abundance data of different groups, strict statistical methods were used to detect the differences in abundance in their fungal communities. Multiple hypothesis tests of rare frequency data and false discovery rate analysis can be used to assess the significance of the observed differences. This analysis was performed at the genus level, and the abundance of five species with the largest difference was plotted (Fig. 3). Fusarium had the most obvious difference in relative abundance among the classified genera in groups BP-vs-BS and HP-vs-HS. The “unclassified genus” had the largest and most obvious difference in relative abundance in group TP-vs-TS. In group XP-vs-XS, Alternaria, Fusarium, Geomyces, Mortierella, and Volutella were the 5 genera with the largest difference.

Analysis of 5 species composition with significant differences between groups. Names of 5 species are on the abscissa and of that the richness are on the ordinate
Environmental Response
Environmental factors (Table S1) could directly reflect the soil fertility, and have significant impact on fungal community structures. Correlations between the fungal community and nutrient elements were displayed using a heatmap (Fig. 4). Only Mycosphaerella and Gibberella were not significantly affected by these nutrient factors. Correspondence analysis shows the correlation between the micro-ecological environment and the fungal community, using straight lines with arrows (Fig. 5). TK, TOC, and TN had the strongest influence on the fungal community. HD samples were sensitive to pH and TP. Nevertheless, samples from XL and TS were Ca2+ sensitive. The correlation between environmental factors and samples from BD differed from that of the other three locations.

Correlation heatmap between species and environmental factors. The environmental factors are marked horizontally at the bottom and species are marked vertically at the right. The values corresponding to each square in the heatmap show the relationship between species and environmental factors. Spearman correlation coefficient r > 0 is positively correlated, and r<0 is negatively correlated. *** Significance test P value < 0.001, ** 0.01 ≥ P ≥ 0.001, * 0.05 ≥ P ≥ 0.01. A P value greater than 0.05 is marked in color white and results with statistical significance are highlighted

Correlations between environmental factors and fungal community. Different colors and shapes represent sample groups in different environments. Arrows represent environmental factors, and angles between environmental factors represent positive and negative correlations between environmental factors and species. Relationships (acute angle: positive correlation; obtuse angle: negative correlation; right angle: no correlation)
Discussion
Previous studies have shown that the peanut pod rot was soil-borne pathogens mixed infection [22, 23] and it is difficult to be completely controlled with chemicals, cultivation methods or even biological techniques [24]. The composition and diversity of soil microbial community structure have a significant impact on plant soil-borne diseases [25,26,27]. Analysis of peanut pod and surrounding soil from the fungal community level may lay a foundation to study the dominant factors and the process of peanut pod rot disease. In this study, the genetic diversity of the fungal communities, extracted from rotted peanut pods and their surrounding soil samples, was measured via next-generation sequencing, and ample information was obtained. The difference in fungal species between rotted peanut pods and corresponding soils was clear. Furthermore, the effects on the fungal community of physicochemical parameters of the soil were subjected to orientation analysis.
Hanlin [28] identified 70 genera of fungi related to peanut shells and seeds in the Southeastern United States, while Wang et al.[29] identified 272 genera in Shandong Province, China. In the present study, the most abundantly identified fungi, relating to peanut pods, were 126 genera in Hebei Province, China. We assume that the fungal species related to peanut pod rot are location-sensitive and vary from place to place. However, the fungal gene database is far from perfect and consequently the “unclassified sequencing reads” which comprise a large proportion of species, might significantly alter the conclusion if the information was available to identify them.
Ascomycota and Basidiomycota dominated our analysis at the phylum level. As the dominant fungi, Ascomycota were frequently observed in peanut roots [30] and were identified as endophytic, which were ubiquitous fungi that inhabit plant tissues, harmlessly [31]. Basidiomycota were also frequently observed in surrounding soils, at high abundance [32]. Zygomycota were found in soil and Sphagnum fuscum plants, at a relatively high abundance [33]. Chytridiomycota were absent from plant tissues but were found at a low level in soil [34]. Our study showed that rotted pod samples contained Ascomycota and Basidiomycota, and the soil samples contained Ascomycota, Basidiomycota, Chytridiomycota, and Zygomycota. This is consistent with the results of previous studies. Overall, the majority of fungal species occur in the soil, through most of their life cycles, some of which are difficult to identify separately [35]. The composition and structure of the field crop drives the ecological succession of the microbial community, and soil fungi are soil context-dependent [36]. Differences in the fungal community, between locations, provided a substantial expression of this spatial heterogeneity. The soil ecosystem is the primary source of the microbial community, but fungal communities are also regulated by the physiological processes in peanuts [37]. The fungal diversity of rotted peanut pods was affected by its corresponding soil community, but was not determined by it.
Fusarium has been found to significantly alter the fungal community of the peanut root system, its absence coincides with a decline in Fusarium-induced rot and the improvement of crop growth [38]. Furthermore, Fusarium is also proven to be correlated with peanut leaf wilt, as a pathogenic fungus [39], it is unclear whether it can also be derived from other plant tissues. Cryptococcus is considered a plant-beneficial fungus, also related to pathogen accumulation [40]. This might explain the relatively less abundance of Cryptococcus and Fusarium observed in soil samples, compared to rotted pod samples.
The basic components of soil fungi environment, including soil pH, organic carbon, nitrogen, and so on, have a certain impact on the composition of soil community. Ectomycorrhizal fungi associated with plant roots are related to organic carbon cycling, consistently increasing soil carbon storage and slowing down carbon cycling by competition for soil nitrogen [41]. Soil carbon content and geographic location determine the composition of the fungal community [42], and fungi species are also related to turnover time and mineral decomposition, which are critical to soil carbon and nitrogen cycling. Soil carbon, nitrogen, and C/N ratio are changed by the composition and structure of the soil fungal community [43].
Trials investigating the connection between potassium and fungal disease revealed that the nutrient element potassium dramatically stimulates epidemics caused by fungi [44]. Such phenomena could help us to understand the influence of soil potassium on the fungal community. Soil phosphorus management also influences the ecological progression of soil microorganisms, without greatly affecting the fungal community of organic plant tissues [45]. The activity of the soil microbial community is affected by the addition of CaCO3, but this is calcium ion-related and not significant [46]. Soil pH is considered to be the primary driver of the bacterial soil community rather than the fungal community [47]. In our study, soil potassium, carbon content, and nitrogen had the strongest influence on the fungal community, and the influences of soil pH, calcium ions, and phosphorus on the fungal community were modest. Interestingly, the correlations between environmental factors and samples differed from locations. The reason for these results might be that soil features were different between different regions, even in the same plots of different locations. The inadequacies of this study is that only limited samples were used to study the diversity of fungi in rhizosphere soil at a single time point, and the statistical analysis of data may be biased. In the further research, it is necessary to track and monitor the diversity and composition of fungal community in different stages of pod rot and in different locations, which will be able to help guide researcher better analyze the law of compound infection and grasp the changes of pathogen species in time.
Change history
14 August 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00284-021-02622-6
References
Fabra A, Castro S, Taurian T, Angelini J, Ibañez F, Dardanelli M, Tonelli M, Bianucci E, Valetti L (2010) Interaction among Arachis hypogaea L. (peanut) and beneficial soil microorganisms: how much is it known? Crit Rev Microbiol 36(3):179–194. https://doi.org/10.3109/10408410903584863
Ziedan E (2006) Manipulating endophytic bacteria for biological control to soil borne diseases of peanut. J Appl Sci Res 2(8):497–502
Wheeler TA, Russell SA, Anderson MG, Serrato-Diaz LM, French-Monar RD, Woodward JE (2016) Management of peanut pod rot I: disease dynamics and sampling. Crop Prot 79:135–142. https://doi.org/10.1016/j.cropro.2015.10.010
Csinos A, Gaines T, Walker M (1984) Involvement of nutrition and fungi in the peanut pod rot complex. Plant Dis 68(1):61–65. https://doi.org/10.1094/PD-68-61
Zharare G, Asher C, Blamey F (2010) Magnesium antagonizes pod-zone calcium and zinc uptake by developing peanut pods. J Plant Nutr 34(1):1–11. https://doi.org/10.1080/01904167.2011.531354
Augusto J, Brenneman TB, Csinos AS (2010) Etiology of peanut pod rot in Nicaragua: II. The role of Pythium myriotylum as defined by applications of gypsum and fungicides. Plant Health Prog 11(1):27. https://doi.org/10.1094/PHP-2010-0215-02-RS
Abdalla ME, Abdel-Fattah GM (2000) Influence of the endomycorrhizal fungus Glomus mosseae on the development of peanut pod rot disease in Egypt. Mycorrhiza 10(1):29–35. https://doi.org/10.1007/s005720050284
Comte J, Fauteux L, del Giorgio PA (2013) Links between metabolic plasticity and functional redundancy in freshwater bacterioplankton communities. Front Microbiol 4:112. https://doi.org/10.3389/fmicb.2013.00112
Muñoz K, Buchmann C, Meyer M, Schmidt-Heydt M, Steinmetz Z, Diehl D, Thiele-Bruhn S, Schaumann G (2017) Physicochemical and microbial soil quality indicators as affected by the agricultural management system in strawberry cultivation using straw or black polyethylene mulching. Appl Soil Ecol 113:36–44. https://doi.org/10.1016/j.apsoil.2017.01.014
Camenzind T, Hättenschwiler S, Treseder KK, Lehmann A, Rillig MC (2018) Nutrient limitation of soil microbial processes in tropical forests. Ecol Monogr 88(1):4–21. https://doi.org/10.1002/ecm.1279
Zhang S, Liu G, Janssen T, Zhang S, Xiao S, Li S, Couvreur M, Bert W (2014) A new stem nematode associated with peanut pod rot in China: morphological and molecular characterization of Ditylenchus arachis n. sp. (Nematoda: Anguinidae). Plant Pathol 63(5):1193–1206. https://doi.org/10.1111/ppa.12183
Schoch C, Seifert K, Huhndorf S, Robert V, Schindel D (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci 109(16):6241–6246. https://doi.org/10.1073/pnas.1117018109
NY/T 85-1988. Method for determination of soil organic matter. Ministry of Agriculture of the PRC. http://www.eshian.com/sat/standard/standardinfodown/7872
NY/T 1121.24-2012. Determination of total nitrogen in soil-automatic kjeldahl apparatus methodMinistry of Agriculture of the PRC. https://www.nongxiyiqi.com/static/resource/20200413/zoXI8Cs80j0NRe2nrZKAZuuIxnkW6OmtlyDykpzA.pdf
GB 9837-1988. Method for determination of soil total phosphorus. Ministry of Agriculture of the PRC. http://file4.foodmate.net/foodvip/biaozhun/2017/zengbu/GB9837-1988.pdf
NY/T 87-1988. Method for determination of total potassium in soil. Ministry of Agriculture of the PRC.https://www.nongyeyq.com/static/upload/file/20200702/1593654898385552.pdf
NY/T 1121.13-2006. Method for determination of total potassium soil exchangeable calcium and magnesium. Ministry of Agriculture of the PRC. http://file4.foodmate.net/foodvip/biaozhun/2017/zengbu/NYT1121.13-2006.pdf
NY/T 1377-2007. Determination of pH in soil. Ministry of Agriculture of the PRC. http://file4.foodmate.net/foodvip/biaozhun/2017/zengbu/NYT1377-2007.pdf
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194. https://doi.org/10.1093/bioinformatics/btr381
Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtssonpalme J, Callaghan TM (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22(21):5271–5277. https://doi.org/10.1111/mec.12481
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75(23):7537. https://doi.org/10.1128/AEM.01541-09
Wheeler TA, Howell CR, Cotton J, Porter D (2005) Pythium species associated with pod rot on west Texas peanuts and in vitro sensitivity of isolates to mefenoxam and azoxystrobin. Peanut Sci 32(1):9–13. https://doi.org/10.3146/0095-3679(2005)32[9:PSAWPR]2.0.CO;2
Sun WM, Feng LN, Guo W, Liu DQ, Meng QF (2012) First report of Neocosmospora striata causing peanut pod rot in China. Plant Dis 96(1):146–146. https://doi.org/10.1094/PDIS-06-11-0461
Besler BA, Grichar WJ, Brewer KD, Baring MR (2003) Assessment of six peanut cultivars for control of Rhizoctonia pod rot when sprayed with Azoxystrobin or Tebuconazole 1. Peanut Sci 30(1):49–52. https://doi.org/10.3146/pnut.30.1.0010
Bakker MG, Glover JD, Mai JG, Kinkel LL (2010) Plant community effects on the diversity and pathogen suppressive activity of soil streptomycetes. Appl Soil Ecol 46(1):35–42. https://doi.org/10.1016/j.apsoil.2010.06.003
Bulluck LR (2002) Organic and synthetic fertility amendments influence soil microbial physical and chemical properties on organic and conventional farms. Appl Soil Ecol 19(2):147–160. https://doi.org/10.1016/s0929-1393(01)00187-1
Gamliel A, Austerweil M, Kritzman G (2000) Non-chemical approach to soilborne pest management-organic amendments. Crop Prot 19(8–10):847–853. https://doi.org/10.1016/s0261-2194(00)00112-5
Hanlin RT (1973) The distribution of peanut fungi in the Southeastern United States. Mycopathol Mycol Appl 49(4):227–241. https://doi.org/10.1007/BF02050716
Wang M, Chen M, Yang Z, Chen N, Chi X, Pan L, Wang T, Yu S, Guo X (2017) Influence of peanut cultivars and environmental conditions on the diversity and community composition of pod rot soil fungi in China. Mycobiology 45(4):392–400. https://doi.org/10.5941/myco.2017.45.4.392
Xu L, Ravnskov S, Larsen J, Nicolaisen M (2012) Linking fungal communities in roots, rhizosphere, and soil to the health status of Pisum sativum. Fems Microbiol Ecol 82(3):736–745. https://doi.org/10.1111/j.1574-6941.2012.01445.x
Arnold AE, Maynard Z, Gilbert GS, Coley PD, Kursar TA (2000) Are tropical fungal endophytes hyperdiverse? Ecol Lett 3(4):267–274. https://doi.org/10.1046/j.1461-0248.2000.00159.x
Hannula S, De Boer W, Van Veen J (2010) In situ dynamics of soil fungal communities under different genotypes of potato, including a genetically modified cultivar. Soil Biol Biochem 42(12):2211–2223. https://doi.org/10.1016/j.soilbio.2010.08.020
Thormann MN, Currah RS, Bayley SE (2001) Microfungi isolated from Sphagnum fuscum from a Southern Boreal Bog in Alberta. Canada Bryologist 104(4):548–559. https://doi.org/10.2307/3244589
Letcher PM, McGee PA, Powell MJ (2004) Distribution and diversity of zoosporic fungi from soils of four vegetation types in New South Wales Australia. Can J Bot 82(10):1490–1500. https://doi.org/10.1139/b04-109
Bridge P, Spooner B (2001) Soil fungi: diversity and detection. Plant Soil 232(1–2):147–154. https://doi.org/10.1023/a:1010346305799
Tedersoo L (2015) Tree diversity and species identity effects on soil fungi, protists and animals are context dependent. ISME J 10:346–362. https://doi.org/10.1038/ismej.2015.116
Sasse J, Martinoia E, Northen T (2018) Feed your friends: do plant exudates shape the root microbiome? Trends Plant Sci 23(1):25–41. https://doi.org/10.1016/j.tplants.2017.09.003
Li X, Boer WD, Zhang YN, Ding C, Zhang T, Wang X (2018) Suppression of soil-borne Fusarium pathogens of peanut by intercropping with the medicinal herb Atractylodes lancea. Soil Biol Biochem 116:120–130. https://doi.org/10.1016/j.soilbio.2017.09.029
Thirumalaisamy P, Dutta R, Jadon KS, Nataraja M, Padvi RD, Rajyaguru R, Yusufzai S (2019) Association and characterization of the Fusarium incarnatum-F. equiseti species complex with leaf blight and wilt of peanut in India. J Gen Plant Pathol 85(2):83–89. https://doi.org/10.1007/s10327-018-0827-y
Li XG, Ding CF, Zhang TL, Wang XX (2014) Fungal pathogen accumulation at the expense of plant-beneficial fungi as a consequence of consecutive peanut monoculturing. Soil Biol Biochem 72:11–18. https://doi.org/10.1016/j.soilbio.2014.01.019
Averill C, Hawkes CV (2016) Ectomycorrhizal fungi slow soil carbon cycling. Ecol Lett 19(8):937–947. https://doi.org/10.1111/ele.12631
Liu J, Sui Y, Yu Z, Shi Y, Chu H, Jin J, Liu X, Wang G (2015) Soil carbon content drives the biogeographical distribution of fungal communities in the black soil zone of northeast China. Soil Biol Biochem 83:29–39. https://doi.org/10.1016/j.soilbio.2015.01.009
Clemmensen KE, Finlay RD, Dahlberg A, Stenlid J, Wardle DA, Lindahl BD (2015) Carbon sequestration is related to mycorrhizal fungal community shifts during long-term succession in boreal forests. New Phytol 205(4):1525–1536. https://doi.org/10.1111/nph.13208
Civitello DJ, Penczykowski RM, Hite JL, Duffy MA, Hall SR (2013) Potassium stimulates fungal epidemics in Daphnia by increasing host and parasite reproduction. Ecology 94(2):380–388. https://doi.org/10.1890/12-0883.1
Beauregard M, Hamel C, St-Arnaud M (2010) Long-term phosphorus fertilization impacts soil fungal and bacterial diversity but not AM fungal community in alfalfa. Microb Ecol 59(2):379–389. https://doi.org/10.1007/s00248-009-9583-z
Rizvi S, Gauquelin T, Gers C, Guérold F, Pagnout C, Baldy V (2012) Calcium-magnesium liming of acidified forested catchments: Effects on humus morphology and functioning. Appl Soil Ecol 62:81–87. https://doi.org/10.1016/j.apsoil.2012.07.014
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microb 75(15):5111–5120. https://doi.org/10.1128/AEM.00335-09
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, M., Sun, W., Cui, S. et al. Analysis of Microbial Diversity and Community Structure of Peanut Pod and Its Surrounding Soil in Peanut Rot Epidemic Area. Curr Microbiol 78, 2173–2182 (2021). https://doi.org/10.1007/s00284-021-02471-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-021-02471-3