
Abstract
Biocontrol strategies using organic substrates such as wood fibers and biocontrol agents such as Trichoderma are currently developed to control soil pathogens such as Fusarium oxysporum. Nonetheless, such biocontrol methods give discording results, notably because microbial communities of organic substrates actually are not taken into account. Therefore, there is a lack of information concerning the variability of microbial composition related to the organic substrate type. Here we studied peat, wood and coir fibers, that are substrates known for their different biocontrol efficiency against Fusarium wilt of cucumber. We analyzed in microcosms the microbial composition of wood fibers, coir fibers and peat, incubated up to 60 days, by using an amplicon-sequencing approach based on 16S rRNA gene for bacteria and the internal transcribed spacer (ITS) for fungi. Diversity was assessed by sequencing the 16S rRNA for bacteria and ITS2 region for fungi. Results showed that bacterial richness was threefold higher for coir fiber and peat than for wood fiber. Fungal richness was three times higher for wood and coir fibers compared to peat. Bacterial and fungal patterns showed a dominance of α- and γ- Proteobacteria and Sordariomycetes for coir fiber; β- and γ-Proteobacteria and Eurotiomycetes for wood fibers; Flavobacteria, Leotiomycetes and Sordariomycetes for peat. In conclusion, results show that substrates have different microbial composition. Finally, for a proper use of a biocontrol strategy is important to take into account the type of substrate.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Producing healthy fruits and vegetables by reducing pesticides uses as well as the maintenance of high crop yields is central in agroecological practices. Biological strategies with integrative approach to protect crop against bio-aggressors were then suggested (Mercier and Manker 2005). Despite promising results with the use of biological control agents belonging to e.g., genus Trichoderma, Pseudomonas or Bacillus to fight against Fusarium oxysporum (Lecomte et al. 2016), the effectiveness of this strategy varies greatly from one production condition to another. Biocontrol indeed requires the optimization of (1) the inoculum density, (2) its formulation and (3) the method for the inoculation of the substrates used for crops. Because these strategies are based on the control much more than on the suppression of the pathogens, the producers must have acquired an in-depth knowledge of (1) the natural resistance of the considered fruits or vegetables and their susceptibility to the pathogens, according to the plant growth stage and culture conditions prevailing in greenhouse (Chaparro et al. 2012), (2) the favorable conditions so that the pathogen is able to attack plants, (3) the biocontrol agents potentially useful to control pathogens. Microorganisms used as biocontrol agents are added to the culture substrates—one of the application of bio augmentation—with the risk that they do not survive (Lebeau 2011). Another strategy relies on the selection of substrates used for crops (Koohakan et al. 2004). The protection against crop diseases varies according to the growing media (Bonanomi et al. 2010). In particularly, wood and coir fibers were recently tested with success for the protection of cucumber against Fusarium oxysporum (Montagne et al. 2016). The specific physicochemical characteristics of these substrates were already demonstrated (Domeño et al. 2011), but little is known regarding the microorganism that colonize these growing media. In view of the impact of the diet on the intestinal microbiota (Turnbaugh et al. 2009), it is interesting to study the indigenous microorganisms of these different organic substrates.
So far the microbial composition of organic substrates (wood fibers, coir fibers and peat) used for cucumber crop is unknown, our work aimed to study and compare their bacterial and fungal compositions. For this, we studied substrates, with different times of incubation, by using an amplicon-sequencing approach based on 16S rRNA gene for bacteria and the internal transcribed spacer (ITS) for fungi.
Experimental
Organic substrates
Three types of substrates were studied: wood fiber (PiF for pine and PoF for poplar), coir fiber (Co) and sphagnum peat (SpP). These substrates induce different plants protection against Fusarium (Montagne et al. 2016). Two distinct incubation processes (A and B) were performed, to test different batches of each substrate (Table 1).
A first incubation “A” was performed over a 3-month period with substrates PiF1s, PiF2d, PoFs, CoP, CoF and SpP. A second incubation “B” (over a 2-month period) was performed on new batches of the same substrates, and two additional substrates were studied: PiF2s and SpPn.
Precisely, pH, organic matter and dry bulk density were initially measured based on different standard methods, NF EN13037 (2000), NF EN13039 (2011), NF EN13041 (2000), respectively.
In details, the incubation protocol was adapted from the XP U44-163 standard (Montagne et al. 2015): 500 ml of substrate were placed at the bottom of a 2–l air-tight jar; 69.02 ml of a KNO3 solution at 4.185 mg/ml were added to each jar to avoid nitrogen being a limiting factor for microbial development. A predetermined volume of distilled water was added to each substrate, and its hydric potential was adjusted to ≈−30 kPa (pF 1.7). A beaker of water was placed in each jar, and then the jars were closed tightly and placed in an incubator at 28 °C for two or three months depending on the incubation A or B. To be close to horticultural conditions (producers use nutrient solution with pH near to 6–7), the pH of all substrates except SpP was set at 6 with a phosphate solution (0.1 M K2HPO4, 0.01 M KH2PO4).
All the substrates were sampled at 30, 60 and 90 days and at 10 and 60 days for the incubations A and B, respectively, and some of them were targeted to apply a community profiling approach (Table 1).
DNA extraction
In total, 0.250 g of dry matter were ground (MM301, Retsch, Germany) in a 25 ml stainless steel bowl with a 1.5 cm diameter stainless steel bead (2 × 1 min, 20 Hz). Then, DNA was extracted using a FastDNA™ SPIN Kit for Soil (MP Biomedicals, USA). Extracted DNA was quantified with SPECTROstar Nano, (BMG LABTECH LVi Plate, Germany).
Amplicon library construction and sequencing
The V3-V4 region of the 16S rRNA gene and the ITS2 region of the fungal internal transcribed spacer were amplified with the primer sets 341f (5′TACCAGGGTATCTAATCCT-3′; Muyzer et al. 1993)/784r (5′-ACGGRAGGCAGCAG-3′; Gamalero et al. 2012) and ITS2_PlaGe (5′-GCTGCGTTCTTCATCGATGC-3′/IT5_PlaGe (5′-GGAAGTAAAAGTCGTAACAAGG-3′; White et al. 1990). PCR were conducted in a final reaction volume of 50 µl containing 0.6 ng/µl of DNA, 0.6 µM of primer 341f/784r or 0.2 µM of primer ITS2_PlaGe/IT5_PlaGe, 200 µM of dNTPs, 0.6 unit/µl of Taq polymerase (DyNAzyme EXT DNA Polymearse, Fisher Scientific), 1 × DyNAzyme EXT Buffer (Fisher Scientific), 2.5 mM of MgCl2 and 500 ng/µl of bovine serum albumin. The following cycling conditions were employed for V3-V4 region: 1 cycle at 94 °C for 60 s, followed by 30 cycles of 94 °C for 60 s, 65 °C for 60 s, 72 °C for 60 s, and a final extension cycle at 72 °C for 10 min (CFX96, C1000 TouchTM, Thermal Cycler, Bio-Rad, United States). For ITS2 amplification, the conditions were: 1 cycle at 95 °C for 15 min, followed by 35 cycles of 95 °C for 30 s, 55 °C for 45 s, 72 °C for 30 s, and a final extension cycle at 72 °C for 7 min. All amplicons were purified with Clean PCR beads (Mokascience) and quantified with a nanodrop (ND-8000). The second amplification was performed with primers containing the Illumina adapters and index using protocol described in Lluch et al. (2015). All amplicons were purified and quantified as previously described. The purified amplicons were then pooled in equimolar concentrations and the final concentration of the library was determined by qPCR using the KAPA Library Quantification Kit. Amplicons libraries were mixed with 15% PhiX control according to Illumina’s protocols. One sequencing run was performed at GeT-PlaGe sequencing facility with a MiSeq reagent kit v2 (500 cycles).
Clustering Miseq reads into operational taxonomic units (OTUs)
The sequencing data were analyzed with Mothur (Schloss et al. 2009) with standard operating procedures described in Kozich et al. (2013). Briefly, 16S rRNA gene sequences were aligned against the 16rRNA gene SILVA database. All sequences that did not align correctly were removed from the data sets. Chimeric sequences were detected with Uchime and removed. Taxonomic affiliation of 16S rRNA gene was performed with a Bayesian classifier against the 16S rRNA gene training set (v9) of the Ribosomal Database Project. Sequences were divided into groups according to their taxonomic units (OTUs) at a 97% identity threshold.
ITS read pairs were combined with Mothur, and the variable ITS2 regions of ITS sequences were extracted with the Perl-based software ITSx. Then sequences were clustered at a 97% identity cutoff using Uclust, and taxonomic affiliation was performed with a Bayesian classifier (80% bootstrap confidence score) against the UNITE database.
Microbial community analyses
In order to enhance the reproducibility of community profiles, only abundant OTUs representing at least 0.1% of the library size were used for microbial community analyses (Barret et al. 2015). Normalization between samples was performed through rarefaction of 8000 sequences per sample. Both α diversity indexes (Simpson inverse) and β diversity were calculated with Mothur (Schloss et al. 2009). Analysis of Similarity (ANOSIM) was performed to assess the effects of different factors on the microbial community structure (P < 0.05). Beta diversity was assessed using Bray–Curtis dissimilarity index. Ordination of the similarity between microbial communities was performed with nonmetric multidimensional scaling (NMDS) plots.
Results and discussion
Diversity and composition of bacteria and fungi were compared between the three types of organic substrates. In detail, we compared richness (number of OUTs), Simpson diversity index, taxonomic composition (microbial classes) and microbial structure (with beta diversity and nonmetric multidimensional scaling).
Bacterial and fungal diversity in substrates and factors involved in the change of the microbial structure
After applying recommended filters, a median of 29,128 16S rRNA gene sequences and 53,390 ITS2 sequences were obtained per sample. According to Good’s coverage estimator, the median coverage was 92% for 16S rRNA gene sequences and closed to saturation (99%) for ITS2 sequences (data not shown). Mean bacterial and fungal richness observed was 80 and 30 (number of OTU), for 16S rRNA gene and ITS2 sequences, respectively, after normalization of abundant OTUs. The distribution and the mean values of richness and diversity are shown for each type of substrate (Table 2) irrespective of the number and the time of incubation. For 16S rRNA gene, coir fibers show the highest number of OTU (124) than peat and wood fibers (108 and 38, respectively). Regarding ITS, wood fibers show the highest number (35), as compared to coir fibers and peat (30 and 10, respectively).
These differences are supported by Simpson diversity indices, bacterial diversity is higher for coir fiber and peat (with an index around 21) than wood fiber (with an index of 7.5) and fungal diversity is around 2.7 for wood and coir fibers and 2.4 for peat. Analysis of similarity of microbial composition (ANOSIM) highlights significant differences in the microbial community structure when you consider the three types of substrates (P < 0.001 for bacterial and fungal community structure). The communities for both incubations (A and B) are significantly different for bacteria (P < 0.001) but not for fungi (P = 0.068). Conversely, the microbial community structure was not impacted by the incubation time of 10 and 60 days (P = 0.585 and P = 0.914, for bacterial and fungal community structure, respectively). The structure of the microbial communities was stabilized in the early stage of incubation (from 10 days). The characteristics of the substrate (type of organic matter, physical structure resulting from the process, etc.) strongly act on the microbial composition. Thus, the manufacturing process has an important role in the defining of microbial structure. Microbial communities depend strongly of the type of substrate. This result can be extrapolated to the effects of diet on the microbiota, observed in the gut or the rumen (Turnbaugh et al. 2009; Russell and Rychlik 2001).
Comparison of taxonomic composition and microbial structure associated with the type of substrates
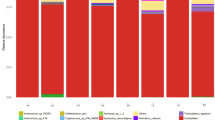
The taxonomic composition of each substrate was then investigated in more details, using bacterial and fungal classes as taxonomic unit (Fig. 1a, b). CoF is characterized by a high proportion of α- and γ-Proteobacteria and Actinobacteria, with 60% and 20%, respectively (Fig. 1a). Pine wood fibers bacterial communities (PiF1s and PiF2d) are mainly composed of α- β- and γ-Proteobacteria and Actinobacteria. The taxonomic composition of PoFs is distinct from PiF1s and PiF2d, with the high prevalence of γ-Proteobacteria (60 and 20% for PoFs and both PiFs, respectively). The presence of Flavobacteria reached 18% in PoFs while showed lower presence in both PiFs. Thus, Pine wood fibers are closer to coir fiber than to Poplar wood fiber (PoFs). PoFs composition is close to SpPn, with Flavobacteria, γ- and α-Proteobacteria as dominant classes. Finally, SpP bacterial community is mainly composed of Actinobacteria. Compared to bacteria, fungal classes (Fig. 1b) are more specific to the substrate. Sordariomycetes dominates reaching 80% presence in coir fiber, while Eurotiomycetes dominates in PiF1s and PiF2d (75 and 95%, respectively). PoFs is mainly characterized by two classes (Incertae-sedis and Leotiomycete, with 90 and 8%, respectively). SpPn is represented by Leotiomycetes, and by Sordariomycetes (50 and 40%, respectively). The class of Agaricomycetes is specific of SpP (represented 40%). Some substrates (e.g., SpP or SpPn) host several well-represented fungal classes, while wood fibers consist in one dominant class. The comparison with class level specifies the microbial particularities of each substrate, for example the influence of tree species (Pine or Poplar) or the pH (SpP or SpPn), and it is expected that the three types of substrate (CoF, PiF and SpP) allow the development of specific microorganisms.

Relative abundances of phylogenetic microbial classes in substrates, for 16S marker (a) and for ITS marker (b). Mean % of total reads. Unclassified was removed for fungal classes. Note that the representative classes were different between substrates. The type of substrate (coir, wood fibers and peat) and to a lesser extent the tree species (PiF and PoF) and pH (SpP and SpPn) impact representative classes. Note the similarity of the microbial compositions of two wood fibers, PiF1s and PiF2d, with Proteobacteria and Eurotiomycetes dominance
To gain more insight into the influence of the type of substrate on the microbial community structure, we performed β-diversity analyses using Bray–Curtis dissimilarity index. Biological replicates for each condition (represented by −1 and −2) are very close (Fig. 2a). The incubation and the time of incubation seem to weakly influence the microbial structure. The bacterial community structure differs according to the type of substrate. The bacterial community structure in wood differs strongly from coir and peat libraries (Fig. 2a). The same differentiation is observed for the fungal communities (Fig. 2b), with differences according to the type of the substrate, and peat community structure was more different than wood or coir community structures.

Influence of substrates on the bacterial (a) or fungal (b) β-diversity by using Nonmetric MultiDimensional Scaling (NMDS) ordination of Bray–Curtis dissimilarity matrix obtained with 16S or ITS Operational Taxonomic Units (OTUs), respectively. Each dot represents a microbial library observed in one sample. Mothur software and R software were used—stress value is 0.1519532 for bacteria and 0.1518726 for fungi. Projection on the graph of the main bacterial (a) or fungal (b) orders explains the substrate distribution on the β-diversity Nonmetric MultiDimensional Scaling. Vectors in the bi-plot overlay were constructed from a matrix containing the relative abundances of each bacterial or fungal order. Only correlations ≤0.05 were included. The angle and length of the vector indicate the direction and strength of the variable. Note that bacterial and fungal libraries differ according to the type of substrate (Wood fiber, coir fiber and peat). With the projection of orders involved in this representation, we can explain this substrate-dependency with Pseudomonadales, Burkholderiales and Eurotiales specificity of wood fibers, for example
According to the projection of orders making substrates repartition (Fig. 2a), orders of Pseudomonadales and Burkholderiales seem to specify wood fibers. These orders belong to γ- and β-Proteobacteria. Order of Sphingobacteriales is specific to peat (Fig. 1a). Regarding fungal repartition (Fig. 2b), Eurotiales, Diaporthales and Pezizales orders are specific to wood fibers, coir fibers and peat, respectively. These orders belong to the Ascomycota division and Pezizomycotina subdivision, but the class is different according to the substrate (Eurotiomycetes, Sordariomycetes and Pezizomycetes classes, respectively).
Bacterial phyla observed in our organic substrates (Bacteroidetes, Actinobacteria, Proteobacteria, Acidobacteria and Firmicutes) are frequently observed in soils (Fierer et al. 2007). β- and γ-Proteobacteria and Actinobacteria are stimulated in presence of fresh organic matter (Bernard et al. 2007). The order of Burkholderiales is specific to wood fiber substrates (Fig. 2a), and the class of Actinobacteria are more especially observed in pine wood soil, possibly linked to low pH and high C/N ratio (Kuramae et al. 2012). In wood fibers (data not shown), the genus Pseudomonas dominates. Numerous Pseudomonas sp. belonging to Plant Growth-Promoting Rhizobacteria (PGPR) are known to improve the plant nutrition (Laslo et al. 2012) and to activate the plant defenses system (Faessel et al. 2008). Moreover, all substrates of our study have high rates of cellulose and lignin (about 45 and 30%, respectively, Montagne et al. 2015). Coir fiber (CoF) contains more lignin than wood fibers (which contain more cellulose and hemi-cellulose). This involves a synergistic action of bacteria synthesizers of pectinase and cellulase and fungi decomposers of cellulose and lignin such as Ascomycota and Basidiomycota (Schellenberger et al. 2010 and de Boer et al. 2005), which dominate in our organic substrates. Moreover, Actinobacteria is the only bacterial phylum able to hydrolyze lignin, like fungi of the Basidiomycota phylum.
Some microorganisms could outcompete fungal opportunists (Bardin et al. 2004). As pointed out by Vivant et al. (2013), a high fungal diversity (and low bacterial diversity) for wood fibers may constitute a barrier against fungal pathogens. Penicillium genus of Eurotiales order is also present in wood fibers (data not shown). Penicillium genus was studied for its competitiveness against Fusarium oxysporum f. sp. radicis-cucumerinum (Sabuquillo et al. 2010). In a previous study, Montagne et al. (2016) observed a systematic decrease of Fusarium oxysporum f. sp. radicis-cucumerinum attack on cucumber when cultivated on wood fiber substrate. Moreover, the protection obtained with coir fibers was lower and dependent of climatic conditions and the level of plant’s stress. The dominance of Eurotiomycetes (in particular the Eurotiales order) and with β- and γ-Proteobacteria (in particular the Burkholderiales and Pseudomonadales orders) may explain the wood fibers protection. On the contrary, the variable level of protection obtained with coir fibers could be explained by the variations in the bacterial community structures.
Conclusion
Actinobacteria, Proteobacteria, Bacteroidetes, Ascomycota and Basidiomycota dominate in substrates. Microbial composition depends on the type of substrates (wood fibers, coir fibers and peat). Wood fibers show microbial particularities with a low bacterial diversity, high fungal diversity and dominance of Eurotiomycetes (85%) and Proteobacteria (90%) classes. As the food consumption is defining intestinal microbiota, the choice of growing media for soilless crop is decisive determining the microorganisms present in the substrate. The presence of specific microorganisms when using wood fibers can be a reason explaining the substrate-induced crop’s protection. Here, we showed that better understanding the microbial properties of organic substrates allows to achieve major advances in the fields of plant health and biological control. Further studies manipulating microbial communities in such substrates are needed to elucidate the relationship between different bacterial and fungal groups and their potential biocontrol.
References
Bardin SD, Huang HC, Moyer JR (2004) Control of Pythium damping-off of sugar beet by seed treatment with crop straw powders and a biocontrol agent. Biol Control 29:453–460. doi:10.1016/j.biocontrol.2003.09.001
Barret M, Briand M, Bonneau S, Préveaux A, Valière S, Bouchez O, Hunault G, Simoneau P, Jacques M-A (2015) Emergence Shapes the Structure of the Seed Microbiota. Appl Environ Microbiol 81:1257–1266. doi:10.1128/AEM.03722-14
Bernard L, Mougel C, Maron P-A, Nowak V, Lévêque J, Henault C, Haichar FEZ, Berge O, Marol C, Balesdent J, Gibiat F, Lemanceau P, Ranjard L (2007) Dynamics and identification of soil microbial populations actively assimilating carbon from 13C-labelled wheat residue as estimated by DNA- and RNA-SIP techniques. Environ Microbiol 9:752–764. doi:10.1111/j.1462-2920.2006.01197.x
Bonanomi G, Antignani V, Capodilupo M (2010) Identifying the characteristics of organic soil amendments that suppress soilborne plant diseases. Soil Biol Biochem 42:136–144. doi:10.1016/j.soilbio.2009.10.012
Chaparro JM, Sheflin AM, Manter DK, Vivanco JM (2012) Manipulating the soil microbiome to increase soil health and plant fertility. Biol Fertil Soils 48:489–499. doi:10.1007/s00374-012-0691-4
de Boer W, Folman LB, Summerbell RC, Boddy L (2005) Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev 29:795–811. doi:10.1016/j.femsre.2004.11.005
Domeño I, Irigoyen I, Muro J (2011) Comparison of traditional and improved methods for estimating the stability of organic growing media. Sci Hortic 130:335–340. doi:10.1016/j.scienta.2011.07.012
Faessel L, Nassr N, Lebeau T, Walter B (2008) Effects of the Plant Defence Inducer, Acibenzolar-S-Methyl, on Hypocotyl Rot of Soybean Caused by Rhizoctonia solani AG-4. J Phytopathol 156:236–242. doi:10.1111/j.1439-0434.2007.01367.x
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364. doi:10.1890/05-1839
Gamalero E, Cesaro P, Cicatelli A, Todeschini V, Musso C, Castiglione S, Fabiani A, Lingua G (2012) Poplar clones of different sizes, grown on a heavy metal polluted site, are associated with microbial populations of varying composition. Sci Total Environ 425:262–270. doi:10.1016/j.scitotenv.2012.03.012
Koohakan P, Ikeda H, Jeanaksorn T, Tojo M, Kusakari S-I, Okada K, Sato S (2004) Evaluation of the indigenous microorganisms in soilless culture: occurrence and quantitative characteristics in the different growing systems. Sci Hortic 101:179–188. doi:10.1016/j.scienta.2003.09.012
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi:10.1128/AEM.01043-13
Kuramae EE, Yergeau E, Wong LC, Pijl AS, van Veen JA, Kowalchuk GA (2012) Soil characteristics more strongly influence soil bacterial communities than land-use type. FEMS Microbiol Ecol 79:12–24. doi:10.1111/j.1574-6941.2011.01192.x
Laslo É, György É, Mara G, Tamás É, Ábrahám B, Lányi S (2012) Screening of plant growth promoting rhizobacteria as potential microbial inoculants. Crop Prot 40:43–48. doi:10.1016/j.cropro.2012.05.002
Lebeau T (2011) Bioaugmentation for in situ soil remediation: how to ensure the success of such a process. In: Singh A, Parmar N, Kuhad RC (eds) Bioaugmentation, biostimulation and biocontrol, soil biology. Springer, Berlin, pp 129–186. doi:10.1007/978-3-642-19769-7_7
Lecomte C, Alabouvette C, Edel-Hermann V, Robert F, Steinberg C (2016) Biological control of ornamental plant diseases caused by Fusarium oxysporum: a review. Biol Control 101:17–30. doi:10.1016/j.biocontrol.2016.06.004
Lluch J, Servant F, Païssé S, Valle C, Valière S, Kuchly C, Vilchez G, Donnadieu C, Courtney M, Burcelin R, Amar J, Bouchez O, Lelouvier B (2015) The characterization of novel tissue microbiota using an optimized 16S metagenomic sequencing pipeline. PLoS ONE 10:1–22. doi:10.1371/journal.pone.0142334
Mercier J, Manker DC (2005) Biocontrol of soil-borne diseases and plant growth enhancement in greenhouse soilless mix by the volatile-producing fungus Muscodor albus. Crop Prot 24:355–362. doi:10.1016/j.cropro.2004.09.004
Montagne V, Charpentier S, Cannavo P, Capiaux H, Grosbellet C, Lebeau T (2015) Structure and activity of spontaneous fungal communities in organic substrates used for soilless crops. Sci Hortic 192:148–157. doi:10.1016/j.scienta.2015.06.011
Montagne V, Capiaux H, Cannavo P, Charpentier S, Renaud S, Liatard E, Grosbellet C, Lebeau T (2016) Protective effect of organic substrates against soil-borne pathogens in soilless cucumber crops. Sci Hortic 206:62–70. doi:10.1016/j.scienta.2016.04.035
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Russell JB, Rychlik JL (2001) Factors that alter rumen microbial ecology. Science 292:1119–1122. doi:10.1126/science.1058830
Sabuquillo P, De Cal A, Melgarejo P (2010) Development of a dried Penicillium oxalicum conidial formulation for use as a biological agent against Fusarium wilt of tomato: selection of optimal additives and storage conditions for maintaining conidial viability. Biol Control 54:221–229. doi:10.1016/j.biocontrol.2010.05.010
Schellenberger S, Kolb S, Drake HL (2010) Metabolic responses of novel cellulolytic and saccharolytic agricultural soil Bacteria to oxygen. Environ Microbiol 12:845–861. doi:10.1111/j.1462-2920.2009.02128.x
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Horn DJV, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi:10.1128/AEM.01541-09
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14-6ra14. doi:10.1126/scitranslmed.3000322
Vivant A-L, Garmyn D, Maron P-A, Nowak V, Piveteau P (2013) Microbial diversity and structure are drivers of the biological barrier effect against Listeria monocytogenes in soil. PLoS ONE 8:1–11. doi:10.1371/journal.pone.0076991
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols 38:315–322
Acknowledgements
We wish to thank the Florentaise firm, the ANRT (Association Nationale de la Recherche et de la Technologie—CIFRE convention No. 2012/1062) for funding this work. We are particularly grateful to the NED team (UMR1289 TANDEM) and the GeT-PLaGE plateform in Toulouse using Illumina Miseq technology (Génopole Toulouse-Midi-pyrénées, INRA Auzeville, 24 Chemin de Borde Rouge-Auzeville, CS52627, 31326 Castanet-Tolosan, France).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest declared.
Rights and permissions
About this article

Cite this article
Montagne, V., Capiaux, H., Barret, M. et al. Bacterial and fungal communities vary with the type of organic substrate: implications for biocontrol of soilless crops. Environ Chem Lett 15, 537–545 (2017). https://doi.org/10.1007/s10311-017-0628-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10311-017-0628-0