
Abstract
The complement cascade is a key arm of the immune system that protects the host from exogenous and endogenous toxic stimuli through its ability to potently regulate inflammation, phagocytosis, and cell lysis. Due to recent clinical trial successes and drug approvals for complement inhibitors, there is a resurgence in targeting complement as a therapeutic approach to prevent ongoing tissue destruction in several diseases. In particular, neuromuscular diseases are undergoing a recent focus, with demonstrated links between complement activation and disease pathology. This review aims to provide a comprehensive overview of complement activation and its role during the initiation and progression of neuromuscular disorders including myasthenia gravis, amyotrophic lateral sclerosis, and Duchenne muscular dystrophy. We will review the preclinical and clinical evidence for complement in these diseases, with an emphasis on the complement-targeting drugs in clinical trials for these indications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The complement system is an evolutionarily ancient effector of innate and adaptive humoral immunity. The system is a proteolytic cascade comprising of blood-circulating soluble proteins and membrane-bound receptors or regulators [1], that act collectively to detect and recognise microbial or danger-associated molecular patterns. This subsequently leads to complement activation to protect the host from noxious stimuli and eliminate its target, through the ability to regulate inflammation, chemotaxis, and phagocytosis, via the activation of complement receptors on innate and adaptive immune cells. Furthermore, complement can also eliminate pathogens directly through cytolytic activity of the membrane attack complex (MAC (C5b-9); [2]). Although the complement system is important for self-defence and maintaining homeostasis, given its non-specific nature, excessive and uncontrolled activation of the complement system is frequently observed in plethora of acute and chronic diseases including neuromuscular disorders. With increasing interest in targeting complement as a therapeutic approach to ameliorate tissue destruction in neuromuscular disease, this review aims to provide an overview of complement activation and its role during the initiation and progression of neuromuscular disorders including myasthenia gravis, amyotrophic lateral sclerosis, and Duchenne muscular dystrophy.
Neuromuscular junction
The neuromuscular junction is a highly specialised chemical synapse between the nerve terminal of a motor neuron and the muscle fibre it innervates (Fig. 1). The electrical impulses (i.e. action potentials) generated from motor neurons travel down the motor nerves to the muscle fibre, which initiates the opening of transmembrane voltage-gated calcium ion channels on the presynaptic membrane [3]. Increased calcium influx into the motor nerve terminal triggers the release of acetylcholine and agrin, proteins involved in muscle contraction into the synaptic cleft. The synaptic cleft is a space between the presynaptic terminal and the postsynaptic muscle membrane and is comprised of an extracellular matrix called the synaptic basal lamina [4]. This basal lamina is composed of numerous molecules that are important for the organisation, alignment, and structural integrity of the neuromuscular junction. The postsynaptic muscle membrane (also referred to as the motor end-plate) is characterised by a high degree of folding, with nicotinic acetylcholine receptors (AChR) clustered on top of these folds [5]. Binding of acetylcholine to these receptors leads to the opening of the sodium channels located at the base of these folds, causing membrane depolarisation triggering muscle contraction [6]. Acetylcholinesterase, an enzyme that breaks down acetylcholine, is also present in the synaptic basal lamina, that effectively terminates synaptic transmission [7, 8]. Several other proteins including rapsyn, agrin, muscle-specific kinase (MuSK), and low-density lipoprotein receptor–related protein 4 (LRP4) are also found at the motor end-plate as complexes, that cause AChR clustering, which is essential in the formation and maintenance of neuromuscular units [9, 10].

The neuromuscular junction. Presynaptic nerve terminal activation leads to influx of Ca2+ through voltage-gated Ca2+ channels (VGCC), causing the secretion of ACh into the synaptic cleft. AChRs present at the postsynaptic membrane bind to the released ACh, causing Na+ influx through voltage-gated Na+ channels, inducing depolarisation of the postsynaptic membrane, and stimulating muscle contraction. Agrin, also released following an action potential at the nerve terminal, binds to its receptor, LRP4/MuSK, driving AChR clustering and maintenance of neuromuscular units. ACh, acetylcholine; AChE, acetylcholinersterase; AChR, acetylcholine receptor; LRP4, low-density lipoprotein receptor–related protein 4; MuSK, muscle-specific kinase; VGCC, voltage-gated Ca2+ channel. Figure adapted from [23, 94]. Images obtained and adapted from “Neuromuscular Junction”, by BioRender.com (2021); retrieved from https://app.biorender.com/biorender-templates
Complement cascade
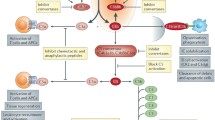
The complement system is an important component of our immune system. Complement activation provides a first line of defence against endogenous and exogenous threats in the form of foreign pathogens and noxious self-derived molecules. Complement also plays a role in the adaptive immune response, where, for example, it acts as an effector system for primary and secondary antibody responses of B cells, and their maturation within the germinal centre for positive selection [11, 12]. The cascade is activated via three different pathways: the classical, lectin, and alternative, where each pathway has a different initiation mechanism. All activation pathways ultimately lead to the generation of biologically active anaphylatoxins (C3a and C5a), opsonins (C1q, C3b, and C4b), and the formation of the MAC. The classical pathway is initiated by the C1 complex comprising C1q, C1r, and C1s, which sense and bind to antigen–antibody complexes with IgG1, IgG2, IgG3, or IgM. However, C1q has also been shown to bind directly to pathogen surfaces in the absence of antibodies [13]. Additionally, C1q can interact and bind to molecules such as actin, adiponectin, amyloid fibrils, C-reactive protein, and prion proteins, all of which can cause an inflammatory response [13]. The lectin pathway is triggered primarily by carbohydrate groups on surfaces of certain pathogens and senescent or apoptotic cells, through the recognition molecules mannose-binding lectin (MBL), ficolins, and collectin-11 [14]. Lectin pathway initiator molecules form complexes with MBL serine proteases (MASP1 and MASP2), leading to the formation of a C3 convertase and activation of the same downstream pathway as in the classical pathway. Unlike the classical and lectin pathways, the alternative pathway is spontaneously and continuously active at a low rate and its activation is amplified in response to bacteria and foreign surfaces, leading to the spontaneous hydrolysis of C3. The formation of C3(H2O) will generate an initial fluid-phase alternative pathway C3 convertase, which can cleave C3 to generate additional C3a and C3b. The C3b deposited on activating surfaces will bind to protease factor B and subsequently cleaved by factor D to generate the final alternative pathway C3 convertase, which will cleave more C3 and thus forming an amplification loop. The C3-mediated amplification by the alternative pathway assists with normal surveillance of the surrounding host environment [15].
The three complement activation pathways all converge on the central complement molecule, C3, with its activation leading to the generation of its cleavage fragments C3a and C3b. The cleavage product, C3b, is an opsonin that will bind to the membrane of pathogens and altered host or foreign surfaces for phagocytic removal [16]. Equally important, C3b is also an essential factor in generating C5 convertases that preferentially cleave complement component C5, which initiates terminal complement activation [17]. The activation of C5 forms two major effectors of complement activation, C5a and C5b. C5a is considered one of the most potent pro-inflammatory molecules, and exhibits wide-ranging biological functions through activity at two receptors; the classical receptor C5aR1, and the alternate receptor C5aR2 [18]. The second active fragment, C5b, associates with the plasma proteins C6 and C7, initiating a conformational change to form a hydrophobic membrane binding site in the lipophilic complex [19]. Once the complex is inserted and locked tightly onto a cell membrane, it will recruit C8 and as many as twelve C9 molecules, which will induce major conformational changes and embed into the membrane to form a transmembrane pore. This permits calcium and sodium influx, consequently leading to cell death via osmotic lysis, as well as triggering inflammatory and other activation pathways [19].
Given the potential for a non-specific nature of the complement cascade, it is tightly controlled through several complement regulatory proteins in the circulation and on cell surfaces, which act to prevent ongoing activation of complement pathways progressing to tissue damage. Fluid-phase classical and lectin pathway regulators include C1 esterase inhibitor (C1-INH) and C4b-binding protein (C4BP). C1-INH regulates complement activation via inactivating C1r, C1s, MASP-1, and MASP2 [16, 20], while C4BP binds to convertases containing C4b and mediates their degradation [16, 20]. Soluble alternative pathway regulators include Factor H and properdin, where Factor H controls tick-over activation and the amplification loop by binding to hydrolysed C3 and C3b, while properdin is a positive regulator of complement activation by binding to and stabilising the alternative pathway C3 convertase [16, 20]. The membrane of host cells also expresses complement regulators to keep their activity under control. For example, the complement regulatory protein CD55 is expressed on host cell surfaces and inactivates C3 and C5 convertases through the dissociation of Bb or C2b [21]. CD59 is also a key regulatory protein, which prevents MAC formation on cells by inhibiting C9 polymerisation [22].
Myasthenia gravis
Myasthenia gravis (MG) is a rare autoimmune disease that affects the neuromuscular junction, whereby nicotinic AChR is depleted at the postsynaptic membrane, compromising neuromuscular transmission [23]. The condition is driven by a B-cell-mediated overproduction of autoantibodies targeting multiple components of the postsynaptic end-plate. The majority of MG patients (~ 80%) possess autoantibodies directed against AChR, with the remaining 20% possessing antibodies directed against MuSK, LRP4, or agrin [24, 25]. The binding of autoantibodies to these proteins at the motor end-plates in generalised MG patients manifests in weakness and fatigability of bulbar, ocular, and skeletal muscles [26]. In patients with AChR antibodies, synaptic transmission at the neuromuscular junction is primarily reduced by autoantibodies binding to AChR through several pathogenic mechanisms. These primarily include (i) inhibiting the binding of acetylcholine to AChRs; (ii) accelerating the internalisation and subsequent degradation of AChRs; and (iii) classical pathway complement activation leading to the formation of MAC on the postsynaptic membrane [23]. Hence, complement activation is considered one of the key effectors of AChR antibody-induced MG (Fig. 2).

Proposed pathological mechanisms driving anti-AChR-positive myasthenia gravis. In myasthenia gravis, synaptic transmission at the neuromuscular junction is reduced through three pathogenic mechanisms. This includes (1) production of AChR autoantibodies that bind and prevent the interaction of ACh at AChRs; (2) acceleration of the normal internalisation and degradation of surface AChRs that are cross-linked by autoantibodies; and (3) classical complement pathway activation induced by C1-complex-antibody interaction, leading to the formation of MAC/C5b-9 on the postsynaptic membrane and dysfunction of the neuromuscular junction. All pathways lead to reduced muscle contraction. ACh, acetylcholine; AChR, acetylcholine receptor; MAC, membrane attack complex. Figure adapted from [23, 94]. Images obtained and adapted from “Neuromuscular Junction”, by BioRender.com (2021); retrieved from https://app.biorender.com/biorender-templates
Complement activation in myasthenia gravis
In AChR-positive generalised MG, complement is activated by AChR antibodies, that are primarily of the IgG1 and IgG3 subclass [27]. After binding to AChRs, these antibodies interact with C1q of the classical complement pathway [28]. This initiates the classical complement cascade through C1r and subsequently C1s activation, ultimately leading to the formation of a C5 convertase that initiates MAC deposition at the neuromuscular junction [28]. MAC formation reduces the integrity of the postsynaptic membrane, minimising membrane surface area, AChR numbers, and voltage-gated sodium ion channels. As a result, neuromuscular transmission is impaired leading to muscle weakness that is a characteristic phenotype of MG [29].
The first evidence that implicated a potential role for complement in MG pathogenesis came from the early 1960s, where changes in various complement protein levels including C3, C4, and terminal components were observed in MG patient serum [30, 31]. These data implied complement components could be involved in the MG pathogenic process; however interestingly, complement levels were not correlated to severity of muscle weakness [32, 33]. Additional immunohistochemistry and electron microscopy studies of the MG neuromuscular junction further suggested a role for complement, as C3 and MAC deposition were identified at the postsynaptic membrane and at degenerating junctional folds in the synaptic space [34,35,36,37,38]. Coinciding with the potential role of complement activation in MG pathogenesis, sera from MG patients was found to induce complement-mediated lysis of cultured myotubes, cementing its role in inducing focal lysis to the muscle [39]. More recently, another study demonstrated that the complement regulator properdin is decreased in MG serum, and its levels to be negatively associated with MG severity [40]. Overall, there is overwhelming clinical data that suggests complement is a major effector pathway in MG patients, and likely contributes significantly to its pathology.
Clinical findings of complement involvement in MG have also been observed in animal models. Animal models allow for the more precise investigation of the functional role of complement in MG and the ability to test different therapeutic strategies. Despite the limitations inherent in using rodents to model human disease, animal models of MG are useful in that they retain many of the pathological phenotypes observed in human MG patients. These include the presence of AChR autoantibodies, complement factors, and IgG deposition at the neuromuscular junction, and a reduction of AChRs at the junctional folds [41]. Animal models of experimentally acquired myasthenia gravis (EAMG) are achieved either by administering AChR antibodies (passive model) or immunising the animal with purified AChR or peptide fragments of AChR (active model; [42,43,44]). EAMG animals acquire muscle weakness, fatigue, and show a decreased response to repetitive nerve stimulation, similar to what is observed in human patients [45]. Substantial evidence from EAMG studies supports a role for complement activation in the initiation and progression of postsynaptic membrane damage. Animals in passive or active EAMG models present with antibody and complement deposition at the neuromuscular junction [46, 47]. Immunofluorescence studies have also correlated this complement deposition with the loss of AChR from neuromuscular junctional folds [46, 47].
Upstream complement activation in experimentally acquired myasthenia gravis
Direct evidence for the contribution of early complement pathway factors in EAMG pathology has been obtained from several studies using mice deficient in specific upstream/proximal components of the complement system. Studies have shown that animals that lack C3 or C4 have significantly lower incidence of active EAMG than their wild-type counterparts, in parallel with reductions in IgG, C3, and MAC deposition at the neuromuscular junction [28]. In contrast to studies investigating the complement components C3 and C4, genetic deletion of the lectin component MBL showed no difference in the susceptibility to the development of EAMG by AChR immunisation [48]. These mice showed similar amounts of IgG, C3, and MAC deposition at the neuromuscular junction suggesting that the lectin pathway is not critically involved in the development of EAMG [48]. To support the role of proximal complement factors in EAMG pathology, several pharmacological approaches for upstream complement blockade have been studied, to evaluate if these therapies can effectively inhibit EAMG induction or improve symptoms in experimental animals. Inhibiting C1q or C2 using an anti-C1q antibody and small interfering RNAs (siRNA) for C2 significantly reduced the incidence of EAMG induced by AChR immunisation and improved muscle strength and survival in mice following EAMG induction [49, 50]. Reductions in C3 and MAC deposition at the neuromuscular junction were also observed along with increased AChR levels [49, 50]. Taken together, these studies indicate that early complement activation through the classical activation pathway in response to AChR autoantibodies plays a key role in driving the disease process.
Terminal complement activation in experimentally acquired myasthenia gravis
In addition to the upstream complement pathways, many studies have also demonstrated that the terminal component of the complement system plays a direct detrimental role in EAMG pathology. Indeed, animals lacking C5 or C6 showed similar results to genetic deletion of upstream components, where genetic deletion of C5 led to markedly lower incidence of disease and reduced the loss of AChR in the muscle for active EAMG, while C6 deficiency led to resistance to passive EAMG induction [51, 52]. Importantly, C6 deficient animals administered human C6 during the induction of EAMG, developed pathology similar to that observed in wild-type animals, suggesting MAC formation is crucial and required for neuromuscular junction damage. Interestingly, in contrast to studies investigating C5 and C6, a study that utilised knockout mice for the effector receptor, C5aR1, showed no differences in the susceptibility to the development of EAMG by AChR immunisation or IgG, C3, and MAC deposition at the neuromuscular junction [53]. This suggests that C5a–C5aR1 signalling is not critically involved in the development of EAMG [53]. To further support terminal complement involvement in MG pathogenesis, multiple studies demonstrated that inhibiting C5 or C6 using anti-C5 or C6 antibodies and the recombinant C5 inhibitor rEV576 prevented development of EAMG by passive transfer of AChR antibodies, and also improved muscle weakness when administered after EAMG induction [54,55,56]. The accumulation of MAC components C6 and C9 was reduced at the neuromuscular junction, blocking the formation of pathogenic MAC on the postsynaptic membrane. A more recent study utilising siRNA for C5 in both a rodent and a non-human primate MG model showed efficient and prolonged suppression of liver C5 expression. Silencing C5 expression in the liver significantly reduced disease severity in both the passive and active EAMG animals, leading to improvements in muscle weakness. This plethora of studies strongly indicate that in addition to the initiating classical components of complement, the effector MAC is crucial in driving the neuromuscular junction damage observed in MG and offers a distinct potential therapeutic target.
Complement regulators in experimentally acquired myasthenia gravis
Additional support for complement-driven pathology in the development of MG is observed in mice lacking specific complement regulators. CD55 is a complement regulator that inhibits C3 and C5 convertases and thus reduces the formation of the MAC. Multiple studies confirmed that genetic deletion of CD55 in mice increases the susceptibility to EAMG [57,58,59]. In CD55 knockout mice, EAMG severity was exacerbated with a greater deposition of C3 observed at the neuromuscular junction, greater reduction of AChR levels, and increased junctional damage compared with control animals [57,58,59]. Interestingly, another study investigating the complement regulator CD59, which inhibits MAC formation directly, showed similar EAMG severity and complement deposition at the neuromuscular junction compared to wild-type animals [57, 59, 60]. Regardless of this negative study, studies utilising complement-deficient animals strongly support that complement activation is a key driver of EAMG pathology, and encourages therapeutic development into targeting the complement system for human MG. In addition to these studies in knockout mice, application of recombinant or purified complement regulators has also been evaluated as potential therapeutics for MG. Administration of human complement receptor 1 (sCR1), which inhibits both the classical and alternative complement pathways, was shown to reduce clinical phenotypes in EAMG induced by AChR immunisation [61]. Other complement regulators that have been investigated are the complement receptor 1–related gene/protein y (CRRY), and CD55, which both inhibit C3 convertases. Separate studies utilised CRRY and CD55 regulators, where CRRY was coupled to the Fc region of IgG2a to prolong circulating half-life, and CD55 coupled to a single-chain antibody directed towards the AChR α-subunit, to assist in its delivery to the neuromuscular junction [62, 63]. Both approaches significantly improved muscle weakness, prevented EAMG induction, and decreased the deposition of C3 and C9 at the neuromuscular junction [62, 63].
Overall, support for a pathogenic role of complement in MG has been well documented in several preclinical EAMG models as detailed above. There is substantial evidence from EAMG animals that suggests induction of the classical complement pathway, and in particular the formation of MAC is required for the induction of EAMG symptoms due to the postsynaptic membrane damage caused by MAC. Hence, inhibiting MAC formation through different strategies at different levels in the classical complement cascade can improve EAMG symptoms. This is further supported in MG patients where MAC deposition at the neuromuscular junction is associated with damaged motor end-plates and muscle weakness. These compelling animal and clinical data led to clinical studies which targeted C5 in MG patients as detailed below.
Clinical trial evidence for complement inhibition in myasthenia gravis
Current common treatment options for MG include acetylcholinesterase inhibitors, corticosteroids, or steroid-sparing immunosuppressive treatments, as well as short-term immunomodulatory therapies, such as plasma exchange, therapeutic apheresis, or intravenous immunoglobulins [64, 65]. Approximately 10–15% of patients fail to respond adequately to these commonly available treatments, or experience intolerable side effects to immunosuppressive treatments, and these patients are considered to have refractory MG [66, 67]. Due to the high degree of disease burden for these patients, there is an unmet clinical need to find more effective and specific immunosuppression, and to reduce adverse effects. As detailed above, animal and clinical data strongly implicate complement-mediated destruction of the neuromuscular junction as a major cause of MG pathology, which propelled complement as an attractive therapeutic target. Indeed, inhibition of the complement cascade has now been extensively studied at the C5 level in patients with generalised MG. Eculizumab (Soliris) is the first specific complement-targeting drug approved for complement-mediated diseases. It is a humanised IgG2 and IgG4 monoclonal antibody, that binds to C5 with high affinity, inhibiting its cleavage into C5a and C5b and ultimately preventing MAC formation [68]. The efficacy of eculizumab in MG was initially assessed for 14 refractory MG patients with AChR autoantibodies in a randomised, double-blind, placebo-controlled Phase II trial (Study C08-001), where a beneficial effect due to therapy was demonstrated in these patients [69]. This further led to a randomised, double-blind, placebo-controlled multicentre Phase III REGAIN trial (ECU-MG-301) in 125 refractory MG patients, with an open-label extension study (ECU-MG-302), which also demonstrated safety with significant and rapid improvement in symptoms [70, 71]. Currently, eculizumab is approved for AChR antibody–mediated refractory MG in the USA, Europe, and Japan.
Although eculizumab is showing promise in MG, it is ineffective for patients harbouring rare C5 mutations in the eculizumab binding site, is parenterally administered, and still incurs a high financial cost compared to other therapies for generalised MG [72, 73]. Hence, additional treatment options for targeting C5 and MAC are of great interest. Zilucoplan, is a small macrocyclic peptide that binds to C5 with high affinity and specificity and inhibits MAC formation by preventing C5 cleavage and thus the first step of MAC assembly (i.e. C5b and C6 interaction). The binding site of Zilucoplan on C5 is distinct from eculizumab, as it still shows efficacy in patients resistant to eculizumab. The efficacy of Zilucoplan in MG was assessed in 44 AChR antibody–positive generalised MG patients in a Phase II double-blind, placebo-controlled trial (NCT03315130). Similar, to eculizumab, Zilucoplan treatment showed significant improvements in primary and secondary endpoints, and a Phase III study (RAISE; NCT04115293) is currently underway for patients with MG.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a heterogenous, late-onset neurodegenerative disorder, that is characterised by the degeneration of both upper and lower α-motor neurons in the motor cortex, brain stem, and spinal cord. This leads to muscle weakness and paralysis and eventual death due to respiratory failure. The initial presentation of ALS varies between patients, where some patients have onset of limb weakness, and some with dysphagia (difficulty swallowing) and/or dysarthria (difficulty talking). The majority of ALS cases are sporadic with unknown aetiology and no common environmental risk factors. However, a subset of cases are familial, where they are associated with gene mutations that have a wide range of functions [74]. The two causes of ALS are however indistinguishable based on their clinical and pathological features, with both presenting with muscle atrophy, weakness, and spasticity [75]. There are several theories that have been hypothesised regarding the cause of muscle denervation in ALS including the unwanted heightened activity of the complement system [74, 75]. Although the complement system in the pathology of ALS has been reviewed previously [76,77,78,79], this review will summarise evidence for complement’s specific involvement in the neuromuscular unit.
Complement activation in amyotrophic lateral sclerosis skeletal muscle
A substantial body of research has been performed on ALS patients and animal models of ALS, investigating the levels of complement components in the blood, motor cortex, spinal cord, and skeletal muscle [80,81,82,83,84,85]. Although the majority of these studies have focussed on complement activation in the circulation and central nervous system, there are now emerging studies that have investigated complement in skeletal muscle ALS pathology. Complement activation with deposition of C1q, C3/C3b, and MAC has been observed in the neuromuscular junction of patients with ALS [86]. Increased deposition of complement factors C1q, C3/C3b, and MAC is demonstrated on the motor end-plates of intercostal muscles from ALS patients, while no deposition is observed in healthy controls [86]. The complement regulators CD55 and CD59 are also detected at the motor end-plates of ALS patient muscles [86], suggesting that the muscle may be attempting to neutralise complement activation to prevent further complement activity and MAC deposition progressing to muscle denervation. Additionally, this study also provided evidence that these components were deposited on motor end-plates, prior to denervation, suggesting that it may play a key role in the initial pathological process leading to the degradation of the ALS neuromuscular junction.
In addition to clinical evidence implicating the complement system in the skeletal muscle, many ALS animal models have also confirmed the role of different complement pathways in the skeletal muscle throughout disease progression. There have been numerous reports implicating complement involvement, where marked up-regulation of C1q, C3, C4, factor B, C5a, C5aR1 and the regulators CD55 and CD59a at mRNA and protein levels are detected in skeletal muscle of multiple ALS mouse models including SOD1G93A and TDP43Q331K transgenic mice [85, 87]. Increases in C5aR1 expression in the skeletal muscle is primarily due to the infiltrating macrophages, as genetically deleting C5ar1 from SOD1G93A ALS mice, dramatically decreases the number of infiltrating macrophages in line with extending survival and improved muscle strength [85, 88]. Additionally, C5aR1 antagonist (PMX205) treatment in SOD1G93A mice extends survival, improves motor functions (i.e. hind-limb grip strength), and slows disease progression [82]. This indicates that C5a–C5aR1 signalling may accelerate ALS disease progression through the increased recruitment of pro-inflammatory macrophages to sites of neuromuscular denervation. Collectively, these findings suggest that complement activation within the skeletal muscle could be a key driver of pathology seen in ALS progression (Fig. 3).

Proposed complement-mediated pathophysiology in the neuromuscular unit of amyotrophic lateral sclerosis patients. SOD1 and TDP43 aggregates from motor neurons and muscles are released into the synaptic cleft, which activates complement and leads to the generation of C5a and C5b from C5 cleavage. C5b leads to the formation of MAC/C5b-9 on the postsynaptic membrane causing dysfunction/destruction of the neuromuscular junction. C5a binds to C5aR1 leading to infiltration and activation of macrophages, which assists in motor neuron degeneration and ultimately neuromuscular denervation leading to paralysis. Images obtained and adapted from “Neuromuscular Junction”, by BioRender.com (2021); retrieved from https://app.biorender.com/biorender-templates
Similar to MG, the substantial clinical and preclinical evidence for complement in the pathophysiology of ALS has prompted the initiation of several clinical trials using anti-complement therapies. To the best of our knowledge, there are three active clinical trials investigating complement inhibition in ALS patients. The first of these is ravulizumab (Ultomiris), a recombinant humanised monoclonal antibody similar to eculizumab that binds the complement protein C5 with high affinity. The efficacy of ravulizumab is being assessed in 382 sporadic and familial ALS patients in a Phase III double-blind, placebo-controlled multicentre trial (NCT04248465). Another clinical trial is also assessing the efficacy of zilucoplan, a C5 inhibitor in 160 ALS patients in a Phase II/III double-blind, placebo-controlled trial (NCT04436497). Lastly, a study is being conducted to evaluate the efficacy and safety of the C3 inhibitor pegcetacopan, in 228 sporadic ALS patients in a Phase II double-blind, placebo-controlled multicentre trial (NCT04579666). All three trials are estimated to be completed between 2022 and 2024.
Potential role of complement in Duchenne muscular dystrophy
Duchenne muscular dystrophy is a severe, progressive, muscle-wasting disease caused by mutations in the DMD gene on the X chromosome, that prevents the production of the muscle isoform of dystrophin [89]. Duchenne muscular dystrophy occurs primarily in males and the initial symptoms are difficulties in walking with waddling gait, difficulties in standing, and frequent falls. Patients present with these symptoms around 2–3 years of age and become wheelchair-bound around 10–12 years of age. Patients then require assisted ventilation at around 20 years of age and will die between 20 and 40 years of age from cardiac or respiratory failure [89]. Dystrophin deficiency in the skeletal muscle of Duchenne muscular dystrophy patients results in altered mechanical functions that maintain muscle cell structural integrity and contractile activity. This contributes to membrane fragility, necrosis of muscle fibres, progressive muscle wasting, and inflammation [90]. In contrast to numerous studies investigating the role of complement cascade in MG and ALS, there have only been scant studies investigating complement in disease induction and underlying muscle pathology in Duchenne muscular dystrophy. However, a prior study examining 66 biopsy specimens containing necrotic muscle fibres including Duchenne muscular dystrophy demonstrated deposition of C3 and C9 on these necrotic fibres [91]. Furthermore, one of the prominent characteristics within muscles of Duchenne muscular dystrophy patients is the infiltration of macrophages and neutrophils, which are the predominant effector myeloid cells for C5a signalling [92]. Indeed, inhibition of C5a using a specific C5a L-aptamer (NOX-D21) in a mouse model of Duchenne muscular dystrophy was recently shown to rescue muscle deficit phenotypes [90], further cementing complement’s potential role in driving disease pathology. By contrast, a separate study demonstrated that genetic ablation of upstream C3 did not have any significant effects on muscle pathology [93]. This is surprisingly consistent with numerous studies in ALS, where the terminal complement pathway, rather than C3 or earlier complement factors, is identified as the key driver of muscle pathology [77, 94]. Similar to investigations in MG and ALS, future studies in Duchenne muscular dystrophy should also focus on the role of specific terminal complement pathway inhibition in disease pathology, which may lead to clinical trials for anti-complement disease-modifying strategies for patients.
Future directions towards therapeutic application of complement-targeted drugs in neuromuscular disorders
Tight regulation and appropriate timing of complement activation are essential for a healthy neuromuscular junction. There is now a spotlight on complement in neuromuscular disorders given the emerging evidence that dysregulation of complement plays a significant role in the pathogenesis of many neuromuscular disorders including MG and ALS. The role of complement in generalised MG is well established in patients and animal models, and appears to represent a key pathogenic effector of generalised MG (Fig. 2). As current commonly available treatment options for MG often fail, and/or cause significant adverse effects in subsets of patients, the future for complement inhibitors in this disease is bright as they promise to increase specificity, efficacy, and safety of generalised MG treatment. In particular, inhibition at the level of C5 appears to be most relevant for MG, due to the selective blockade one of the key mechanisms of action of the anti-AChR antibodies (i.e. terminal pathway activation and MAC formation). Given the key role identified for MAC in this disease [94], and an apparent lack of role for C5a receptors [90], it would be of interest to identify whether specifically targeting MAC formation could lead to improved therapeutic precision for MG, in the hopes of further reducing disease burden and possible side effects. In ALS, there may be differing roles for complement at different stages of the disease. Early in the disease process complement may act to limit tissue damage by clearing protein aggregates and degenerating synapses, through targeted opsonisation and phagocytosis. However, later in the disease process, junctional damage and protein aggregates overwhelm this normally protective response, resulting an inflammatory environment that appears driven in part through terminal complement activities that accelerates disease progression (Fig. 3). Hence, targeting this terminal complement pathway in ALS could provide significant benefits to patients. For conditions like Duchenne muscular dystrophy and other rarer neuromuscular diseases, there has been little attention given to complement’s role in disease pathophysiology. However, given the promising data from recent MG clinical trials, it would be a potential fruitful area of research and potential clinical application for complement inhibitors. In summary, there is a promising future for complement therapeutics in neuromuscular diseases, and it is hoped that targeted complement inhibition could provide a lasting impact on patients, across multiple conditions in the years to come.
References
Kolev M, Le Friec G, Kemper C (2014) Complement–tapping into new sites and effector systems. Nat Rev Immunol 14(12):811–820
Hess C, Kemper C (2016) Complement-mediated regulation of metabolism and basic cellular processes. Immunity 45(2):240–254
Desaki J, Uehara Y (1981) The overall morphology of neuromuscular junctions as revealed by scanning electron microscopy. J Neurocytol 10(1):101–110
Sanes JR (2003) The basement membrane/basal lamina of skeletal muscle. J Biol Chem 278(15):12601–12604
De Harven E, Coers C (1959) Electron microscope study of the human neuromuscular junction. J Biophys Biochem Cytol 6(1):7–10
Engel AG, Shen XM, Selcen D, Sine SM (2015) Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 14(5):461
Bernard V, Girard E, Hrabovska A, Camp S, Taylor P, Plaud B, Krejci E (2011) Distinct localization of collagen Q and PRiMA forms of acetylcholinesterase at the neuromuscular junction. Mol Cell Neurosci 46(1):272–281
Ohno K, Brengman J, Tsujino A, Engel AG (1998) Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci U S A 95(16):9654–9659
Barik A, Lu Y, Sathyamurthy A, Bowman A, Shen C, Li L, Xiong WC, Mei L (2014) LRP4 is critical for neuromuscular junction maintenance. J Neurosci 34(42):13892–13905
Ruegg MA, Bixby JL (1998) Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci 21(1):22–27
Blank M, Shoenfeld Y (2007) B cell targeted therapy in autoimmunity. J Autoimmun 28(2–3):62–68
Cumpelik A, Heja D, Hu Y, Varano G, Ordikhani F, Roberto MP, He Z, Homann D, Lira SA, Dominguez-Sola D, Heeger PS (2021) Dynamic regulation of B cell complement signaling is integral to germinal center responses. Nat Immunol 22(6):757–768
Dalakas MC, Alexopoulos H, Spaeth PJ (2020) Complement in neurological disorders and emerging complement-targeted therapeutics. Nat Rev Neurol 16(11):601–617
Shastri A, Bonifati DM, Kishore U (2013) Innate immunity and neuroinflammation. Mediators Inflamm 2013:342931
Ricklin D, Hajishengallis G, Yang K, Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11(9):785–797
Ricklin D, Reis ES, Lambris JD (2016) Complement in disease: a defence system turning offensive. Nat Rev Nephrol 12(7):383–401
Daha MR, Fearon DT, Austen KF (1976) C3 requirements for formation of alternative pathway C5 convertase. J Immunol 117(2):630–634
Manthey HD, Woodruff TM, Taylor SM, Monk PN (2009) Complement component 5a (C5a). Int J Biochem Cell Biol 41(11):2114–2117
Hadders MA, Bubeck D, Roversi P, Hakobyan S, Forneris F, Morgan BP, Pangburn MK, Llorca O, Lea SM, Gros P (2012) Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep 1(3):200–207
Zipfel PF, Skerka C (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9(10):729–740
Hourcade DE, Mitchell L, Kuttner-Kondo LA, Atkinson JP, Medof ME (2002) Decay-accelerating factor (DAF), complement receptor 1 (CR1), and factor H dissociate the complement AP C3 convertase (C3bBb) via sites on the type A domain of Bb. J Biol Chem 277(2):1107–1112
Navenot JM, Villanova M, Lucas-Heron B, Malandrini A, Blanchard D, Louboutin JP (1997) Expression of CD59, a regulator of the membrane attack complex of complement, on human skeletal muscle fibers. Muscle Nerve 20(1):92–96
Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S (2016) Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat Rev Neurol 12(5):259–268
Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren J (2019) Myasthenia gravis. Nat Rev Dis Primers 5(1):30
Vincent A, Huda S, Cao M, Cetin H, Koneczny I, Rodriguez Cruz PM, Jacobson L, Viegas S, Jacob S, Woodhall M, Nagaishi A, Maniaol A, Damato V, Leite MI, Cossins J, Webster R, Palace J, Beeson D (2018) Serological and experimental studies in different forms of myasthenia gravis. Ann N Y Acad Sci 1413(1):143–153
Gilhus NE (2017) Eculizumab: a treatment option for myasthenia gravis? Lancet Neurol 16(12):947–948
Tuzun E, Christadoss P (2013) Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun Rev 12(9):904–911
Tuzun E, Scott BG, Goluszko E, Higgs S, Christadoss P (2003) Genetic evidence for involvement of classical complement pathway in induction of experimental autoimmune myasthenia gravis. J Immunol 171(7):3847–3854
Conti-Fine BM, Milani M, Kaminski HJ (2006) Myasthenia gravis: past, present, and future. J Clin Invest 116(11):2843–2854
Nastuk WL, Plescia OJ, Osserman KE (1960) Changes in serum complement activity in patients with myasthenia gravis. Proc Soc Exp Biol Med 105:177–184
Strauss AJ, van der Geld HW, Kemp PG Jr, Exum ED, Goodman HC (1965) Immunological concomitants of myasthenia gravis. Ann N Y Acad Sci 124(2):744–766
Barohn RJ, Brey RL (1993) Soluble terminal complement components in human myasthenia gravis. Clin Neurol Neurosurg 95(4):285–290
Romi F, Kristoffersen EK, Aarli JA, Gilhus NE (2005) The role of complement in myasthenia gravis: serological evidence of complement consumption in vivo. J Neuroimmunol 158(1–2):191–194
Engel AG, Lambert EH, Howard FM (1977) Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin Proc 52(5):267–280
Engel AG, Sahashi K, Fumagalli G (1981) The immunopathology of acquired myasthenia gravis. Ann N Y Acad Sci 377:158–174
Nakano S, Engel AG (1993) Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology 43(6):1167–1172
Rash JE, Albuquerque EX, Hudson CS, Mayer RF, Satterfield JR (1976) Studies of human myasthenia gravis: electrophysiological and ultrastructural evidence compatible with antibody attachment to acetylcholine receptor complex. Proc Natl Acad Sci U S A 73(12):4584–4588
Sahashi K, Engel AG, Lambert EH, Howard FM Jr (1980) Ultrastructural localization of the terminal and lytic ninth complement component (C9) at the motor end-plate in myasthenia gravis. J Neuropathol Exp Neurol 39(2):160–172
Ashizawa T, Appel SH (1985) Complement-dependent lysis of cultured rat myotubes by myasthenic immunoglobulins. Neurology 35(12):1748–1753
Ozawa Y, Uzawa A, Yasuda M, Kojima Y, Oda F, Himuro K, Kawaguchi N, Kuwabara S (2021) Changes in serum complements and their regulators in generalized myasthenia gravis. Eur J Neurol 28(1):314–322
Baggi F, Antozzi C, Toscani C, Cordiglieri C (2012) Acetylcholine receptor-induced experimental myasthenia gravis: what have we learned from animal models after three decades? Arch Immunol Ther Exp (Warsz) 60(1):19–30
Baggi F, Annoni A, Ubiali F, Milani M, Longhi R, Scaioli W, Cornelio F, Mantegazza R, Antozzi C (2004) Breakdown of tolerance to a self-peptide of acetylcholine receptor alpha-subunit induces experimental myasthenia gravis in rats. J Immunol 172(4):2697–2703
Losen M, Martinez-Martinez P, Molenaar PC, Lazaridis K, Tzartos S, Brenner T, Duan RS, Luo J, Lindstrom J, Kusner L (2015) Standardization of the experimental autoimmune myasthenia gravis (EAMG) model by immunization of rats with Torpedo californica acetylcholine receptors–Recommendations for methods and experimental designs. Exp Neurol 270:18–28
Tuzun E, Berrih-Aknin S, Brenner T, Kusner LL, Le Panse R, Yang H, Tzartos S, Christadoss P (2015) Guidelines for standard preclinical experiments in the mouse model of myasthenia gravis induced by acetylcholine receptor immunization. Exp Neurol 270:11–17
Christadoss P, Poussin M, Deng C (2000) Animal models of myasthenia gravis. Clin Immunol 94(2):75–87
Engel AG, Sakakibara H, Sahashi K, Lindstrom JM, Lambert EH, Lennon VA (1979) Passively transferred experimental autoimmune myasthenia gravis. Sequential and quantitative study of the motor end-plate fine structure and ultrastructural localization of immune complexes (IgG and C3), and of the acetylcholine receptor. Neurology 29(2):179–88
Sahashi K, Engel AG, Linstrom JM, Lambert EH, Lennon VA (1978) Ultrastructural localization of immune complexes (IgG and C3) at the end-plate in experimental autoimmune myasthenia gravis. J Neuropathol Exp Neurol 37(2):212–223
Li J, Qi H, Tuzun E, Allman W, Yilmaz V, Saini SS, Deymeer F, Saruhan-Direskeneli G, Christadoss P (2009) Mannose-binding lectin pathway is not involved in myasthenia gravis pathogenesis. J Neuroimmunol 208(1–2):40–45
Huda R, Tuzun E, Christadoss P (2013) Complement C2 siRNA mediated therapy of myasthenia gravis in mice. J Autoimmun 42:94–104
Tuzun E, Li J, Saini SS, Yang H, Christadoss P (2007) Pros and cons of treating murine myasthenia gravis with anti-C1q antibody. J Neuroimmunol 182(1–2):167–176
Chamberlain-Banoub J, Neal JW, Mizuno M, Harris CL, Morgan BP (2006) Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats. Clin Exp Immunol 146(2):278–286
Christadoss P (1988) C5 gene influences the development of murine myasthenia gravis. J Immunol 140(8):2589–2592
Qi H, Tuzun E, Allman W, Saini SS, Penabad ZR, Pierangeli S, Christadoss P (2008) C5a is not involved in experimental autoimmune myasthenia gravis pathogenesis. J Neuroimmunol 196(1–2):101–106
Biesecker G, Gomez CM (1989) Inhibition of acute passive transfer experimental autoimmune myasthenia gravis with Fab antibody to complement C6. J Immunol 142(8):2654–2659
Soltys J, Kusner LL, Young A, Richmonds C, Hatala D, Gong B, Shanmugavel V, Kaminski HJ (2009) Novel complement inhibitor limits severity of experimentally myasthenia gravis. Ann Neurol 65(1):67–75
Zhou Y, Gong B, Lin F, Rother RP, Medof ME, Kaminski HJ (2007) Anti-C5 antibody treatment ameliorates weakness in experimentally acquired myasthenia gravis. J Immunol 179(12):8562–8567
Kaminski HJ, Kusner LL, Richmonds C, Medof ME, Lin F (2006) Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp Neurol 202(2):287–293
Lin F, Kaminski HJ, Conti-Fine BM, Wang W, Richmonds C, Medof ME (2002) Markedly enhanced susceptibility to experimental autoimmune myasthenia gravis in the absence of decay-accelerating factor protection. J Clin Invest 110(9):1269–1274
Morgan BP, Chamberlain-Banoub J, Neal JW, Song W, Mizuno M, Harris CL (2006) The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin Exp Immunol 146(2):294–302
Tuzun E, Saini SS, Morgan BP, Christadoss P (2006) Complement regulator CD59 deficiency fails to augment susceptibility to actively induced experimental autoimmune myasthenia gravis. J Neuroimmunol 181(1–2):29–33
Piddlesden SJ, Jiang S, Levin JL, Vincent A, Morgan BP (1996) Soluble complement receptor 1 (sCR1) protects against experimental autoimmune myasthenia gravis. J Neuroimmunol 71(1–2):173–177
Hepburn NJ, Chamberlain-Banoub JL, Williams AS, Morgan BP, Harris CL (2008) Prevention of experimental autoimmune myasthenia gravis by rat Crry-Ig: a model agent for long-term complement inhibition in vivo. Mol Immunol 45(2):395–405
Kusner LL, Satija N, Cheng G, Kaminski HJ (2014) Targeting therapy to the neuromuscular junction: proof of concept. Muscle Nerve 49(5):749–756
Gilhus NE, Verschuuren JJ (2015) Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol 14(10):1023–1036
Mantegazza R, Bonanno S, Camera G, Antozzi C (2011) Current and emerging therapies for the treatment of myasthenia gravis. Neuropsychiatr Dis Treat 7:151–160
Silvestri NJ, Wolfe GI (2014) Treatment-refractory myasthenia gravis. J Clin Neuromuscul Dis 15(4):167–178
Suh J, Goldstein JM, Nowak RJ (2013) Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med 86(2):255–260
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nurnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368(23):2169–2181
J.F. Howard, Jr., R.J. Barohn, G.R. Cutter, M. Freimer, V.C. Juel, T. Mozaffar, M.L. Mellion, M.G. Benatar, M.E. Farrugia, J.J. Wang, S.S. Malhotra, J.T. Kissel, M.G.S. Group (2013) A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve 48(1):76–84
J.F. Howard, Jr., K. Utsugisawa, M. Benatar, H. Murai, R.J. Barohn, I. Illa, S. Jacob, J. Vissing, T.M. Burns, J.T. Kissel, S. Muppidi, R.J. Nowak, F. O’Brien, J.J. Wang, R. Mantegazza, R.S. Group (2017) Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol 16(12):976–986
Muppidi S, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, Jacob S, Vissing J, Burns TM, Kissel JT, Nowak RJ, Andersen H, Casasnovas C, de Bleecker JL, Vu TH, Mantegazza R, O’Brien FL, Wang JJ, Fujita KP, Howard JF Jr, G. Regain Study (2019) Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve 60(1):14–24
Dhillon S (2018) Eculizumab: a review in generalized myasthenia gravis. Drugs 78(3):367–376
Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, Noji H, Kitamura K, Eto T, Takahashi T, Masuko M, Matsumoto T, Wano Y, Shichishima T, Shibayama H, Hase M, Li L, Johnson K, Lazarowski A, Tamburini P, Inazawa J, Kinoshita T, Kanakura Y (2014) Genetic variants in C5 and poor response to eculizumab. N Engl J Med 370(7):632–639
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17085
Brown RH Jr, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377(16):1602
Carpanini SM, Torvell M, Morgan BP (2019) Therapeutic inhibition of the complement system in diseases of the central nervous system. Front Immunol 10:362
Lee JD, Coulthard LG, Woodruff TM (2019) Complement dysregulation in the central nervous system during development and disease. Semin Immunol 45:101340
Parker SE, Hanton AM, Stefanou SN, Noakes PG, Woodruff TM, Lee JD (2019) Revisiting the role of the innate immune complement system in ALS. Neurobiol Dis 127:223–232
Kjaeldgaard AL, Pilely K, Olsen KS, Pedersen SW, Lauritsen AO, Moller K, Garred P (2018) Amyotrophic lateral sclerosis: the complement and inflammatory hypothesis. Mol Immunol 102:14–25
Heurich B, El Idrissi NB, Donev RM, Petri S, Claus P, Neal J, Morgan BP, Ramaglia V (2011) Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J Neuroimmunol 235(1–2):104–109
Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, Woodruff TM, Noakes PG (2013) Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 10:119
Lee JD, Kumar V, Fung JN, Ruitenberg MJ, Noakes PG, Woodruff TM (2017) Pharmacological inhibition of complement C5a–C5a1 receptor signalling ameliorates disease pathology in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Br J Pharmacol 174(8):689–699
Mantovani S, Gordon R, Macmaw JK, Pfluger CM, Henderson RD, Noakes PG, McCombe PA, Woodruff TM (2014) Elevation of the terminal complement activation products C5a and C5b–9 in ALS patient blood. J Neuroimmunol 276(1–2):213–218
Sta M, Sylva-Steenland RM, Casula M, de Jong JM, Troost D, Aronica E, Baas F (2011) Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis 42(3):211–220
Wang HA, Lee JD, Lee KM, Woodruff TM, Noakes PG (2017) Complement C5a–C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Skelet Muscle 7(1):10
Bahia El Idrissi N, Bosch S, Ramaglia V, Aronica E, Baas F, Troost D (2016) Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J Neuroinflammation 13(1):72
Lee JD, Levin SC, Willis EF, Li R, Woodruff TM, Noakes PG (2018) Complement components are upregulated and correlate with disease progression in the TDP-43(Q331K) mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 15(1):171
Woodruff TM, Lee JD, Noakes PG (2014) Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc Natl Acad Sci U S A 111(1):E3-4
Mercuri E, Bonnemann CG, Muntoni F (2019) Muscular dystrophies. Lancet 394(10213):2025–2038
Hyzewicz J, Tanihata J, Kuraoka M, Nitahara-Kasahara Y, Beylier T, Ruegg UT, Vater A, Takeda S (2017) Low-intensity training and the C5a complement antagonist NOX-D21 rescue the mdx phenotype through modulation of inflammation. Am J Pathol 187(5):1147–1161
Engel AG, Biesecker G (1982) Complement activation in muscle fiber necrosis: demonstration of the membrane attack complex of complement in necrotic fibers. Ann Neurol 12(3):289–296
Rosenberg AS, Puig M, Nagaraju K, Hoffman EP, Villalta SA, Rao VA, Wakefield LM, Woodcock J (2015) Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med 7(299):299rv4
Han R, Frett EM, Levy JR, Rader EP, Lueck JD, Bansal D, Moore SA, Ng R, Beltran-Valero de Bernabe D, Faulkner JA, Campbell KP (2010) Genetic ablation of complement C3 attenuates muscle pathology in dysferlin-deficient mice. J Clin Invest 120(12):4366–4374
Howard JF Jr (2018) Myasthenia gravis: the role of complement at the neuromuscular junction. Ann N Y Acad Sci 1412(1):113–128
Funding
This work was supported by the National Health and Medical Research Council of Australian (grant #2000095 to JDL and TMW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
TMW holds patents and patent applications relating to the use of complement inhibitors for neurological disease.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the Special issue on: Complement & Disease: Out of the Shadow into the Spotlight — Guest Editors: Daniel Ricklin & Richard B. Pouw
Rights and permissions
About this article

Cite this article
Lee, J.D., Woodruff, T.M. The emerging role of complement in neuromuscular disorders. Semin Immunopathol 43, 817–828 (2021). https://doi.org/10.1007/s00281-021-00895-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-021-00895-4