
Abstract
The epidermis closely interacts with nerve endings, and both epidermis and nerves produce substances for mutual sustenance. Neuropeptides, like substance P (SP) and calcitonin gene-related protein (CGRP), are produced by sensory nerves in the dermis; they induce mast cells to release vasoactive amines that facilitate infiltration of neutrophils and T cells. Some receptors are more important than others in the generation of itch. The Mas-related G protein-coupled receptors (Mrgpr) family as well as transient receptor potential ankyrin 1 (TRPA1) and protease activated receptor 2(Par2) have important roles in itch and inflammation. The activation of MrgprX1 degranulates mast cells to communicate with sensory nerve and cutaneous cells for developing neurogenic inflammation. Mrgprs and transient receptor potential vanilloid 4 (TRPV4) are crucial for the generation of skin diseases like rosacea, while SP, CGRP, somatostatin, β-endorphin, vasoactive intestinal peptide (VIP), and pituitary adenylate cyclase-activating polypeptide (PACAP) can modulate the immune system during psoriasis development. The increased level of SP, in atopic dermatitis, induces the release of interferon (IFN)-γ, interleukin (IL)-4, tumor necrosis factor (TNF)-α, and IL-10 from the peripheral blood mononuclear leukocytes. We are finally starting to understand the intricate connections between the skin neurons and resident skin cells and how their interaction can be key to controlling inflammation and from there the pathogenesis of diseases like atopic dermatitis, psoriasis, and rosacea.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The old term “neurodermitis,” which indicates atopic eczema or allergic dermatitis, defines a close relationship between nerves and skin. It reflects the clinical observation that the development and the progression of allergic dermatitis is sensitive to emotional stress and environmental stimulation. It has been a longstanding clinical observation that chronic inflammatory skin disorders such as atopic dermatitis (AD), psoriasis, and rosacea are exacerbated by stress [1]. “Neurogenic inflammation” describes a mechanism by which sensory nerves contribute to inflammation [2]. In 1876, Stricker observed the phenomenon that cutaneous blood flow was increased in innervated areas when the corresponding dorsal roots were stimulated [3]. Together with similar findings [4], this phenomenon was defined as neurogenic vasodilation [4]. Later, it led to the concept of neurogenic inflammation, which describes the vasodilation and protein extravasation caused by inflammatory neuropeptides [5].
Both the somatosensory nervous system and the immune system are essential for the host defense against potential harmful infection and tissue damage [6]. While the immune system, which is the traditional host defense system, protects the host by combacting infective agents and restores tissue integrity, the somatosensory nervous system helps to avoid the noxious stimuli by removing the danger. There are abundant nociceptors in the skin which cover and protect the host from the outer environment. They respond to any noxious stimuli instantaneously and transduce them to electrical activity to produce sensation and reflex. Nociceptor neurons can transmit the action potentials antidromically, from the branch points to the periphery, as well as orthodromic input from the periphery to the central nervous system (CNS), which is called axon reflex [7]. Thus, the neuronal mediators are released from the depolarized axon terminals to the stimulated area, enabling a rapid response, well before the immune system is activated [6].
Skin mechanisms of neurogenic inflammation
Nervous system in skin
One of the major roles of the skin is to sense and respond to signals from the outer environment as well as protect our bodies. Abundant nerve fibers, including autonomic and sensory nerves, are densely distributed over all skin layers. They can communicate with different cell populations in different layers of the skin by releasing various types of neuropeptides. Almost all cutaneous cells express functional receptors for neuropeptides, through which they receive signals from the nervous system. In return, skin cells produce neuropeptides and neurotrophins, which in turn stimulate nerve fibers. This exchange creates a positive bidirectional feedback loop able to augment inflammatory response [8,9,10,11,12]. The finding that various kinds of chronic inflammatory skin disorders, such as AD and psoriasis, have common features of increased neurotrophin expression and peptidergic nerve fibers supports this pathophysiologic phenomenon [8].
In the epidermis, neuropeptides released from the nerve fibers stimulate keratinocytes to produce proinflammatory cytokines such as interleukin (IL)-1α, IL-6, and IL-8 [13,14,15,16]. On langerhans cells (LCs) in the epidermis, neuropeptide substance P (SP) enhances their migration and antigen presentation, leading to promotion of allergic sensitization [17,18,19]. In the dermis, sensory nerve fibers are intermingled with noradrenergic and acetylcholinergic nerve fibers containing additional neuropeptides such as neuropeptide Y (NPY) or vasoactive intestinal peptide (VIP). Sensory nerve fibers are commonly found in close contact with mast cells, blood vessels, or hair follicles in the dermis. Dermal mast cells have a particularly close relationship with the nervous system in terms of neurogenic inflammation. Neuropeptide SP released from the sensory nerve endings induces mast cell degranulation and subsequent proinflammatory effects of mediators such as histamine [8, 9, 20]. In turn, histamine, released from mast cells, evokes the release of neuropeptides acting on the histamine receptors on the sensory nerve endings, which establish a bidirectional loop between mast cells and sensory nerves. Moreover, SP induces vascular endothelial growth factor (VEGF) release from mast cells, which promotes endothelial cell proliferation and vascularization, facilitating the inflammatory process. Fibroblasts in the dermis also express receptors for SP as well as SP production, both of which are enhanced after exposure to SP or interferon (IFN)-γ [21, 22]. Thus, neuropeptides and neurotrophins contribute to the exaggeration of the inflammatory process in acute skin inflammation which overexpresses SP, nerve growth factor (NGF), and IFN-γ and later contribute to fibrosis in chronic skin inflammation [5].
Sensory nerve and neuropeptides
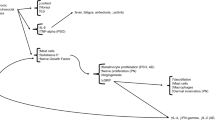
Neurogenic inflammation is mediated by the release of neuropeptides such as SP and calcitonin gene-related protein (CGRP). When sensory nerves are stimulated by certain stimuli, they release biologically active neuropeptides to transfer signals. SP and CGRP are the classic neuropeptides which act directly on the vascular endothelial cells and smooth muscle cells, thereby mediating vascular effects [23, 24]. SP increases vascular permeability with subsequent plasma extravasation and edema [23, 24]. The release of SP increases intercellular adhesion molecules (ICAMs) and vascular cell adhesion molecules (VCAMs) on vascular epithelial cells [25] and induces VEGF release from mast cells [26, 27], which facilitate hypervascularization and infiltration of inflammatory cells. CGRP is a potent microvascular vasodilator which contributes to the majority of the neurogenic vasodilation and is involved in recruitment of inflammatory cells [28, 29]. It was shown that CGRP enhanced LC antigen presentation on Th2 responses, while inhibiting presentation for the Th1 response, thereby shifting LCs toward Th2 responses [30]. Both SP and CGRP act through their subsequent G-protein-coupled receptor (GPCR), neurokinin (NK)-1 receptor for SP, and the CGRP receptor complex for CGRP [31, 32]. Recently, an NK-1 antagonist, aprepitant, was demonstrated to inhibit itch in AD mouse models and showed efficacy in chronic pruritus in humans [33]. The selective CGRP receptor antagonist as well as anti-CGRP antibodies have been developed and are currently under clinical trial showing promising results for migraine in which CGRP is the critical player in the pathogenesis [34]. Like CGRP, pituitary adenylate cyclase-activating polypeptide (PACAP) and VIP also inhibit LC antigen presentation for the generation of Th1 cells while enhancing presentation for Th2 responses. Also, PACAP and VIP enhance presentation for differentiation of Th17 cells, thereby shifting Th cells toward Th17 as well as Th2 responses [35] (Fig. 1).

Densely distributed nerve fibers in skin communicate with mast cells, endothelial cells, keratinocytes, Langerhans cells, and fibroblasts. By releasing neuropeptides, such as substance P (SP) or calcitonin gene-related receptor protein (CGRP), neurons activate skin cells that, in return, release histamine or proinflammatory cytokines—which activate sensory nerve terminals generating a bidirectional positive feedback loop that results in increased inflammation. Abbreviations: transient receptor potential cation channel subfamily V member 1 (TRPV1); transient receptor potential ankyrin 1 (TRPA1); G protein coupled receptors (GPCR); protease activated-receptor 2 (PAR-2); protease activated receptor 4 (PAR4) A13 receptor family (A13, X)
PARs and Mrgprs
The release of neuropeptides from the sensory nerve is triggered by a rise in the cytosolic Ca2+ concentration [36]. Cutaneous sensory nerves express GPCRs in addition to voltage-gated Ca channels, the activation of which increase cytosolic Ca2+ concentration. There are five specific GPCRs that are mainly involved in cutaneous neurogenic inflammation, which includes protease-activated receptors 2 and 4 (PAR-2 and PAR-4) and Mas-related G-coupled protein receptors C11, A3, and X (MrgprC11, MrgprA3, and MrgprX) [37,38,39,40,41]. Calcium channels such as nociceptive transient receptor potential vanilloid 1 (TRPV1) and transient receptor potential ankyrin 1 (TRPA1) co-localize with them [42]. PAR-2 is involved in pruritus and various skin diseases such as atopic dermatitis [43, 44] while PAR-4 is involved in edema formation, leukocyte recruitment, and analgesia [45,46,47,48,49]. Mrgprs are shown to be involved in histamine-independent itch pathways such as chloroquine-induced [50] or bovine adrenal medulla (BAM) 8–22-induced pruritus [51]. In the Mrgpr family, there are nine subfamilies including MrgprA to MrgprH and MrgprX [52]. Among them, MrgprA3, C11, and X1 are known to be involved in peripheral itch transduction and scratch behavior. MrgprX1 is expressed on mast cells while MrgprA3 and C11 are located on the sensory nerves. This is the only case discovered until now that Mrgpr is expressed in non-neuronal cells [53]. Mrgpr activation on mast cells strongly evokes scratch behavior to itch which subsequently results in skin barrier disruption and loss of immune homeostasis in skin. MrgprA3 and C11, co-localized with various neuropeptides, can sensitize TRPV1 and TRPA1 channels in sensory neurons and induce cellular secretion of neuropeptides [50, 51]. The activation of MrgprX1 degranulates mast cells to communicate with sensory nerve and cutaneous cells for developing neurogenic inflammation [54].
TRP channels
Cationic channels expressed on the sensory nerve endings include some TRP channels, which are involved in neuropeptide release. TRPV1 is a nociceptive cationic channel responsive to high temperature (> 43 °C), and capsaicin is its natural agonist [55]. When TRPV1 is activated by these direct activators, Ca2+ influx is initiated and neuropeptides such as SP and CGRP are released to induce neurogenic inflammation. Like PAR and Mrgpr, TRPV1-mediated Ca2+ influx in skin can regulate proinflammatory gene expression to affect immune cells, in addition to neuropeptide release. In addition to the sensory nerve, TRPV1 is also found in cutaneous cell functioning as a sensor for pain and chemical stimuli, including keratinocytes, mast cells, dendritic cells, sebocytes, dermal blood vessels, hair follicles, and sweat glands [56]. In endothelial cells and smooth muscle cells, TRPV1-mediated Ca2+ influx induces vasodilation by releasing nitric oxide (NO). Meanwhile, TRPA1 is a ligand-gated non-selective Ca2+ channel which responds to cold thermal sensation (< 17 °C), contrary to TRPV1. TRPA1 is localized to approximately 60–75% of sensory C fibers, which are also TRPV1-positive [57]. Topical application of cinnamaldehyde, a TRPA1 agonist, in human skin induces significantly increased itch sensation, which implies a central role for TRPA1 in the itch mechanism [58]. TRPA1 has been shown to play a critical role in itch, including endothelin (ET)-1-mediated itch and chloroquine-induced itch [51, 58, 59] while TRPV1 has shown a contradictory role in itch [60,61,62]. TRPA1 has been widely investigated on its role in chronic skin inflammation. In addition to thermal stimuli, several inflammatory mediators such as growth factors, bradykinins, proteases and thymic stromal lymphopoietin (TSLP) have been found to act on TRPA1 indirectly [63,64,65]. TSLP, a central cytokine in Th2-mediated inflammation such as AD, has recently been found to activate TRPA1 by binding a specific receptor, the TSLP receptor (TSLPR), on the sensory nerve in the skin of atopic dermatitis patients [66]. In addition, TRPA1 plays an important role in Th2 cell-dependent itch mediated by the IL-31 receptor expressed on sensory nerves. In a mouse model of AD of transgenic mice overexpressing IL-13, itching was significantly reduced in TRPA1 antagonist-treated mice [67]. Therefore, TRP channels, especially TRPA1, are considered to act like a “gatekeeper” which mediates cytokine signaling of cutaneous inflammation into sensory nerve activation [68,69,70].
Skin diseases with neurogenic inflammation
Neurogenic inflammation in rosacea
Rosacea is a chronic inflammatory skin disorder which is represented by facial flushing, telangiectasia, and inflammatory papules and pustules on the central location of the face. It has heterogeneous clinical manifestations depending on subtypes: erythematotelangiectatic rosacea (ETR) which has non-transient episodes of flushing and persistent central facial erythema, papulopustular rosacea (PPR) which has transient papules and pustules in addition to the characteristics seen in ETR, phymatous rosacea which has a thickened skin with irregular surface nodularity, and lastly, ocular rosacea which accompanies characteristic ophthalmic symptoms [71]. Although the pathogenesis of rosacea is not fully elucidated, dysregulation of the innate immune system, imbalance of commensal skin microbiota, and abnormal neurovascular signaling are considered to be implicated in the development of rosacea. Trigger factors of rosacea such as exposure to sunlight, heat, or cold; alcohol; spicy foods; or exercise can activate peripheral sensory nerve endings, which implies the particular role of neurogenic inflammation in the pathogenesis of rosacea [71].
Affected rosacea skin has a significantly lower threshold for heat and chemicals compared to non-affected skin, which defines it as sensitive skin [72]. The density of the sensory neuron is increased in the ETR subtype [73]. In addition, the density of TRP ion channels is increased on the sensory neurons and blood vessels as well as immune cells in all subtypes of rosacea [74, 75]. Dermal immunolabeling of TRPV2 and TRPV3 and gene expression of TRPV1 are significantly increased in ETR. PPR showed an enhanced immunoreactivity for TRPV2 and TRPV4, and phymatous rosacea for TRPV3 and TRPV4 [74]. Each subtype of TRPV has different functions, respectively: TRPV1 has a role in vasoregulation and nociception and activated by capsaicin, heat, and inflammation; TRPV2 in innate immunity, nociception, inflammation, vasoregulation, and heat sensing; and both TRPV3 and TRPV4 in heat sensing [76,77,78]. Beyond TRPV1–4, TRPA1 has been shown to be related to pathogenesis of rosacea. TRPA1 is activated by spices such as cinnamaldehyde and mustard oil as well as thermal stimuli. In mouse experiments, topical cinnamaldehyde induced vasodilation in a TRPA1-dependent mechanism, which could be involved in the flushing phenomenon in rosacea patients [79]. TRPA1 can also sense oxidants, which could support the role of reactive oxygen species (ROS) in the development of rosacea [80]. In rat neurons, TRPA1 is co-localized with PAR-2 which can be activated by proteases to induce inflammation in the human skin [63]. Therefore, it is supposed that the increased amount of serine protease in rosacea might induce TRPA1-mediated inflammation via upregulation of PAR [63].
Meanwhile, neuropeptides such as PACAP, SP, VIP, and CGRP are increased in rosacea [81, 82]. VIP and PACAP, as well as CGRP, play as potent vasodilators, acting on the smooth muscle cells in arterioles, while SP is critical for edema via the NK-1 receptor on postcapillary venules in rosacea [83]. PACAP can also stimulate NO release from endothelial cells which results in indirect vasodilation [84]. Neuropeptides also activate mast cells to release histamine which induces vasodilation and tryptase which is a chemotactic agent for fibroblasts and matrix metalloproteinases (MMPs), contributing to fibrosis in rosacea [85, 86]. In addition, neuropeptides stimulate IL-1β production and activate leukocyte migration via upregulation of VCAMs in rosacea [25, 87]. There is literature that shows promising efficacy of intradermal botulinum toxin injection for treating refractory erythema and flushing in patients with rosacea, which needs further investigation [88].
Neurogenic inflammation in psoriasis
Psoriasis is one of the common chronic inflammatory skin disorders with the prevalence ranging from 0.5 to 11.4% in adults worldwide [89]. It is characterized by hyperproliferation of abnormally differentiated keratinocytes and cutaneous immune cell infiltration including T cells, dendritic cells, and neutrophils. Clinically, psoriasis manifests as well-demarcated red indurated plaques with silvery thick scales over any body area, especially on the prominence such as elbows or knees. The pathogenesis of psoriasis has been rapidly evolving in recent years, in which the IL-23/Th17 cell axis plays a major role in close interaction with keratinocytes [90]. However, there has been multiple literature reporting on clinical symptom changes in psoriatic patients after having acquired central or peripheral nerve damage. The patients showed spontaneous clearance or improvement of the skin lesion which was limited to the area affected with nerve damage while the non-affected area did not [91,92,93,94,95,96,97,98,99]. Similarly, in a murine model of psoriasis, cutaneous denervation by traumatic nerve injury resulted in reduction of clinical symptoms of psoriasis [100]. These observations imply that the nervous system may be critical for the pathogenesis of psoriasis.
Immunohistochemical studies in psoriatic patients display an altered expression of various neuropeptides and of their receptors, as well as a marked proliferation of the cutaneous nerve in the skin [101]. These neuropeptides include SP, CGRP, somatostatin, β-endorphin, VIP, and PACAP, which can modulate the immune system during psoriasis development [101]. SP initiates the inflammatory process, leading to proliferation of specific T-lymphocytes and mast cell degranulation, in the early stages of psoriasis [102, 103]. CGRP has a role as a potent vasodilator in the pathogenesis of cutaneous inflammation in psoriasis, and synergizes with SP [104]. VIP modulates mast cell degranulation and the production of proinflammatory cytokines, such as IL-6, IL-8, and RANTES (regulated upon activation, normal T cell expressed and secreted, also known as CCL5), in addition to vasodilation, all of which are involved in the pathogenesis of psoriasis [105]. Aberrant expression of these neuropeptides is especially important for pruritus in psoriasis, which is present in 60–90% of patients with psoriasis [106]. There is a significant correlation between the number of SP-positive nerve fibers and NK-2 receptor-immunoreactive cells in the psoriatic skin lesion and intensity of pruritus [107]. Psoriasis patients with pruritus also showed higher expression of receptors for SP and CGRP compared to non-pruritic patients, while the immunoreactivity of SP, CGRP, VIP, and PACAP did not show significant difference [108]. In addition, the expression of NGF and its receptors is upregulated in pruritic lesions of psoriasis skin and correlated with the intensity of pruritus [108, 109]. NGF plays a role in modulating nerve innervation and neuropeptide release. It is mitogenic to endothelial cells, activates lymphocytes, degranulates mast cells, and induces keratinocyte hyperproliferation, all of which constitute the development of psoriasis [110, 111]. On the contrary, semaphorin-3A, which inhibits neuronal outgrowth of sensory C fibers, is downregulated in the dermis of pruritic psoriasis skin lesion and negatively correlated with pruritus [112, 113]. Thereby, upregulated NGF with downregulated semaphorin A might contribute to the hyperinnervation of sensory C fibers in psoriatic lesion which clinically induces pruritus.
The clinical trials of botulinum toxin A administration to treat psoriasis by inhibiting neuropeptide release has been reported in a few studies. Zanchi et al. reported significant efficacy of botulinum toxin A injection in the patients of inverse psoriasis, a variant of psoriasis which affects the intertriginous area [114]. The patients showed favorable clinical improvement although it is possible that the observed improvement is due to reduced sweating and maceration in the flexural area due to the anti-hydrotic effect of botulinum toxin and not from the inhibition of neurogenic inflammation. A study using murine model of plaque psoriasis showed marked reduction of acanthosis and lymphocyte infiltration after botulinum toxin A injection [115]. However, recent clinical trials of botulinum toxin A injection on the patients with plaque psoriasis did not show significant efficacy compared with control [116].
Neurogenic inflammation in atopic dermatitis
AD is a chronic relapsing inflammatory skin disease which is characterized by skin barrier disruption and immunological alteration. Clinically, it manifests as eczematous skin eruptions with severe pruritus with continued flares and remission in chronic course. AD most frequently occurs in infancy or childhood with 10–20% prevalence worldwide, which decreases to 2–3% in adulthood [117]. Although the etiology of AD is not fully elucidated, it is considered a multifactorial disorder with genetic and environmental background. However, one of the key histological findings of AD is the excessive density of cutaneous sensory nerve fibers in skin lesion, which implies the role of innervation and neuropeptides in the pathogenesis of AD [118, 119]. The skin of AD lesion is hyper-innervated with increased SP- and CGRP-positive nerve fibers in the epidermis and papillary dermis with increased mast cell-nerve fiber contacts, compared to the non-lesional skin [120,121,122]. NGF and its receptor are highly upregulated in the keratinocytes of AD patients compared with healthy keratinocytes, which contribute to neurite overgrowth and the increased proportion of CGRP-positive neurite length [119, 123]. NGF levels are also increased in plasma of AD patients and correlate with clinical severity and eosinophil counts [124]. In the NC/Nga AD mouse model, the topical high-affinity NGF receptor inhibitors improved clinical symptoms and decreased the epidermal density of the nerve fibers [125]. In addition to NGF, neurotrophin-4 production is increased in the epidermis of AD lesion [126] and the brain-derived neurotrophic factor (BDNF) level is also elevated in plasma and eosinophils from AD patients, which is chemotactic for eosinophils [125]. On the other hand, the production of semaphorin 3A, the epidermal axon repulsion factor, is decreased in atopic keratinocytes, which consequently contributes to the hyper-innervation in AD skin together with increased neurotrophins [127]. The alteration of epidermal Sema3A and NGF levels with the modulation of epidermal innervation was demonstrated after phototherapy in AD patients [118]. Nerve fiber sprouting has also been observed in the skin lesions of patients with nummular eczema and allergic contact eczema [120, 128]. The plasma levels of neuropeptide SP are increased in AD patients, and remain elevated even after AD remission [129]. The increased level of SP, in AD, induces the release of IFN-γ, IL-4, TNF-α, and IL-10 from the peripheral blood mononuclear leukocytes [130, 131].
The plasma levels of CGRP are not elevated in AD patients although they are significantly higher in AD patients with intense pruritus compared to the AD patients without pruritus [129]. CGRP upregulates IL-13 and human leukocyte antigen (HLA)-DR expression in circulating cutaneous lymphocyte-associated antigen (CLA)-positive T cells in AD patients, which does not in healthy controls [132]. CGRP also increases the IL-13/IFN-γ ratio after culture, which supports its immunomodulatory ability in AD [132].
In a mouse model of AD, stress deteriorated AD symptoms with increased neurogenic inflammation presented by mast cell degranulation, interstitial neuropeptidergic dense core granules, mast cell apoptosis, and endothelial gapping [133]. However, in mice lacking the NK-1 SP receptor, AD worsening was not observed, underlining the importance of NK-1 receptors on the sensorial terminations. Interestingly, the total CD4+ cell number was not changed by stress but the cytokine profile shifted toward Th2 in the skin, which is allergy-relevant. Taken together, stress exacerbates AD via SP-dependent neurogenic inflammation and subsequent shifting of local cytokine milieu toward Th2 [133]. In accordance with these findings, SP-induced scratch behavior in mice is mediated by NK-1 receptor activation [134, 135]. The administration of NK-1 receptor antagonist BIIF 1139 CL decreased scratching behavior in mouse models [136]. Aprepitant (Emend™), a selective high-affinity NK-1 receptor antagonist which was originally developed for the prevention of chemotherapy-induced emesis, significantly improved pruritus in patients with chronic pruritus including AD [33, 137, 138]. A mouse model of AD showed that systemic aprepitant administration decreased both the serum IgE levels and the density of SP-positive nerve fibers in lesional skin [139, 140]. Thus, pharmacologic interference of SP-mediated neurogenic inflammation can be a promising alternative therapeutic target in the treatment of recalcitrant AD.
Neurogenic inflammation in prurigo nodularis
Prurigo nodularis (PN) is a chronic skin condition characterized by intensely pruritic lichenified or excoriated papules or nodules. It is considered as a localized form of chronic dermatitis representing a cutaneous reaction pattern to repetitive scratching or rubbing due to pruritus. Many patients of PN have a personal or family background of atopic dermatitis and elevated serum immunoglobulin E (IgE) level. Systemic diseases which potentially cause pruritus such as uremia and other pruritic skin conditions including insect bites and scabies can also trigger PN [141]. The histology of PN frequently shows neural hyperplasia in dermal nerves as well as hyperkeratosis, irregular acanthosis, fibrosis of papillary dermis with vertically arranged collagen fibers, and non-specific inflammatory cell infiltration [142]. It is increasingly accepted that such neural proliferation and neurogenic inflammation play an important role in initiating and maintaining chronic pruritus possibly leading to PN, although its exact pathogenesis is not fully elucidated.
Previous studies about PN showed that NGF and CGRP are main mediators implicated in these processes [143, 144]. An electron microscopy study demonstrated that CGRP-immunoreactive nerve fibers were increased in number in the dermis of PN lesions and were co-localized with mast cells and eosinophils which were also increased in PN compared to normal skin. On the contrary, in the area without nerve fibers, there was neither eosinophil nor mast cells [144]. This indicates the involvement of a close interaction between the neuropeptide CGRP and cutaneous immune cells such as mast cells or eosinophils in the pathogenesis of PN. CGRP is an essential mediator of vasodilation in the skin except for the adrenergic and cholinergic neurotransmitters, which may contribute to vasodilation observed in PN. CGRP can activate mast cells directly through CGRP receptors on the mast cell surface, which may lead to the bidirectional positive feedback loop between nerve fibers and mast cells [20]. CGRP, together with SP, increases eosinophil chemotaxis, activation, and survival [43]. Meanwhile, eosinophils can produce NGF themselves. NGF, which is primarily a neurotrophic factor, also has a proinflammatory effect directly or indirectly, by enhancing neuropeptide release. NGF, in turn, can activate eosinophils to release proinflammatory mediators. NGF is also associated with TRK1 activation resulting in increased TRPV1 expression on nerve fibers and subsequent release of SP and CGRP, thereby establishing a vicious cytokine “pro-itch” cycle [43]. This is supported by a immunohistochemistry study that shows that NGF- and tyrosine kinase A (trkA)-immunoreactive cells are increased in the dermis of PN lesion [143]. However, like the CGRP-immunoreactive nerve fibers, these cells are observed in the dermis, not in the epidermis. Although the main source of cutaneous NGF is keratinocytes, it is assumed that NGF-producing dermal cells, such as mast cells, eosinophils, and lymphocytes, can be the source of increased NGF in PN [143].
Future challenges in skin neurogenic inflammation
Much time has passed since the term “neurodermatitis” was first coined in 1876. Many phenomena have since been described in great detail, and the term “stress,” when applied to skin inflammation, has been translated into molecular pathways and is not anymore just a psychoanalytic definition. Now we know that the epidermis closely interacts with nerve endings and that both epidermis and nerves produce substances for mutual sustenance. Neuropeptides, like SP and CGRP, are produced by sensory nerves in the dermis; they induce mast cells to release vasoactive amines that facilitate infiltration of neutrophils and T cells. We know that some receptors are more important than others in the generation of itch. Mrgprs as well as TRPA1 and Par-2 [37,38,39,40,41] have important roles in itch and inflammation. The activation of MrgprX1 degranulates mast cells to communicate with sensory nerve and cutaneous cells for developing neurogenic inflammation [54]. Mostly importantly, we now know that Mrgprs and TRPV4 are crucial in rosacea [145], while SP, CGRP, somatostatin, β-endorphin, VIP, and PACAP can modulate the immune system during psoriasis development [101] and the increased level of SP, in AD, induces the release of IFN-γ, IL-4, TNF-α, and IL-10 from the peripheral blood mononuclear leukocytes [130, 131].
We are finally starting to understand the intricate connections between the different skin cell types while new challenges are rising. The borders of our skin are no longer marked by the limits of the epidermis but extended to communities of bacteria that live in symbiosis with us. Our microbiome can influence nerve endings, epidermis reactivity [146], and even the maturation of cells that are essential to pruritus such as mast cells [147, 148].
The essential role that the peripheral nerve system plays in shaping skin inflammation suggests that many skin diseases reflect an imbalance between the function of the epidermis, dermis, and the sensory nerves. An abnormal skin microbiome, along with the presence of pathogens, will likely add an additional layer of complexity. Continued research studies are required to better understand these most recent complex clinical interactions.
References
Huynh M, Gupta R, Koo JY (2013) Emotional stress as a trigger for inflammatory skin disorders. Semin Cutan Med Surg 32(2):68–72
Chen Y, Lyga J (2014) Brain-skin connection: stress, inflammation and skin aging. Inflamm Allergy Drug Targets 13(3):177–190
Stricker (1876) Mikroskopische Studien iiber Wachstum und Wechsel der Haai'e,. In: Ebner V (ed) manual of human embriology, vol xxiv. Sitz. Her. d. K. Akad. d. Wiss., Wien,
Bayliss WM, Starling EH (1901) The movements and innervation of the small intestine. J Physiol 26(3–4):125–138. https://doi.org/10.1113/jphysiol.1901.sp000827
Peters EM (2012) The neuroendocrine-immune connection regulates chronic inflammatory disease in allergy. Chem Immunol Allergy 98:240–252. https://doi.org/10.1159/000336527
Chiu IM, von Hehn CA, Woolf CJ (2012) Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci 15(8):1063–1067. https://doi.org/10.1038/nn.3144
Szolcsanyi J (1996) Capsaicin-sensitive sensory nerve terminals with local and systemic efferent functions: facts and scopes of an unorthodox neuroregulatory mechanism. Prog Brain Res 113:343–359
Liezmann C, Klapp B, Peters EM (2011) Stress, atopy and allergy: a re-evaluation from a psychoneuroimmunologic persepective. Derm Endocrinol 3(1):37–40. https://doi.org/10.4161/derm.3.1.14618
Peters EM, Ericson ME, Hosoi J, Seiffert K, Hordinsky MK, Ansel JC, Paus R, Scholzen TE (2006) Neuropeptide control mechanisms in cutaneous biology: physiological and clinical significance. J Investig Dermatol 126(9):1937–1947. https://doi.org/10.1038/sj.jid.5700429
Botchkarev VA, Yaar M, Peters EM, Raychaudhuri SP, Botchkareva NV, Marconi A, Raychaudhuri SK, Paus R, Pincelli C (2006) Neurotrophins in skin biology and pathology. J Investig Dermatol 126(8):1719–1727. https://doi.org/10.1038/sj.jid.5700270
Roosterman D, Goerge T, Schneider SW, Bunnett NW, Steinhoff M (2006) Neuronal control of skin function: the skin as a neuroimmunoendocrine organ. Physiol Rev 86(4):1309–1379. https://doi.org/10.1152/physrev.00026.2005
Cevikbas F, Steinhoff A, Homey B, Steinhoff M (2007) Neuroimmune interactions in allergic skin diseases. Curr Opin Allergy Clin Immunol 7(5):365–373. https://doi.org/10.1097/ACI.0b013e3282a644d2
Park YM, Kim CW The effects of substance P and vasoactive intestinal peptide on interleukin-6 synthesis in cultured human keratinocytes. J Dermatol Sci 22(1):17–23. https://doi.org/10.1016/S0923-1811(99)00038-9
Song IS, Bunnett NW, Olerud JE, Harten B, Steinhoff M, Brown JR, Sung KJ, Armstrong CA, Ansel JC (2000) Substance P induction of murine keratinocyte PAM 212 interleukin 1 production is mediated by the neurokinin 2 receptor (NK-2R). Exp Dermatol 9(1):42–52
Burbach GJ, Kim KH, Zivony AS, Kim A, Aranda J, Wright S, Naik SM, Caughman SW, Ansel JC, Armstrong CA The neurosensory tachykinins substance P and neurokinin A directly induce keratinocyte nerve growth factor. J Investig Dermatol 117(5):1075–1082. https://doi.org/10.1046/j.0022-202x.2001.01498.x
Dallos A, Kiss M, Polyánka H, Dobozy A, Kemény L, Husz S Effects of the neuropeptides substance P, calcitonin gene-related peptide, vasoactive intestinal polypeptide and galanin on the production of nerve growth factor and inflammatory cytokines in cultured human keratinocytes. Neuropeptides 40(4):251–263. https://doi.org/10.1016/j.npep.2006.06.002
Nakano Y (2004) Stress-induced modulation of skin immune function: two types of antigen-presenting cells in the epidermis are differentially regulated by chronic stress. Br J Dermatol 151(1):50–64. https://doi.org/10.1111/j.1365-2133.2004.05980.x
Beresford L, Orange O, Bell EB, Miyan JA (2004) Nerve fibres are required to evoke a contact sensitivity response in mice. Immunology 111(1):118–125
Joachim RA, Handjiski B, Blois SM, Hagen E, Paus R, Arck PC Stress-induced neurogenic inflammation in murine skin skews dendritic cells towards maturation and migration. Am J Pathol 173(5):1379–1388. https://doi.org/10.2353/ajpath.2008.080105
Rosa AC, Fantozzi R (2013) The role of histamine in neurogenic inflammation. Br J Pharmacol 170(1):38–45. https://doi.org/10.1111/bph.12266
Liu JY, Hu JH, Zhu QG, Li FQ, Sun HJ (2006) Substance P receptor expression in human skin keratinocytes and fibroblasts. Br J Dermatol 155(4):657–662. https://doi.org/10.1111/j.1365-2133.2006.07408.x
Bae SJ, Matsunaga Y, Takenaka M, Tanaka Y, Hamazaki Y, Shimizu K, Katayama I (2002) Substance P induced preprotachykinin-a mRNA, neutral endopeptidase mRNA and substance P in cultured normal fibroblasts. Int Arch Allergy Immunol 127(4):316–321 57749
Brain SD, Williams TJ (1989) Interactions between the tachykinins and calcitonin gene-related peptide lead to the modulation of oedema formation and blood flow in rat skin. Br J Pharmacol 97(1):77–82
Saria A (1984) Substance P in sensory nerve fibres contributes to the development of oedema in the rat hind paw after thermal injury. Br J Pharmacol 82(1):217–222
Lindsey KQ, Caughman SW, Olerud JE, Bunnett NW, Armstrong CA, Ansel JC (2000) Neural regulation of endothelial cell-mediated inflammation. J Investig Dermatol Symp Proc 5(1):74–78. https://doi.org/10.1046/j.1087-0024.2000.00013.x
Castellani ML, Galzio RJ, Felaco P, Tripodi D, Toniato E, De Lutiis MA, Conti F, Fulcheri M, Conti C, Theoharides TC, Caraffa A, Antinolfi P, Felaco M, Tete S, Pandolfi F, Shaik-Dasthagirisaheb YB (2010) VEGF, substance P and stress, new aspects: a revisited study. J Biol Regul Homeost Agents 24(3):229–237
Kohara H, Tajima S, Yamamoto M, Tabata Y (2010) Angiogenesis induced by controlled release of neuropeptide substance P. Biomaterials 31(33):8617–8625. https://doi.org/10.1016/j.biomaterials.2010.07.079
Mishima T, Ito Y, Hosono K, Tamura Y, Uchida Y, Hirata M, Suzsuki T, Amano H, Kato S, Kurihara Y, Kurihara H, Hayashi I, Watanabe M, Majima M (2011) Calcitonin gene-related peptide facilitates revascularization during hindlimb ischemia in mice. Am J Phys Heart Circ Phys 300(2):H431–H439. https://doi.org/10.1152/ajpheart.00466.2010
Zhou Z, Hu CP, Wang CJ, Li TT, Peng J, Li YJ (2010) Calcitonin gene-related peptide inhibits angiotensin II-induced endothelial progenitor cells senescence through up-regulation of klotho expression. Atherosclerosis 213(1):92–101. https://doi.org/10.1016/j.atherosclerosis.2010.08.050
Ding W, Stohl LL, Wagner JA, Granstein RD (2008) Calcitonin gene-related peptide biases Langerhans cells toward Th2-type immunity. J Immunol (Baltimore, Md : 1950) 181(9):6020–6026
McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM (1998) RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393(6683):333–339. https://doi.org/10.1038/30666
Garret C, Carruette A, Fardin V, Moussaoui S, Peyronel JF, Blanchard JC, Laduron PM (1991) Pharmacological properties of a potent and selective nonpeptide substance P antagonist. Proc Natl Acad Sci U S A 88(22):10208–10212
Stander S, Siepmann D, Herrgott I, Sunderkotter C, Luger TA (2010) Targeting the neurokinin receptor 1 with aprepitant: a novel antipruritic strategy. PLoS One 5(6):e10968. https://doi.org/10.1371/journal.pone.0010968
Wrobel Goldberg S, Silberstein SD (2015) Targeting CGRP: a new era for migraine treatment. CNS drugs 29(6):443–452. https://doi.org/10.1007/s40263-015-0253-z
Ding W, Manni M, Stohl LL, Zhou XK, Wagner JA, Granstein RD (2012) Pituitary adenylate cyclase-activating peptide and vasoactive intestinal polypeptide bias Langerhans cell Ag presentation toward Th17 cells. Eur J Immunol 42(4):901–911. https://doi.org/10.1002/eji.201141958
Jans R, Sartor M, Jadot M, Poumay Y (2004) Calcium entry into keratinocytes induces exocytosis of lysosomes. Arch Dermatol Res 296(1):30–41. https://doi.org/10.1007/s00403-004-0469-0
Zhao P, Metcalf M, Bunnett NW (2014) Biased signaling of protease-activated receptors. Front Endocrinol 5:67. https://doi.org/10.3389/fendo.2014.00067
Chen Y, Yang C, Wang ZJ (2011) Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience 193:440–451. https://doi.org/10.1016/j.neuroscience.2011.06.085
Steinhoff M, Neisius U, Ikoma A, Fartasch M, Heyer G, Skov PS, Luger TA, Schmelz M (2003) Proteinase-activated receptor-2 mediates itch: a novel pathway for pruritus in human skin. J Neurosci Off J Soc Neurosci 23(15):6176–6180
Fu Q, Cheng J, Gao Y, Zhang Y, Chen X, Xie J (2015) Protease-activated receptor 4: a critical participator in inflammatory response. Inflammation 38(2):886–895. https://doi.org/10.1007/s10753-014-9999-6
Cocks TM, Moffatt JD (2000) Protease-activated receptors: sentries for inflammation? Trends Pharmacol Sci 21(3):103–108
Gouin O, Lebonvallet N, L'Herondelle K, Le Gall-Ianotto C, Buhe V, Plee-Gautier E, Carre JL, Lefeuvre L, Misery L (2015) Self-maintenance of neurogenic inflammation contributes to a vicious cycle in skin. Exp Dermatol 24(10):723–726. https://doi.org/10.1111/exd.12798
Mollanazar NK, Smith PK, Yosipovitch G (2016) Mediators of chronic pruritus in atopic dermatitis: getting the itch out? Clin Rev Allergy Immunol 51(3):263–292. https://doi.org/10.1007/s12016-015-8488-5
Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, Dubus P, Hovnanian A (2009) Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med 206(5):1135–1147. https://doi.org/10.1084/jem.20082242
Tourdot BE, Conaway S, Niisuke K, Edelstein LC, Bray PF, Holinstat M (2014) Mechanism of race-dependent platelet activation through the protease-activated receptor-4 and Gq signaling axis. Arterioscler Thromb Vasc Biol 34(12):2644–2650. https://doi.org/10.1161/atvbaha.114.304249
Vellani V, Kinsey AM, Prandini M, Hechtfischer SC, Reeh P, Magherini PC, Giacomoni C, McNaughton PA (2010) Protease activated receptors 1 and 4 sensitize TRPV1 in nociceptive neurones. Mol Pain 6:61. https://doi.org/10.1186/1744-8069-6-61
Karanjia R, Spreadbury I, Bautista-Cruz F, Tsang ME, Vanner S (2009) Activation of protease-activated receptor-4 inhibits the intrinsic excitability of colonic dorsal root ganglia neurons. Neurogastroenterol Motil 21(11):1218–1221. https://doi.org/10.1111/j.1365-2982.2009.01353.x
Asfaha S, Cenac N, Houle S, Altier C, Papez MD, Nguyen C, Steinhoff M, Chapman K, Zamponi GW, Vergnolle N (2007) Protease-activated receptor-4: a novel mechanism of inflammatory pain modulation. Br J Pharmacol 150(2):176–185. https://doi.org/10.1038/sj.bjp.0706975
Houle S, Papez MD, Ferazzini M, Hollenberg MD, Vergnolle N (2005) Neutrophils and the kallikrein-kinin system in proteinase-activated receptor 4-mediated inflammation in rodents. Br J Pharmacol 146(5):670–678. https://doi.org/10.1038/sj.bjp.0706371
Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng HJ, Geng Y, Undem BJ, Kollarik M, Chen ZF, Anderson DJ, Dong X (2009) Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell 139(7):1353–1365. https://doi.org/10.1016/j.cell.2009.11.034
Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, Bautista DM (2011) TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor-mediated itch. Nat Neurosci 14(5):595–602. https://doi.org/10.1038/nn.2789
Solinski HJ, Gudermann T, Breit A (2014) Pharmacology and signaling of MAS-related G protein-coupled receptors. Pharmacol Rev 66(3):570–597. https://doi.org/10.1124/pr.113.008425
Bader M, Alenina N, Andrade-Navarro MA, Santos RA (2014) MAS and its related G protein-coupled receptors, Mrgprs. Pharmacol Rev 66(4):1080–1105. https://doi.org/10.1124/pr.113.008136
Solinski HJ, Petermann F, Rothe K, Boekhoff I, Gudermann T, Breit A (2013) Human Mas-related G protein-coupled receptors-X1 induce chemokine receptor 2 expression in rat dorsal root ganglia neurons and release of chemokine ligand 2 from the human LAD-2 mast cell line. PLoS One 8(3):e58756. https://doi.org/10.1371/journal.pone.0058756
Boillat A, Alijevic O, Kellenberger S (2014) Calcium entry via TRPV1 but not ASICs induces neuropeptide release from sensory neurons. Mol Cell Neurosci 61:13–22. https://doi.org/10.1016/j.mcn.2014.04.007
Stander S, Moormann C, Schumacher M, Buddenkotte J, Artuc M, Shpacovitch V, Brzoska T, Lippert U, Henz BM, Luger TA, Metze D, Steinhoff M (2004) Expression of vanilloid receptor subtype 1 in cutaneous sensory nerve fibers, mast cells, and epithelial cells of appendage structures. Exp Dermatol 13(3):129–139. https://doi.org/10.1111/j.0906-6705.2004.0178.x
Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112(6):819–829
Hojland CR, Andersen HH, Poulsen JN, Arendt-Nielsen L, Gazerani P (2015) A human surrogate model of itch utilizing the TRPA1 agonist trans-cinnamaldehyde. Acta Derm Venereol 95(7):798–803. https://doi.org/10.2340/00015555-2103
Liang J, Ji Q, Ji W (2011) Role of transient receptor potential ankyrin subfamily member 1 in pruritus induced by endothelin-1. Neurosci Lett 492(3):175–178. https://doi.org/10.1016/j.neulet.2011.02.009
Liu T, Xu ZZ, Park CK, Berta T, Ji RR (2010) Toll-like receptor 7 mediates pruritus. Nat Neurosci 13(12):1460–1462. https://doi.org/10.1038/nn.2683
Kim SJ, Park GH, Kim D, Lee J, Min H, Wall E, Lee CJ, Simon MI, Lee SJ, Han SK (2011) Analysis of cellular and behavioral responses to imiquimod reveals a unique itch pathway in transient receptor potential vanilloid 1 (TRPV1)-expressing neurons. Proc Natl Acad Sci U S A 108(8):3371–3376. https://doi.org/10.1073/pnas.1019755108
Yun JW, Seo JA, Jeong YS, Bae IH, Jang WH, Lee J, Kim SY, Shin SS, Woo BY, Lee KW, Lim KM, Park YH (2011) TRPV1 antagonist can suppress the atopic dermatitis-like symptoms by accelerating skin barrier recovery. J Dermatol Sci 62(1):8–15. https://doi.org/10.1016/j.jdermsci.2010.10.014
Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H, Noguchi K (2007) Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest 117(7):1979–1987. https://doi.org/10.1172/jci30951
Malin S, Molliver D, Christianson JA, Schwartz ES, Cornuet P, Albers KM, Davis BM (2011) TRPV1 and TRPA1 function and modulation are target tissue dependent. J Neurosci 31(29):10516–10528. https://doi.org/10.1523/jneurosci.2992-10.2011
Wang S, Dai Y, Fukuoka T, Yamanaka H, Kobayashi K, Obata K, Cui X, Tominaga M, Noguchi K (2008) Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain 131(5):1241–1251. https://doi.org/10.1093/brain/awn060
Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, Pellegrino M, Estandian DM, Bautista DM (2013) The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 155(2):285–295. https://doi.org/10.1016/j.cell.2013.08.057
Oh MH, Oh SY, Lu J, Lou H, Myers AC, Zhu Z, Zheng T (2013) TRPA1-dependent pruritus in IL-13-induced chronic atopic dermatitis. J Immunol (Baltimore, Md : 1950) 191(11):5371–5382. https://doi.org/10.4049/jimmunol.1300300
Bautista DM, Pellegrino M, Tsunozaki M (2013) TRPA1: a gatekeeper for inflammation. Annu Rev Physiol 75:181–200. https://doi.org/10.1146/annurev-physiol-030212-183811
Gouin O, L'Herondelle K, Lebonvallet N, Le Gall-Ianotto C, Sakka M, Buhe V, Plee-Gautier E, Carre JL, Lefeuvre L, Misery L, Le Garrec R (2017) TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: pro-inflammatory response induced by their activation and their sensitization. Protein Cell. https://doi.org/10.1007/s13238-017-0395-5
Kodji X, Aubdool AA, Brain SD (2016) Evidence for physiological and pathological roles for sensory nerves in the microvasculature and skin. Curr Res Transl Med 64(4):195–201. https://doi.org/10.1016/j.retram.2016.09.002
Two AM, Wu W, Gallo RL, Hata TR (2015) Rosacea: part I. Introduction, categorization, histology, pathogenesis, and risk factors. J Am Acad Dermatol 72(5):749–758; quiz 759-760. https://doi.org/10.1016/j.jaad.2014.08.028
Guzman-Sanchez DA, Ishiuji Y, Patel T, Fountain J, Chan YH, Yosipovitch G (2007) Enhanced skin blood flow and sensitivity to noxious heat stimuli in papulopustular rosacea. J Am Acad Dermatol 57(5):800–805. https://doi.org/10.1016/j.jaad.2007.06.009
Schwab VD, Sulk M, Seeliger S, Nowak P, Aubert J, Mess C, Rivier M, Carlavan I, Rossio P, Metze D, Buddenkotte J, Cevikbas F, Voegel JJ, Steinhoff M (2011) Neurovascular and neuroimmune aspects in the pathophysiology of rosacea. J Investig Dermatol Symp Proc 15(1):53–62. https://doi.org/10.1038/jidsymp.2011.6
Sulk M, Seeliger S, Aubert J, Schwab VD, Cevikbas F, Rivier M, Nowak P, Voegel JJ, Buddenkotte J, Steinhoff M (2012) Distribution and expression of non-neuronal transient receptor potential (TRPV) ion channels in rosacea. J Investig Dermatol 132(4):1253–1262. https://doi.org/10.1038/jid.2011.424
Gerber PA, Buhren BA, Steinhoff M, Homey B (2011) Rosacea: the cytokine and chemokine network. J Investig Dermatol Symp Proc 15(1):40–47. https://doi.org/10.1038/jidsymp.2011.9
Ni Raghallaigh S, Powell FC (2014) Epidermal hydration levels in patients with rosacea improve after minocycline therapy. Br J Dermatol 171(2):259–266. https://doi.org/10.1111/bjd.12770
Hachem JP, Houben E, Crumrine D, Man MQ, Schurer N, Roelandt T, Choi EH, Uchida Y, Brown BE, Feingold KR, Elias PM (2006) Serine protease signaling of epidermal permeability barrier homeostasis. J Investig Dermatol 126(9):2074–2086. https://doi.org/10.1038/sj.jid.5700351
Spoendlin J, Voegel JJ, Jick SS, Meier CR (2013) Risk of rosacea in patients with diabetes using insulin or oral antidiabetic drugs. J Investig Dermatol 133(12):2790–2793. https://doi.org/10.1038/jid.2013.225
Pozsgai G, Bodkin JV, Graepel R, Bevan S, Andersson DA, Brain SD (2010) Evidence for the pathophysiological relevance of TRPA1 receptors in the cardiovascular system in vivo. Cardiovasc Res 87(4):760–768. https://doi.org/10.1093/cvr/cvq118
Graepel R, Fernandes ES, Aubdool AA, Andersson DA, Bevan S, Brain SD (2011) 4-oxo-2-nonenal (4-ONE): evidence of transient receptor potential ankyrin 1-dependent and -independent nociceptive and vasoactive responses in vivo. J Pharmacol Exp Ther 337(1):117–124. https://doi.org/10.1124/jpet.110.172403
Baylie RL, Brayden JE (2011) TRPV channels and vascular function. Acta Physiol (Oxford, England) 203(1):99–116. https://doi.org/10.1111/j.1748-1716.2010.02217.x
Helfrich YR, Maier LE, Cui Y, Fisher GJ, Chubb H, Fligiel S, Sachs D, Varani J, Voorhees J (2015) Clinical, histologic, and molecular analysis of differences between erythematotelangiectatic rosacea and telangiectatic photoaging. JAMA Dermatol 151(8):825–836. https://doi.org/10.1001/jamadermatol.2014.4728
Greeno EW, Mantyh P, Vercellotti GM, Moldow CF (1993) Functional neurokinin 1 receptors for substance P are expressed by human vascular endothelium. J Exp Med 177(5):1269–1276
Seeliger S, Buddenkotte J, Schmidt-Choudhury A, Rosignoli C, Shpacovitch V, von Arnim U, Metze D, Rukwied R, Schmelz M, Paus R, Voegel JJ, Schmidt WE, Steinhoff M (2010) Pituitary adenylate cyclase activating polypeptide: an important vascular regulator in human skin in vivo. Am J Pathol 177(5):2563–2575. https://doi.org/10.2353/ajpath.2010.090941
Muto Y, Wang Z, Vanderberghe M, Two A, Gallo RL, Di Nardo A (2014) Mast cells are key mediators of cathelicidin-initiated skin inflammation in rosacea. J Investig Dermatol 134(11):2728–2736. https://doi.org/10.1038/jid.2014.222
Madva EN, Granstein RD (2013) Nerve-derived transmitters including peptides influence cutaneous immunology. Brain Behav Immun 34:1–10. https://doi.org/10.1016/j.bbi.2013.03.006
Shi X, Wang L, Li X, Sahbaie P, Kingery WS, Clark JD (2011) Neuropeptides contribute to peripheral nociceptive sensitization by regulating interleukin-1beta production in keratinocytes. Anesth Analg 113(1):175–183. https://doi.org/10.1213/ANE.0b013e31821a0258
Park KY, Hyun MY, Jeong SY, Kim BJ, Kim MN, Hong CK (2015) Botulinum toxin for the treatment of refractory erythema and flushing of rosacea. Dermatology (Basel, Switzerland) 230(4):299–301. https://doi.org/10.1159/000368773
Michalek IM, Loring B, John SM (2017) A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol JEADV 31(2):205–212. https://doi.org/10.1111/jdv.13854
Di Cesare A, Di Meglio P, Nestle FO (2009) The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Investig Dermatol 129(6):1339–1350. https://doi.org/10.1038/jid.2009.59
Veale D, Farrell M, Fitzgerald O (1993) Mechanism of joint sparing in a patient with unilateral psoriatic arthritis and a longstanding hemiplegia. Br J Rheumatol 32(5):413–416
Sowell JK, Pippenger MA, Crowe MJ (1993) Psoriasis contralateral to hemiparesis following cerebrovascular accident. Int J Dermatol 32(8):598–599
Sethi S, Sequeira W (1990) Sparing effect of hemiplegia on scleroderma. Ann Rheum Dis 49(12):999–1000
Joseph T, Kurian J, Warwick DJ, Friedmann PS (2005) Unilateral remission of psoriasis following traumatic nerve palsy. Br J Dermatol 152(1):185–186. https://doi.org/10.1111/j.1365-2133.2005.06330.x
Farber EM, Lanigan SW, Boer J (1990) The role of cutaneous sensory nerves in the maintenance of psoriasis. Int J Dermatol 29(6):418–420
Raychaudhuri SP, Farber EM (1993) Are sensory nerves essential for the development of psoriatic lesions? J Am Acad Dermatol 28(3):488–489
Dewing SB (1971) Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol 104(2):220–221
Reyter I, Woodley D (2004) Widespread unilateral plaques in a 68-year-old woman after neurosurgery. Arch Dermatol 140(12):1531–1536. https://doi.org/10.1001/archderm.140.12.1531-e
Chowdhury MM, Hedges R, Lanigan SW (2000) Unilateral resolution of palmar eczema and hyperhidrosis complicated by Horner's syndrome following ipsilateral endoscopic cervical sympathectomy. Br J Dermatol 143(3):653–654
Ostrowski SM, Belkadi A, Loyd CM, Diaconu D, Ward NL (2011) Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Investig Dermatol 131(7):1530–1538. https://doi.org/10.1038/jid.2011.60
Saraceno R, Kleyn CE, Terenghi G, Griffiths CE (2006) The role of neuropeptides in psoriasis. Br J Dermatol 155(5):876–882. https://doi.org/10.1111/j.1365-2133.2006.07518.x
Payan DG, Brewster DR, Goetzl EJ (1983) Specific stimulation of human T lymphocytes by substance P. J Immunol (Baltimore, Md : 1950) 131(4):1613–1615
Maggi CA (1997) The effects of tachykinins on inflammatory and immune cells. Regul Pept 70(2–3):75–90
O'Halloran DJ, Bloom SR (1991) Calcitonin gene related peptide. BMJ (Clinical research ed) 302(6779):739–740
Kakurai M, Fujita N, Murata S, Furukawa Y, Demitsu T, Nakagawa H (2001) Vasoactive intestinal peptide regulates its receptor expression and functions of human keratinocytes via type I vasoactive intestinal peptide receptors. J Investig Dermatol 116(5):743–749. https://doi.org/10.1046/j.0022-202x.2001.doc.x
Amatya B, Wennersten G, Nordlind K (2008) Patients’ perspective of pruritus in chronic plaque psoriasis: a questionnaire-based study. J Eur Acad Dermatol Venereol JEADV 22(7):822–826. https://doi.org/10.1111/j.1468-3083.2008.02591.x
Amatya B, El-Nour H, Holst M, Theodorsson E, Nordlind K (2011) Expression of tachykinins and their receptors in plaque psoriasis with pruritus. Br J Dermatol 164(5):1023–1029. https://doi.org/10.1111/j.1365-2133.2011.10241.x
Chang SE, Han SS, Jung HJ, Choi JH (2007) Neuropeptides and their receptors in psoriatic skin in relation to pruritus. Br J Dermatol 156(6):1272–1277. https://doi.org/10.1111/j.1365-2133.2007.07935.x
Nakamura M, Toyoda M, Morohashi M (2003) Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br J Dermatol 149(4):718–730
Raychaudhuri SK, Raychaudhuri SP, Weltman H, Farber EM (2001) Effect of nerve growth factor on endothelial cell biology: proliferation and adherence molecule expression on human dermal microvascular endothelial cells. Arch Dermatol Res 293(6):291–295
Bischoff SC, Dahinden CA (1992) Effect of nerve growth factor on the release of inflammatory mediators by mature human basophils. Blood 79(10):2662–2669
Taneda K, Tominaga M, Negi O, Tengara S, Kamo A, Ogawa H, Takamori K (2011) Evaluation of epidermal nerve density and opioid receptor levels in psoriatic itch. Br J Dermatol 165(2):277–284. https://doi.org/10.1111/j.1365-2133.2011.10347.x
Kou K, Nakamura F, Aihara M, Chen H, Seto K, Komori-Yamaguchi J, Kambara T, Nagashima Y, Goshima Y, Ikezawa Z (2012) Decreased expression of semaphorin-3A, a neurite-collapsing factor, is associated with itch in psoriatic skin. Acta Derm Venereol 92(5):521–528. https://doi.org/10.2340/00015555-1350
Zanchi M, Favot F, Bizzarini M, Piai M, Donini M, Sedona P (2008) Botulinum toxin type-A for the treatment of inverse psoriasis. J Eur Acad Dermatol Venereol JEADV 22(4):431–436. https://doi.org/10.1111/j.1468-3083.2007.02457.x
Ward NL, Kavlick KD, Diaconu D, Dawes SM, Michaels KA, Gilbert E (2012) Botulinum neurotoxin A decreases infiltrating cutaneous lymphocytes and improves acanthosis in the KC-Tie2 mouse model. J Investig Dermatol 132(7):1927–1930. https://doi.org/10.1038/jid.2012.60
Todberg T, Zachariae C, Bregnhoj A, Hedelund L, Bonefeld KK, Nielsen K, Iversen L, Skov L (2017) The effect of botulinum neurotoxin A in patients with plaque psoriasis—an exploratory trial. J Eur Acad Dermatol Venereol JEADV 32:e81–e82. https://doi.org/10.1111/jdv.14536
Leung DY, Bieber T (2003) Atopic dermatitis. Lancet (London, England) 361(9352):151–160. https://doi.org/10.1016/s0140-6736(03)12193-9
Tominaga M, Tengara S, Kamo A, Ogawa H, Takamori K (2009) Psoralen-ultraviolet A therapy alters epidermal Sema3A and NGF levels and modulates epidermal innervation in atopic dermatitis. J Dermatol Sci 55(1):40–46. https://doi.org/10.1016/j.jdermsci.2009.03.007
Dou YC, Hagstromer L, Emtestam L, Johansson O (2006) Increased nerve growth factor and its receptors in atopic dermatitis: an immunohistochemical study. Arch Dermatol Res 298(1):31–37. https://doi.org/10.1007/s00403-006-0657-1
Jarvikallio A, Harvima IT, Naukkarinen A (2003) Mast cells, nerves and neuropeptides in atopic dermatitis and nummular eczema. Arch Dermatol Res 295(1):2–7. https://doi.org/10.1007/s00403-002-0378-z
Tobin D, Nabarro G, Baart de la Faille H, van Vloten WA, van der Putte SC, Schuurman HJ (1992) Increased number of immunoreactive nerve fibers in atopic dermatitis. J Allergy Clin Immunol 90(4 Pt 1):613–622
Ostlere LS, Cowen T, Rustin MH (1995) Neuropeptides in the skin of patients with atopic dermatitis. Clin Exp Dermatol 20(6):462–467
Roggenkamp D, Falkner S, Stab F, Petersen M, Schmelz M, Neufang G (2012) Atopic keratinocytes induce increased neurite outgrowth in a coculture model of porcine dorsal root ganglia neurons and human skin cells. J Investig Dermatol 132(7):1892–1900. https://doi.org/10.1038/jid.2012.44
Yamaguchi J, Aihara M, Kobayashi Y, Kambara T, Ikezawa Z (2009) Quantitative analysis of nerve growth factor (NGF) in the atopic dermatitis and psoriasis horny layer and effect of treatment on NGF in atopic dermatitis. J Dermatol Sci 53(1):48–54. https://doi.org/10.1016/j.jdermsci.2008.08.011
Takano N, Sakurai T, Ohashi Y, Kurachi M (2007) Effects of high-affinity nerve growth factor receptor inhibitors on symptoms in the NC/Nga mouse atopic dermatitis model. Br J Dermatol 156(2):241–246. https://doi.org/10.1111/j.1365-2133.2006.07636.x
Grewe M, Vogelsang K, Ruzicka T, Stege H, Krutmann J (2000) Neurotrophin-4 production by human epidermal keratinocytes: increased expression in atopic dermatitis. J Investig Dermatol 114(6):1108–1112. https://doi.org/10.1046/j.1523-1747.2000.00974.x
Tominaga M, Ogawa H, Takamori K (2008) Decreased production of semaphorin 3A in the lesional skin of atopic dermatitis. Br J Dermatol 158(4):842–844. https://doi.org/10.1111/j.1365-2133.2007.08410.x
Kinkelin I, Motzing S, Koltenzenburg M, Brocker EB (2000) Increase in NGF content and nerve fiber sprouting in human allergic contact eczema. Cell Tissue Res 302(1):31–37
Salomon J, Baran E (2008) The role of selected neuropeptides in pathogenesis of atopic dermatitis. J Eur Acad Dermatol Venereol JEADV 22(2):223–228. https://doi.org/10.1111/j.1468-3083.2007.02399.x
Kim KH, Park KC, Chung JH, Choi HR (2003) The effect of substance P on peripheral blood mononuclear cells in patients with atopic dermatitis. J Dermatol Sci 32(2):115–124
Gordon DJ, Ostlere LS, Holden CA (1997) Neuropeptide modulation of Th1 and Th2 cytokines in peripheral blood mononuclear leucocytes in atopic dermatitis and non-atopic controls. Br J Dermatol 137(6):921–927
Antunez C, Torres MJ, Lopez S, Rodriguez-Pena R, Blanca M, Mayorga C, Santamaria-Babi LF (2009) Calcitonin gene-related peptide modulates interleukin-13 in circulating cutaneous lymphocyte-associated antigen-positive T cells in patients with atopic dermatitis. Br J Dermatol 161(3):547–553. https://doi.org/10.1111/j.1365-2133.2009.09318.x
Pavlovic S, Daniltchenko M, Tobin DJ, Hagen E, Hunt SP, Klapp BF, Arck PC, Peters EM (2008) Further exploring the brain-skin connection: stress worsens dermatitis via substance P-dependent neurogenic inflammation in mice. J Investig Dermatol 128(2):434–446. https://doi.org/10.1038/sj.jid.5701079
Scholzen T, Armstrong CA, Bunnett NW, Luger TA, Olerud JE, Ansel JC (1998) Neuropeptides in the skin: interactions between the neuroendocrine and the skin immune systems. Exp Dermatol 7(2–3):81–96
Andoh T, Nagasawa T, Satoh M, Kuraishi Y (1998) Substance P induction of itch-associated response mediated by cutaneous NK1 tachykinin receptors in mice. J Pharmacol Exp Ther 286(3):1140–1145
Ohmura T, Hayashi T, Satoh Y, Konomi A, Jung B, Satoh H (2004) Involvement of substance P in scratching behaviour in an atopic dermatitis model. Eur J Pharmacol 491(2–3):191–194. https://doi.org/10.1016/j.ejphar.2004.03.047
Dando TM, Perry CM (2004) Aprepitant: a review of its use in the prevention of chemotherapy-induced nausea and vomiting. Drugs 64(7):777–794
Duval A, Dubertret L (2009) Aprepitant as an antipruritic agent? N Engl J Med 361(14):1415–1416. https://doi.org/10.1056/NEJMc0906670
Lee JH, Cho SH (2011) Korean red ginseng extract ameliorates skin lesions in NC/Nga mice: an atopic dermatitis model. J Ethnopharmacol 133(2):810–817. https://doi.org/10.1016/j.jep.2010.11.020
Liu B, Escalera J, Balakrishna S, Fan L, Caceres AI, Robinson E, Sui A, McKay MC, McAlexander MA, Herrick CA, Jordt SE (2013) TRPA1 controls inflammation and pruritogen responses in allergic contact dermatitis. FASEB J 27(9):3549–3563. https://doi.org/10.1096/fj.13-229948
Doyle JA, Connolly SM, Hunziker N, Winkelmann RK (1979) Prurigo nodularis: a reappraisal of the clinical and histologic features. J Cutan Pathol 6(5):392–403
Cowan MA (1964) Neurohistological changes in prurigo nodularis. Arch Dermatol 89:754–758
Johansson O, Liang Y, Emtestam L (2002) Increased nerve growth factor- and tyrosine kinase A-like immunoreactivities in prurigo nodularis skin—an exploration of the cause of neurohyperplasia. Arch Dermatol Res 293(12):614–619. https://doi.org/10.1007/s00403-001-0285-8
Liang Y, Jacobi HH, Reimert CM, Haak-Frendscho M, Marcusson JA, Johansson O (2000) CGRP-immunoreactive nerves in prurigo nodularis—an exploration of neurogenic inflammation. J Cutan Pathol 27(7):359–366
Mascarenhas NL, Wang Z, Chang YL, Di Nardo A (2017) TRPV4 mediates mast cell activation in cathelicidin-induced rosacea inflammation. J Investig Dermatol 137(4):972–975. https://doi.org/10.1016/j.jid.2016.10.046
Williams MR, Gallo RL (2017) Evidence that human skin microbiome dysbiosis promotes atopic dermatitis. J Investig Dermatol 137(12):2460–2461. https://doi.org/10.1016/j.jid.2017.09.010
Wang Z, Mascarenhas N, Eckmann L, Miyamoto Y, Sun X, Kawakami T, Di Nardo A (2017) Skin microbiome promotes mast cell maturation by triggering stem cell factor production in keratinocytes. J Allergy Clin Immunol 139(4):1205–1216 e1206. https://doi.org/10.1016/j.jaci.2016.09.019
Igawa S, Di Nardo A (2017) Skin microbiome and mast cells. Transl Res 184(6):68–76
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Neurogenic Inflammation - Guest Editors: Tony Yaksh and Anna Di Nardo
Rights and permissions
About this article

Cite this article
Choi, J.E., Di Nardo, A. Skin neurogenic inflammation. Semin Immunopathol 40, 249–259 (2018). https://doi.org/10.1007/s00281-018-0675-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-018-0675-z