
Abstract
Bone marrow transplantation (BMT) serves as the only curative treatment for patients with β-thalassemia major; however, hemostatic changes have been observed in these BMT patients. Aggregability of thalassemic red blood cells (RBCs) and increased red blood cell-derived microparticles (RMPs) expressing phosphatidylserine (PS) are thought to participate in thromboembolic events by initially triggering platelet activation. To our knowledge, there has been no report providing quantitation of these circulating PS-expressing RBCs and RMPs in young β-thalassemia patients after BMT. Whole blood from each subject was fluorescently labeled to detect RBC markers (CD235a) and annexin-V together with the known number TruCount™ beads. PS-expressing RBCs, RMPs, and activated platelets were identified by flow cytometry. In our randomized study, we found the decreased levels of three aforementioned factors compared to levels in patients receiving regular blood transfusion (RT). This study showed that BMT in β-thalassemia patients decreases the levels of circulating PS-expressing RBCs, their MPs, and procoagulant platelets when compared to patients who received RT. Normalized levels of these coagulation markers may provide the supportive evidence of the effectiveness of BMT for curing thalassemia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
β-Thalassemia is a group of chronic hemolytic anemias caused by the absence or insufficient production of β-globin chains that form tetramers with α-globin chains. Patients with β-thalassemia major receive regular blood transfusions (RTs) to maintain their hemoglobin levels and mitigate anemic condition; however, this treatment is not curative [1, 2]. At present, bone marrow transplantation (BMT) serves as the only cure for patients with certain hematological diseases, including β-thalassemia major, homozygous β-thalassemia, and hemoglobin E/β-thalassemia. Young patients are offered treatment with BMT if matched donor stem cells are available [3,4,5].
Hemostatic changes and thrombotic events are frequently observed in patients undergoing BMT. As a consequence of abnormal hemoglobin synthesis in β-thalassemia, the α-globin chains precipitate and form the inclusion bodies within red blood cells (RBCs) causing oxidative stress and perturbing the normal asymmetric composition of the lipid membrane. Phosphatidylserine (PS), which is normally localized in the inner membrane leaflet, switches to the outer membrane and this can be detected by probing with annexin-V in a calcium-dependent manner [6,7,8,9]. PS-externalizing RBCs play an important role as inducers of a hypercoagulable state in thalassemia by triggering chronic platelet activation [9]. In addition, PS can be recognized and bound by coagulation factors, especially tissue factor and factor VII, facilitating further proteolysis, activation of factor X, and generation of thrombin [9]. These PS-expressing RBCs are reported to be increased in hemolytic anemias such as sickle cell disease [10, 11], paroxysmal nocturnal hemoglobinuria [12], and thalassemia [13].
Furthermore, microparticles (MPs) (smaller than 1 μm in diameter) shed from RBCs, called red blood cell-derived microparticles (RMPs), have been documented [14,15,16]. RMPs also bear PS on their surface and are described as the second most frequent of circulating MPs after platelet-derived MP [17]. In thalassemia, RMPs are elevated in patients who have undergone splenectomy compared those in non-splenectomized patients and might play a role in activating platelets [14, 18]. To our knowledge, RMP levels in patients post-BMT have never been studied. Here, we report the detection in whole blood of hypercoagulation parameters, including PS-exposing RBCs, RMPs, and procoagulant platelet levels, in young β-thalassemic patients who received RT or have undergone BMT.
Subjects, materials, and methods
Subjects
Blood samples were obtained from β-thalassemic patients prior to receiving regular blood transfusion and those following BMT in which engraftment was achieved as revealed by full chimeras of donor cells. The post-BMT duration ranged from 2 months to 2 years. All BMT patients received hematopoietic stem cells from donors who were normal and had no background of being heterozygotes or carriers of thalassemia except for one patient who received BMT from a haploidentical donor with thalassemia trait. Age- and sex-matched healthy volunteers were also recruited for this study. After giving written consent, blood samples were collected in K3EDTA and buffered sodium citrate tubes, carried on ice, and processed within 4 h. This study was approved by the Ethics Committee of the Faculty of Medicine Ramathibodi Hospital (MURA2014/553).
Complete blood count
K3EDTA blood samples were mixed and passed though hematological analyzer (Sysmex Xi-800, Kobe, Japan).
Whole blood staining
To detect PS-expressing RBCs and RMPs, 10 μl of citrate blood were diluted in phosphate-buffered saline (PBS) at 1:10 to reduce spontaneous platelet activation and shedding of platelet-derived microparticles. Then, 10 μl of diluted blood were added to Trucount™ tubes (BD Biosciences, San Jose) containing a known number of counting beads. Ninety microliters of a mixture of saturated monoclonal antibodies comprised of phycoerythrin (PE)-conjugated annexin-V and fluorescein isothiocyanate (FITC)-conjugated anti-CD235a monoclonal antibody (mAb) in 0.2-μm pre-filtered annexin-V binding buffer was added into Trucount tubes (all antibodies were from BD Biosciences), and samples were incubated for 15 min in the dark at room temperature. Three hundred microliters of filtered annexin-V binding buffer were added into each sample tube to bring the final volume to 400 μl.
For determination of activated platelets, 10 μl of diluted blood were incubated in another polystyrene tube with allophycocyanin (APC)-conjugated anti-human CD41a mAb and either PE-conjugated anti-human CD62P mAb or anti-human CD142 mAb. Ninety microliters of PBS were added and samples were incubated for 15 min in the dark at room temperature. Then, 1% paraformaldehyde was added to reach a final volume of 350 μl before flow cytometric analysis using flow cytometer FACS CantoII, (BD Biosciences). Isotype controls were also used.
FACS analysis
All parameters, including forward scatter (FSC), side scatter (SSC), and fluorescence detection channels, were set up in logarithmic scale. Polystyrene beads (Spherotech, Illinois) of 1-μm diameter were used to set the gate for the MP region. The annexin-V binding buffer was run first to identify the noise background. Next, the sample tubes were run and acquisition was stopped when 1500 events of Trucount beads were collected. RBCs that were probed with annexin-V were identified as PS-expressing RBCs, and annexin-V+ MPs bearing CD235a were called RMPs. The absolute numbers of PS-exposing RBCs were calculated from the percentage of PS-exposing RBCs multiplied by the RBC count, whereas the total numbers of RMPs were calculated from the formulae below. The flow cytometric gating strategies are shown in Fig. 1.

Representative flow cytometric gating strategies for identifying a PS-exposing RBCs, RMPs, and b activated platelets. RBC and RMP populations were identified by their CD235a+ and annexin-V+ expression using plots with logarithmic scales of SSC/CD235a and CD235a/annexin-V, respectively. RBCs exposing PS were detected by their annexin-V+ staining while RMPs were identified by their annexin-V+/CD235a+ expression. The population of platelets expressing activation marker and tissue factor were identified by the positivity to CD41a/CD62P or CD142 if used
The population of platelets expressing activation marker (CD62P) and tissue factor (CD142) was investigated by setting the gate around the platelet population, and then determining the percentage of platelets expressing CD62P and CD142 using the characteristic fluorescence profile of CD41a/CD62P or CD41a/CD142 in a bivariant plot.
Statistical analysis
Data were expressed as mean ± SEM. The differences between subject groups were determined by the non-parametric Kruskal-Wallis test and p values <0.05 were considered statistically significant.
Results
Hematologic characteristics
Table 1 shows the hematological parameters and significant differences among subject groups recruited in this cross-sectional study. The anemic conditions and other RBC indices typically found in thalassemic patients are reflected in the RT group. These findings were normalized after BMT. The one exception was the platelet counts, which were significantly lower in the BMT group than in the RT and normal groups.
Details of patients’ disease background, transplantation characteristics, and frequencies of complications are given in Supplementary Table 1. Of the BMT group, 12 were patients with β-thalassemia major while 37 had β-thalassemia/HbE.
PS-externalizing RBC levels
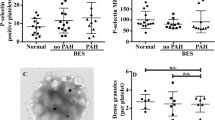
Patients who had BMT had significantly lower percentages (1.8 ± 0.5%; range 0.2–60.9%) and absolute counts (93,973 ± 25,444 cells/μl; range 10,440–869,960 cells/μl) of RBCs expressing PS (Fig. 2a, b) in comparison to normal individuals (0.2 ± 0.01%; range 0.1–0.5%; 9846 ± 1323 cells/μl; range 4030–24,050 cells/μl) and patients who received RT (8.4 ± 2.9%; range 0.2–60.9%; 395,524 ± 116,259 cells/μl; range 5340–2,362,920 cells/μl).

The percentages (a) and absolute counts (b) of PS-exposing RBCs in normal subjects, patients receiving regular transfusion (RT) or after bone marrow transplantation (BMT)
RMP levels
The percentages (17.7 ± 1.6%; range 1.1–43.9%) (Fig. 3a) and the absolute levels (4470 ± 488 events/μl; range 713–16,267 events/μl) (Fig. 3b) of RMPs in the BMT group, though not significant, tended to be less than in normal individuals (23.6 ± 2.3%; range 8.3–32.7%; 2506 ± 381 events/μl; range 380–5319 events/μl) and the RT group (28.1 ± 4.0%; range 0.9–62.2%; 6011 ± 1786 events/μl; range 299–25,298 events/μl).

The percentages (a) and absolute numbers (b) of circulating RMPs in normal subjects, patients receiving regular blood transfusion (RT) or after bone marrow transplantation (BMT)
Activated platelet levels
The percentages of procoagulant platelets in post-BMT thalassemia patients, defined by their expression of CD62P (12.8 ± 1.2%; range 2.6–42.63%) (Fig. 4a) and CD142 (0.3 ± 0.1%; range 0–2.3%) (Fig. 4b), were significantly less than in patients who received RT (CD62P: 17.9 ± 2.3%; range 3.4–40.1% and CD142: 3.3 ± 0.4%; range 0.1–7.1%), but did not differ significantly from percentages in normal individuals (CD62P: 7.3 ± 1.3%; range 1.1–21.6% and CD142: 0.6 ± 0.1%; range 0–1.3%).

The percentages of platelets expressing CD62P (P-selectin) (a) and CD142 (tissue factor) (b) in normal subjects, patients receiving regular blood transfusion (RT) or after bone marrow transplantation (BMT)
Discussion
Allogeneic BMT provides an opportunity for hematopoietic reconstitution in vivo and the correction of the thalassemic phenotype. BMT for patients with β-thalassemia was first successfully performed over three decades ago [19]. The best cell source for BMT is a human leukocyte antigen-identical donor, thereby reducing the transplantation-related complications, including graft-versus host effect or immune rejection, and increasing the rates of disease-free survival [20]. However, BMT patients still require packed RBC transfusions to control the anemic condition, which results from the lack of fully functional erythropoiesis. When these infused RBCs age or are damaged, they externalize PS (from the inner membrane leaflet to the outer side) and are destroyed by the reticuloendothelial system, limiting their lifespan [21,22,23]. In addition, they can shed membranous MPs which also bear PS and other markers of their parental cells. In this context, they carry glycophorin A (CD235a) from RBCs, and so provide a means to access them by multi-parametric flow cytometry. These damaged RBCs and RMPs along with abnormal but intact RBCs are thought to be one of the etiologies of the thrombotic risk experienced by thalassemia patients.
In the present study, we could reliably measure the levels of PS-expressing RBCs and circulating RMPs in young patients with β-thalassemia major by flow cytometric analysis. Flow cytometric quantification of circulating blood cells and their components is a reliable and simple tool offering new insight into the association of RBC abnormality and the hypercoagulable state which occurs in thalassemia [14, 18]. Blood samples used in our investigations were collected before RBC transfusion to avoid confounding due to the RMPs from stored packed RBCs [16]. In our randomized study, we observed the significantly decreased percentages and total counts of PS-externalizing RBCs, shed RMPs, and activated platelets in a post-BMT patient group. Their levels were normalized to those found in normal individuals and significantly lower than the RT patient groups. This suggests that transplantation may correct, in part, the prothrombotic phenotype in β-thalassemia by means of reducing procoagulant risk factors. This is in concordant with the report of Sirachainan et al. that performing BMT in children with severe β-thalassemia can correct the abnormal levels of coagulation markers and anti-coagulation factors toward normal baselines [24]. Tantawy et al. reported increased numbers of RMPs along with platelet-derived MPs in young thalassemia major patients, and positive correlations with vascular dysfunction, aortic stiffness, and pulmonary hypertension in those thalassemic patients [25]. The fact that RBC membrane vesiculates under oxidative stress conditions such as iron overload and the presence of excess unbound globin chains can explain why elevated PS exposure on RBCs and subsequent release of RMPs are found in β-thalassemic patients, particularly those received RT. Severely damaged RBCs are known to have an accumulation of unaffected globin chains in the cytoskeleton and a perturbation of lipid-protein membrane function, provoking the loss of RBC membrane asymmetry and the forming of MPs [6, 26, 27].
For the interpretation of PS-exposing RBC and RMP levels, looking at individual profile changes could be additionally helpful because each patient has different starting levels. In this study, there was no correlation between these two parameters and RBC counts. Therefore, the kinetic levels of RMPs and RBCs expressing PS cannot solely be explained by the different RBC counts. In addition, BMT patients also require platelet transfusion which implies that platelets are consumed in the coagulation activation resulting from endothelial cell damage [28], but platelet activation status has not been reported yet. In the present study, we demonstrated lower frequencies of platelets bearing P-selectin and tissue factor in patients after BMT. Regarding their role in the hypercoagulable state, we recently reported the prothrombotic potential of MPs on promoting platelet aggregation and neutrophil-platelet formation [29], which support the contention of further platelet activation by MPs. Furthermore, we have ruled out the possibility that different pre-conditioning regimens might have impacted on our determined parameters, since no significant differences were found among BMT patients who received various conditioning regimens. This is in line with other reports in which there is no significant effect of pre-conditioning regimens on coagulation profiles nor on MP amounts [30, 31].
In summary, our present data show that BMT effectively decreases the concentrations of procoagulant circulating PS-exposing RBCs, their MPs, and procoagulant platelets compared to treatment with usual blood transfusion. Detection of MPs with other origins could be useful, supplementing the RMPs, broadening the potential diagnostic biomarkers for BMT.
References
Fucharoen S, Winichagoon P (2002) Thalassemia and abnormal hemoglobin. Int J Hematol 76(Suppl 2):83–89
Fucharoen S, Winichagoon P (2011) Haemoglobinopathies in southeast Asia. Indian J Med Res 134:498–506
Hongeng S, Pakakasama S, Chaisiripoomkere W, Chuansumrit A, Sirachainan N, Ungkanont A et al (2004) Outcome of transplantation with unrelated donor bone marrow in children with severe thalassaemia. Bone Marrow Transplant 33(4):377–379
Anurathapan U, Hongeng S, Pakakasama S, Sirachainan N, Songdej D, Chuansumrit A et al (2016) Hematopoietic stem cell transplantation for homozygous beta-thalassemia and beta-thalassemia/hemoglobin E patients from haploidentical donors. Bone Marrow Transplant 51(6):813–818
Issaragrisil S, Kunacheewa C (2016) Matched sibling donor hematopoietic stem cell transplantation for thalassemia. Curr Opin Hematol 23(6):508–514
Rund D, Rachmilewitz E (2005) Beta-thalassemia. N Engl J Med 353(11):1135–1146
Freikman I, Amer J, Cohen JS, Ringel I, Fibach E (2008) Oxidative stress causes membrane phospholipid rearrangement and shedding from RBC membranes—an NMR study. Biochim Biophys Acta 1778(10):2388–2394
Kuypers FA, Yuan J, Lewis RA, Snyder LM, Kiefer CR, Bunyaratvej A et al (1998) Membrane phospholipid asymmetry in human thalassemia. Blood 91(8):3044–3051
Eldor A, Rachmilewitz EA (2002) The hypercoagulable state in thalassemia. Blood 99(1):36–43
Mankelow TJ, Griffiths RE, Trompeter S, Flatt JF, Cogan NM, Massey EJ et al (2015) Autophagic vesicles on mature human reticulocytes explain phosphatidylserine-positive red cells in sickle cell disease. Blood 126(15):1831–1834
de Jong K, Larkin SK, Styles LA, Bookchin RM, Kuypers FA (2001) Characterization of the phosphatidylserine-exposing subpopulation of sickle cells. Blood 98(3):860–867
Basu S, Banerjee D, Ghosh M, Chakrabarti A (2010) Erythrocyte membrane defects and asymmetry in paroxysmal nocturnal hemoglobinuria and myelodysplastic syndrome. Hematology 15(4):236–239
Zahedpanah M, Azarkeivan A, Aghaieepour M, Nikogoftar M, Ahmadinegad M, Hajibeigi B et al (2014) Erythrocytic phosphatidylserine exposure and hemostatic alterations in beta-thalassemia intermediate patients. Hematology 19(8):472–476
Pattanapanyasat K, Noulsri E, Fucharoen S, Lerdwana S, Lamchiagdhase P, Siritanaratkul N et al (2004) Flow cytometric quantitation of red blood cell vesicles in thalassemia. Cytometry B Clin Cytom 57(1):23–31
Xiong Z, Oriss TB, Cavaretta JP, Rosengart MR, Lee JS (2012) Red cell microparticle enumeration: validation of a flow cytometric approach. Vox Sang 103(1):42–48
Rubin O, Crettaz D, Canellini G, Tissot JD, Lion N (2008) Microparticles in stored red blood cells: an approach using flow cytometry and proteomic tools. Vox Sang 95(4):288–297
Pattanapanyasat K, Gonwong S, Chaichompoo P, Noulsri E, Lerdwana S, Sukapirom K et al (2007) Activated platelet-derived microparticles in thalassaemia. Br J Haematol 136(3):462–471
Lamchiagdhase P, Nitipongwanich R, Rattanapong C, Noulsri E, Lerdwana S, Pattanapanyasat K (2004) Red blood cell vesicles in thalassemia. J Med Assoc Thail 87(3):233–238
Thomas ED, Buckner CD, Sanders JE, Papayannopoulou T, Borgna-Pignatti C, De Stefano P et al (1982) Marrow transplantation for thalassaemia. Lancet 2(8292):227–229
Sabloff M, Chandy M, Wang Z, Logan BR, Ghavamzadeh A, Li CK et al (2011) HLA-matched sibling bone marrow transplantation for beta-thalassemia major. Blood 117(5):1745–1750
Boas FE, Forman L, Beutler E (1998) Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci U S A 95(6):3077–3081
Kuypers FA, de Jong K (2004) The role of phosphatidylserine in recognition and removal of erythrocytes. Cell Mol Biol (Noisy-le-Grand, France) 50(o):147–158
Basu S, Banerjee D, Chandra S, Chakrabarti A (2008) Loss of phospholipid membrane asymmetry and sialylated glycoconjugates from erythrocyte surface in haemoglobin E beta-thalassaemia. Br J Haematol 141(1):92–99
Sirachainan N, Thongsad J, Pakakasama S, Hongeng S, Chuansumrit A, Kadegasem P et al (2012) Normalized coagulation markers and anticoagulation proteins in children with severe beta-thalassemia disease after stem cell transplantation. Thromb Res 129(6):765–770
Tantawy AA, Adly AA, Ismail EA, Habeeb NM (2013) Flow cytometric assessment of circulating platelet and erythrocytes microparticles in young thalassemia major patients: relation to pulmonary hypertension and aortic wall stiffness. Eur J Haematol 90(6):508–518
Schrier SL (2002) Pathophysiology of thalassemia. Curr Opin Hematol 9(2):123–126
Voskou S, Aslan M, Fanis P, Phylactides M, Kleanthous M (2015) Oxidative stress in beta-thalassaemia and sickle cell disease. Redox Biol 6:226–239
Jevtic D, Vujic D, Zecevic Z, Veljkovic D, Gazikalovic S, Elezovic I (2010) Coagulation disturbances in paediatric patients with hepatic veno-occlusive disease after stem cells transplantation. Srp Arh Celok Lek 138(Suppl 1):33–38
Klaihmon P, Phongpao K, Kheansaard W, Noulsri E, Khuhapinant A, Fucharoen S et al (2017) Microparticles from splenectomized beta-thalassemia/HbE patients play roles on procoagulant activities with thrombotic potential. Ann Hematol 96(2):189–198
Rank A, Nieuwland R, Toth B, Pihusch V, Delker R, Hiller E et al (2011) Microparticles for diagnosis of graft-versus-host disease after allogeneic stem transplantation. Transplantation 92(2):244–250
Trummer A, De Rop C, Stadler M, Ganser A, Buchholz S (2011) P-selectin glycoprotein ligand-1 positive microparticles in allogeneic stem cell transplantation of hematologic malignancies. Exp Hematol 39(11):1047–1055
Acknowledgements
We thank the staff at the Hematology-Oncology Division, Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, for recruiting subjects and helping in blood sample collection. Thanks also go to Dr. Arthur E. Brown for editing the manuscript. PK and SV were financially supported by the Thailand Research Fund (TRF) RGJ-PhD scholarship. EN thanks the Faculty of Medicine Siriraj Hospital, Mahidol University, for supporting the research project. KP was the recipient of the TRF-Distinguished Research Professor Award (grant contact number DPG5980001).
Author information
Authors and Affiliations
Contributions
PK, SV, EN performed the experiments, analyzed the data and wrote manuscript; SL, UA, NS supervised the project and analyzed the data; SH, KP designed the experiment, wrote, and edited the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Supplementary Table 1
Demographic data of two patient groups. (DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Klaihmon, P., Vimonpatranon, S., Noulsri, E. et al. Normalized levels of red blood cells expressing phosphatidylserine, their microparticles, and activated platelets in young patients with β-thalassemia following bone marrow transplantation. Ann Hematol 96, 1741–1747 (2017). https://doi.org/10.1007/s00277-017-3070-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-017-3070-2