
Abstract
Thromboembolic events including cerebral thrombosis, deep vein thrombosis, and pulmonary embolism are major complications in β-thalassemia. Damaged red blood cells and chronic platelet activation in splenectomized β-thalassemia/HbE patients were associated with increased microparticles (MPs) releases into blood circulation. MPs are small membrane vesicles, which play important roles on coagulation. However, the role of MP in thalassemia is poorly understood. In this study, the effects of splenectomized-MPs on platelet activation and aggregation were investigated. The results showed that isolated MPs from fresh platelet-free plasma of patients and normal subjects directly induce platelet activation, platelet aggregation, and platelet-neutrophil aggregation in a dose-dependent manner. Interestingly, MPs obtained from splenectomized patients are more efficient in induction of platelet activation (P-selectin+) when compared to MPs from normal subjects (P < 0.05), tenfold lower than pathophysiological level, at 1:0.1 platelet MP ratio. Co-incubation of splenectomized-MPs with either normal-, non-splenectomized- or splenectomized-platelets at 1:10 platelet MP ratio increased platelet activation up to 5.1 ± 2.2, 5.6 ± 3.7, and 9.5 ± 3.0%, respectively, when normalized with individual baseline. These findings suggest that splenectomized patients were proned to be activated by MPs, and splenectomized-MPs could play an important role on chronic platelet activation and aggregation, leading to thrombus formation in β-thalassemia/HbE patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microparticles (MPs), also known as microvesicles or ectosomes, are plasma membrane vesicles of less than 1000 nm shed from various cell types including platelets, red blood cells (RBCs), leukocytes, epithelial cells of various tissue origins, and tumor cells [1–3]. Among these, platelet-derived MPs are the most abundant MPs found in the bloodstream, constituting approximately 70 to 90% of the total number of MPs [4, 5]. MPs are released as a consequence of cellular activation, cell senescence, and apoptosis [1, 6, 7]. Although the presence of MPs in the blood is common in normal individuals, an increase in the level of MPs has been shown in patients presenting with various pathological conditions such as cardiovascular disease, thrombosis, sickle cell disease, and thalassemia [6–12]. Our group has previously shown that β-thalassemia/HbE patients had significantly increased amounts of MPs originated from activated platelets, and these MPs were correlated not only with the increased platelet counts but also with the increased procoagulant activity [13]. Characterizations by using proteome analysis revealed that these MPs were mostly from oxidatively damaged RBCs and platelets with altered proteins involving in regulation of cell stress. Production of platelet-derived MPs with abnormal proteome may render thromboembolic complications which are commonly found in β-thalassemia/HbE patients [14]. For these reasons, MPs released could be thus regarded as indicators for cellular injury and are of interest for understanding the mechanisms of pathology of vascular disease.
It has been known that platelets play a critical role in triggering arterial thrombosis and in promoting atherogenesis as well as in participating in inflammation and immune reaction [15, 16]. Platelets normally circulate in a non-activated state, but after vascular tissue injury, they release contents from their intracellular granules that trigger aggregates and eventually induce the formation of a hemostatic plug [15, 17]. Furthermore, the formation of platelet-leukocyte aggregates, and leukocyte activation had been described as an important factor in vascular repair and microcirculatory disturbances in vascular ischemic tissue. Interaction of activated platelets with monocytes and neutrophils via binding between P-selectin expressed on the surface of activated platelets and the leukocyte counter-receptor P-selectin GP ligand-1 (PSGL-1) may impair leukocyte recruitment and activation processes [17, 18]. Elevated levels of platelet expression and platelet-leukocyte aggregates have been associated with increased risk of specific diseases as an early injury marker in patients with acute myocardial infarction [19, 20], and in patients with stroke [21], as well as in patients with thalassemia [22–24].
Since MPs exhibit a variable spectrum of biological information in the form of RNA, components of signaling pathways, bioactive lipids, membrane-anchored receptors, and adhesion molecules, thereby transferring bioactive substances once interact with and taken up by various target cells [17, 18, 25, 26]. However, the procoagulant role of MPs shed from platelets is not well understood. The objective of the present study is to investigate the effect of MPs on platelet activation and platelet-leukocyte formation. We hypothesized that MPs, particularly those obtained from splenectomized β-thalassemia/HbE patients would potentiate platelet aggregation, expression of adhesion molecules, and interaction with leukocytes.
Materials and methods
Subjects
Sixty-three non-blood transfusion dependent β-thalassemia/HbE patients (33 non-splenectomy and 30 splenectomy) and 30 normal subjects with matching in age (24–48 years old) and sex (% male gender; 60, 50, and 40%, respectively), were recruited. Patients who had been splenectomized for more than 5 years performed splenectomy at the ages ranging from seven to 33 years old. The study protocol was approved by the Mahidol University Institutional Review Board (approval number 2014/013.0502) and the Institutional Review Board of the Faculty of Medicine Siriraj Hospital (approval number SI657/2014). Written informed consent was obtained from all individual participants included in the study. All subjects had normal glucose-6-phosphate dehydrogenase levels. There were no evidences of concurrent infection, history of vaso-occlusive episode, or atherosclerotic vascular disease. Patients under treatments with aspirin, antibiotics, anti-depressants, beta-blockers, and anti-platelets were excluded, and none had been hospitalized or transfused within 4 weeks. All patients received folic acid and iron chelator daily. Hemoglobin levels (mean ± SD) in normal subjects, non-splenectomized and splenectomized patients were 13 ± 1.0, 7.3 ± 1.6, and 7.1 ± 1 g/dL, respectively. The mean annual Hb level of non-splenectomized patients was 7.3 ± 0.9 g/dL while for splenectomized patients, it was 6.5 ± 0.7 g/dL. The number of nucleated RBC (213 ± 43 cells/100 white blood cells (WBC)) and platelet count (745 ± 145 × 103 cells/μL) in splenectomized patients were higher than those of non-splenectomized patients (13 ± 6 cells/100 WBC, 211 ± 46 × 103 cells/μL, respectively) (P < 0.05). Serum ferritin levels (mean ± SE) in β-thalassemia/HbE patients (981 ± 279 ng/mL in non-splenectomized patients and 1481 ± 410 ng/mL in splenectomized patients) were significantly increased when compared to normal subjects (116 ± 38 ng/mL in normal subjects). All blood samples were collected at room temperature (RT) and processed within 2–3 h.
Microparticles
MPs were isolated by centrifugation method as previously described [14]. Briefly, platelet poor plasma (PPP) was prepared by a centrifugation of each freshly drawn 3.2% sodium citrate blood sample at 1,500×g for 15 min at RT and then centrifuged at 14,000×g for 2 min at 20 °C to obtain platelet free-plasma (PFP). Afterward, the PFP was re-centrifuged at 14,000×g for another 45 min at 4 °C to collect a pellet of MPs.
To evaluate cellular origin and absolute number of MPs, isolated MPs or whole blood samples were stained with fluorochrome conjugated annexin V (phosphatidylserine, PS) and monoclonal antibodies (mAbs) to glycophorin A (GPA) (CD235a, RBC marker), glycoprotein (GP) IIb/IIIa (CD41a, platelet marker), leukocyte common antigen (CD45, leukocyte marker), and endoglin (CD105, endothelial cell marker) in annexin V binding buffer containing TruCount™ beads (Becton Dickinson Biosciences (BDB), San Diego, CA) for absolute number calculation. MP population was determined by beads with 1 μm in diameter (Spherotech, Lake Forest, IL, USA). All mAbs and reagents were purchased from BDB. Fluorescence signals were determined using FACSCalibur™ flow cytometer (BDB) as previously described [13].
Aggregometric analysis of platelet aggregation
Platelet-rich plasma (PRP) was immediately prepared from fasting fresh blood donors by centrifugation at 180×g for 10 min at RT and then re-centrifuged to collect PPP. The PRP was primed with MPs at platelet to MP ratio of 1:0.1 (physical condition) for 15 min, then, treated with 1.5 μM adenosine diphosphate (ADP) (Sigma, St Louis, MO, USA). Platelet aggregation was determined using aggregometer, model 500CA/560CA whole blood lumi ionized calcium aggregation systems (Chronolog, Havertown, PA, USA). PPP was used as blank. PRP samples treated with 8 μM ADP were used as positive control and those treated with normal saline (NSS) were negative control.
Flow cytometric analysis of platelet-leukocyte aggregation
Diluted whole blood samples from splenectomized patients and normal subjects were incubated with allogenic MPs from splenectomized patients and normal as chessboard at WBCs to MPs ratio 1:1 and 1:10 for 15 min at RT. Untreated blood samples with spontaneous platelet-leukocyte aggregation were used as baseline. After incubation, samples were stained with fluorochrome conjugated with specific mAbs to GPA, GPIIb/IIIa, CD45, and CD11b (activated leukocyte marker, BDB). RBCs were lyzed by FACSlysis solution (BDB) before determination fluorescence signals by FACSCalibur™ flow cytometer (BDB). The percentages of platelet-leukocyte aggregation (GPA−GPIIb/IIa+CD45+CD11b+) were analyzed as previously described [22].
Immunofluorescent analysis of microparticle-platelet-leukocyte aggregation
Isolated MPs were conjugated with PKH67 (green fluorescence) (Sigma) as manufacturer’s recommendation. PKH67 labeled MPs were co-incubated with whole blood at WBC to MP ratio of 1:100 for 30 min at RT. Then, RBCs were lyzed by FACSlysis solution (BDB). After fixing with 4% paraformaldehyde solution and blocking with 3% bovine serum albumin in phosphate buffer saline (PBS), samples were incubated with mouse anti-human GPIIb/IIIa (BDB), and subsequently Alexa Fluor®568 conjugated goat anti-mouse IgG antibody (BDB) and 4′, 6-diamidino-2-phenylindole (DAPI) (Life Technologies, Carlsbad, CA, USA). Fluorescent signals and images were analyzed by NIS-Elements 4.10.0 software, Nikon Eclipse Ti-Clsi4 microscope (Nikon, Melville, NY, USA).
Flow cytometric analysis of platelet activation
Diluted whole blood from both splenectomized and non-splenectomized patients and normal subjects were incubated with allogenic MPs isolated from both splenectomized and non-splenectomized patients and normal as chessboard at platelets to MPs ratio of 1:0.01, 1:0.1, 1:1, and 1:10 for 15 min at RT with or without 0.5 μM ADP. Untreated samples with spontaneous platelet activation were used as baseline. After incubation, samples were stained with fluorochrome conjugated with specific mAbs to GPIIb/IIIa and P-selectin (CD62P, platelet activation marker, BDB). The percentages of platelet activation (GPIIb/IIIa+P-selectin+) were determined using FACSCalibur™ flow cytometer (BDB) as described in the previous study [22].
Statistical analysis
Data were analyzed using SPSS Version 18.0 (IBM, Chicago, USA). Comparisons between parameters were evaluated with a non-parametric Mann-Whitney U Test. The threshold for statistical significance for all comparisons was P < 0.05.
Results
MPs induce ex vivo platelet aggregation
To determine the physiologic response of MPs, platelet aggregation in PRP was measured by aggregometer. Exposure of platelets obtained from normal individuals to low dose agonist, 1.5 μM ADP alone triggers a primary wave of aggregation without degranulation, followed by rapid disaggregation. In the presence of high concentration of agonist, 8 μM ADP, both primary and secondary waves of aggregation were induced, with more than 80% maximum aggregation (Fig. 1). Primed normal-platelets with MPs from β-thalassemia/HbE patients or normal subjects at physiological platelet, MP ratio 1:0.1, showed the first wave aggregation after adding 1.5 μM ADP; then, the aggregation was reduced with a short period before second wave of aggregation appeared with inducing platelet granule secretion. Primed platelets with MPs from both splenectomized patients and normal subjects increased percentages of maximal aggregation at the primary aggregation (56 ± 15 and 67 ± 18%, respectively), when compared to 1.5 μM ADP-treated platelets alone (37 ± 4%) (P < 0.05). These results clearly showed that MPs could induce platelet aggregation. However, aggregometry did not show a significant difference of platelet aggregation when platelets were primed with either splenectomized-MPs or normal-MPs at physiological condition.

Microparticles promote platelet aggregation. Light transmission aggregometry was used to assess aggregation in response to MPs in normal platelet-rich plasma (PRP) stimulated with ADP. Data are shown graphically as a percentage of light transmittance (y-axis) over time (x-axis). Normal PRP treated with 1.5 μM ADP demonstrates only a primary wave of aggregation followed by rapid platelet disaggregation. While primed PRP with MPs obtained from either splenectomized β-thalassemia/HbE patient (P-MP) or normal subject (N-MP) at platelet MP ratio 1:0.1 leads to induce robust platelet aggregation. PRP treated with 8 μM
MPs induce ex vivo platelet-neutrophil aggregation
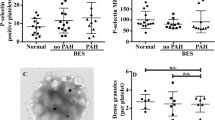
The activated platelets, apart from rapidly adhere to one another (platelet aggregation), can also adhere to leukocytes. Formation of platelet-leukocyte aggregation is an important factor in thrombotic events. The ability of MPs to induce platelet-leukocyte aggregation was determined by co-incubation whole blood with isolated MPs at leukocyte to MP ratio of 1:1 and 1:10. Flow cytometric analysis showed that both splenectomized-MP and normal-MP at leukocyte to MP ratio of 1:10 promoted aggregation of platelet-neutrophil, but not platelet-monocyte and platelet-lymphocyte when compared to the low dose of MPs (leukocyte MP ratio 1:1) (P < 0.05) (Fig. 2a–c). The aggregation of platelet-leukocyte-MP was clearly evident by fluorescent microscope (Fig. 2d). A few platelets that spontaneously aggregated on leukocytes could be observed. After co-incubation of whole blood sample with PKH67-labeled MPs (green fluorescence), the increase of platelet-leukocyte aggregation was found.

Microparticles enhanced platelet-neutrophil aggregation. (a–c) Platelet-leukocyte aggregates were quantified by flow cytometry, GPA−CD45+CD11b+CD41a+, neutrophil (a), monocyte (b), and lymphocyte (c) populations that were discerned through characteristic side scatter. Whole blood was treated with MPs at white blood cell (WBC) MP ratio 1:1 and 1:10. Fold changes of platelet-leukocyte aggregation were calculated with individual baseline. Asterisk indicates significant difference at P < 0.05. BE-S splenectomized β-thalassemia/HbE patients. d MP-platelet-leukocyte aggregation was analyzed in whole blood samples with the absence or presence of PKH67-labeled MPs by using immunofluorescent assay. Whole blood samples were stained for platelet with mouse anti-human GPIIb/IIIa, and subsequently a Alexa Fluor®568 conjugated goat anti-mouse IgG antibody (BDB) and nucleus of leukocytes with DAPI
MPs increased P-selectin expression on platelets
Generally, high amounts of MPs in splenectomized β-thalassemia/HbE patients are strongly associated with high platelet counts and chronic platelet activation, leading to an increased risk of thromboembolic events. The effect of splenectomized-MP on platelets was analyzed by flow cytometry as the technique is highly sensitive, easy, and rapid to perform. Flow cytometric analysis of platelet activation in peripheral blood samples was classified by size (FSC/SSC) and platelet activation markers (GPIIb/IIIa+P-selectin+ platelets).
The increased percentages of platelet activation were observed in MP-treated platelets obtained from all three groups; normal subjects (Fig. 3a (i–iii)), β-thalassemia/HbE patients with non-splenectomy (Fig. 3a (iv–vi)), and splenectomy (Fig. 3a (vii–ix)). Co-incubation between normal-MPs and platelets from normal subjects, non-splenectomized patients, and splenectomized patients at platelet to MP ratio 1:10 resulted in a significantly increased platelet activation, 3.4 ± 1.9, 4.9 ± 2.9, and 5.2 ± 2.7%, respectively, when compared to those composing of platelet to MP ratio 1:0.1 (1.1 ± 0.4, 1.8 ± 0.7, and 1.8 ± 1.0%, respectively), (P < 0.05) (Fig. 3a (i, iv, vii)). Similarly, the significantly increased platelet activation at platelet to MP ratio 1:10 of non-splenectomized-MPs treated platelets from normal subjects (4.5 ± 1.6%) and non-splenectomized patients (5.2 ± 3.1%) was observed when compared to those having platelet to MP ratio 1:0.1 (1.3 ± 0.8 and 2.9 ± 2.5%, respectively), (P < 0.05) (Fig. 3a(ii, v)). Splenectomized-MPs treated platelets from normal subjects (5.1 ± 2.2%) and splenectomized patients (9.5 ± 3.0%) at platelet to MP ratio 1:10 also showed significantly increased percentages of platelet activation comparing to those having platelet to MP ratio 1:0.1 (1.8 ± 0.8 and 5.6 ± 3.1%, respectively), (P < 0.05) (Fig. 3a (iii, ix)). This phenomenon indicated that MPs from all groups were a strong dose dependent inducer for P-selectin expression on platelets.

Peripheral blood MPs induced platelet activation. a Co-incubation between (i–ix) platelet samples from normal subjects (top row), non-splenectomized patients (middle row), and splenectomized patients (bottom row) were treated with isolated MPs from normal subjects (left column), non-splenectomized patients (middle column) and splenectomized patients (right column) at platelet MP ratio 1:0.01 to 1:10. b Isolated MPs from normal subjects, non-splenectomized (BE-NS) and splenectomized β-thalassemia/Hb E (BE-S) patients were co-incubated with individual platelets from each group at platelet MP ratio (i) 1:0.01, (ii) 1:0.1, (iii) 1:1 and (iv) 1:10. Percentages of platelet activation (CD41a+CD62P+) were determined by flow cytometry and normalized percent increase with individual baseline. Asterisk indicates significant difference at P < 0.05
Induction of MPs on platelet activation was based not only on the amounts of MPs but also on the origins of MPs. Clearly, platelet activation from splenectomized-MPs was stronger than MPs from other groups at the lowest platelet to MP ratio of 1:0.01 (Fig. 3b). Platelet activation of splenectomized-MP treated splenectomized-platelets (5.4 ± 3.6%) and non-splenectomized-platelets (4.1 ± 3.0%) were increased significantly when compared to those of normal-MP treatment (1.9 ± 1.2 and 1.0 ± 0.5%, respectively), (P < 0.05). Moreover, efficiency to induce platelet activation of splenectomized-MPs in patients and normal subjects was higher than MPs from other sources at all doses (Fig. 3b (ii–iv).
Platelet defects including hypersensitivity and hyperactivation were also found in β-thalassemia/HbE patients who had normal coagulation profiles (prothrombin time, activated partial thromboplastin time, and thrombin time) (data not shown). The results in splenectomized-MP treated platelets at physiological condition (Fig. 3b (ii)) clearly showed that percentages of platelet activation in β-thalassemia/HbE platelets were higher than those of normal platelets (P < 0.05). Indeed, percentages of platelet activation in splenectomized-MP treated platelets from splenectomized patients at all four different doses were significantly increased when compared to splenectomized-MP treated platelets from normal subjects at the same dose of MPs (P < 0.05) (Fig. 3b).
Additionally, the effects of MPs as agonist to activated platelets were determined. Normal- and splenectomized-platelets treated with a low concentration, 0.5 μM ADP, increased percentages of platelet activation 2.0 ± 0.9 and 2.38 ± 0.33%, respectively (Fig. 4a, no MP). Combination between the low dose of ADP and splenectomized-MPs induced platelet activation from normal- and splenectomized-platelets in a dose-dependent manner (Fig. 4a (ii, iv)). These results are consisting with the additional effect of MPs and ADP examined by aggregometer. The effect in increased platelet activation in splenectomized-MPs was higher than normal-MPs as shown in Fig. 4b.

Microparticles enhanced effect of ADP on platelet activation. a Platelet samples from normal subjects (top row) and splenectomized patients (bottom row) were treated with 0.5 μM ADP alone or combined with isolated MPs from normal subjects (left column) and splenectomized patients (right column) at platelet MP ratio 1:0.01 to 1:10. b Combined 0.5 μM ADP and isolated MPs from normal subjects and splenectomized β-thalassemia/Hb E (BE-S) patients were co-incubated with individual platelets from each group at platelet MP ratio (i) 1:0.01, (ii) 1:0.1, (iii) 1:1 and (iv) 1:10. Percentages of platelet activation (CD41a+CD62P+) were determined by flow cytometry and normalized percent increase with individual baseline. Asterisk indicates significant difference at P < 0.05
Discussion
Thromboembolic complications are commonly found in β-thalassemia, especially splenectomized patients, and are the major causes of death [24]. There have been numerous reports of thromboembolism including cerebral thrombosis, deep venous thrombosis, and pulmonary embolism and recurrent arterial occlusion, with thrombi in small and large pulmonary vessels. Several brain magnetic resonance imaging studies confirm the high prevalence of silent ischemic lesions in patients with β-thalassemia intermedia, especially in splenectomized adults who are transfusion-independent and those with elevated platelet counts [27]. Epidemiology study of thromboembolic events in 8860 β-thalassemia patients, which demonstrated that thromboembolic events occurred in 0.9% of transfusion dependent β-thalassemia and 3.9% of non-transfusion dependent β-thalassemia, suggest that the incidence occurred in patients with non-transfusion dependent β-thalassemia intermedia was 4.38 times more frequently than those patients with regularly transfused β-thalassemia major [28].
Mechanisms of thrombosis in β-thalassemia/HbE are described with association of several factors such as phosphatidylserine (PS) exposure on damaged RBC membrane contribute to thrombin generation [29], increased platelet factor 3-like activity [13], increased coagulation factors, decreased fibrinolysis [30], activation of platelets, leukocytes and vascular endothelium [22, 29], and increased platelet-leukocyte aggregation [22, 23]. Moreover, increased oxidative stress in RBCs and platelets and chronic platelet activation in β-thalassemia/HbE lead to generation of MPs, which have strong procoagulant activity. MPs may contribute to promote thrombus stabilization and platelet plug formation. High levels of circulating MPs in splenectomized β-thalassemia/HbE patients generated from chronic platelet activation and damaged RBCs have been demonstrated [12, 13]. Splenectomy is a therapeutic option for patients with thalassemia who had hypersplenism. Nonetheless, splenectomy could result in the increased amounts of MPs in blood circulation. MPs, especially from splenectomized patients, had high efficiency to form prothombinase complexes [14]. However, direct effect of MPs on platelets that may contribute to an enhanced risk of vascular complications in thalassemia was not emphasized yet. Therefore, this study focuses on the investigation of procoagulant activity of MPs in induction of platelet activation and aggregation in order to understand mechanisms of the lifelong hypercoagulable state in β-thalassemia/HbE.
Clearly, MPs isolated from β-thalassemia/HbE patients could induce platelet aggregation and platelet-neutrophil aggregation. β-Thalassemia/HbE-MPs had enrichment of adhesion molecules such as platelet glycoprotein IIb/IIIa, P-selectin, and CD36 on their surface as parent cells [13]. Moreover, MPs are small in size that could act like a bridge for cell-to-cell interaction leading to thrombus formation. An ex vivo study showed that MPs promoted platelet-neutrophil aggregation via P-selectin and PSGL-1 interaction. The P-selectin-expressing MPs bridge neutrophils that express PSGL-1, even when L-selectin function was blocked under flow shear stresses [31]. However, the mechanism of MP in activation of neutrophil function that may contribute to cell-to-cell interaction, thrombus formation, and proinflammation in thalassemia should be considered.
This study showed that MPs obtained from both normal individuals and β-thalassemia/HbE patients could induce platelet activation. Long storage whole blood, packed red cells, and platelet concentration could spontaneously generate MPs that might cause adverse effect to β-thalassemia/HbE patients, especially splenectomized cases. The increased level of MPs during blood storage had been observed [32]. These MPs also could initiate thrombin generation. Moreover, RBC-derived MPs generated from storage RBCs could scavenge the vasodilator nitric oxide about 1000 times faster than with intact RBCs [33]. All these indicated that MPs generated during blood storage could increase risk of thrombosis, suggesting that caution in storage and preparation of blood donors for transfusion should be considered. Therefore, short storage or washing blood component might be required for quality of blood transfusion.
Although effects of MPs on induced platelet aggregation and platelet-neutrophil aggregation were not significantly different between MPs from β-thalassemia/HbE patients and normal subjects, MPs from splenectomized β-thalassemia/HbE patients still play a strong agonist to activate P-selectin expression on platelets when compared to MPs from other groups. It is indicated that splenectomized β-thalassemia/HbE-MPs could carry some bioactive molecules such as adhesion molecules, bioactive lipids, and miRNAs that lead to the regulation of vascular system. Proteomic analysis of MPs in our previous study showed that soluble N-ethylmaleimide-sensitive factor attachment protein (α-SNAP) levels were increased in MPs from splenectomized β-thalassemia/HbE patients compared to normal subjects. α-SNAP is a member of transporting protein that plays role in platelet exocytosis and regulation of vascular endothelial cadherin expression [14]. Moreover, dose-dependent induction of platelet activation by MPs was observed. This suggested that the effect of MPs on platelet functions of splenectomized β-thalassemia/HbE patients was resulted from both number and intrinsic properties of the splenectomized β-thalassemia/HbE-MPs.
In summary, MPs have a strong effect in enhancing platelet activation, platelet aggregation, and platelet-neutrophil aggregation. The increased levels of MPs in splenectomized β-thalassemia/HbE patients lead to hypercoagulable state and the higher risk of thromboembolic events. However, the strategy of a comprehensive MP biology and its mechanism to cellular response on homeostasis in β-thalassemia/HbE is necessary to verification.
Abbreviations
- MPs:
-
microparticles
- PPP:
-
platelet-poor plasma
- PRP:
-
platelet-rich plasma
- PS:
-
phosphatidylserine
- PSGL-1:
-
P-selectin glycoprotein ligand-1
References
Burnier L, Fontana P, Kwak BR, Angelillo-Scherrer A (2009) Cell-derived microparticles in haemostasis and vascular medicine. Thromb Haemost 101(3):439–451
Freyssinet JM, Toti F (2010) Formation of procoagulant microparticles and properties. Thromb Res 125(Suppl 1):S46–S48. doi:10.1016/j.thromres.2010.01.036
Rak J (2010) Microparticles in cancer. Semin Thromb Hemost 36(8):888–906. doi:10.1055/s-0030-1267043
Horstman LL, Ahn YS (1999) Platelet microparticles: a wide-angle perspective. Crit Rev Oncol Hematol 30(2):111–142
Berckmans RJ, Nieuwland R, Boing AN, Romijn FP, Hack CE, Sturk A (2001) Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb Haemost 85(4):639–646
Piccin A, Murphy WG, Smith OP (2007) Circulating microparticles: pathophysiology and clinical implications. Blood Rev 21(3):157–171. doi:10.1016/j.blre.2006.09.001
Simak J, Gelderman MP (2006) Cell membrane microparticles in blood and blood products: potentially pathogenic agents and diagnostic markers. Transfus Med Rev 20(1):1–26. doi:10.1016/j.tmrv.2005.08.001
van Beers EJ, Schaap MC, Berckmans RJ, Nieuwland R, Sturk A, van Doormaal FF, Meijers JC, Biemond BJ (2009) Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica 94(11):1513–1519. doi:10.3324/haematol.2009.008938
Tomer A, Harker LA, Kasey S, Eckman JR (2001) Thrombogenesis in sickle cell disease. J Lab Clin Med 137(6):398–407. doi:10.1067/mlc.2001.115450
Pattanapanyasat K, Noulsri E, Fucharoen S, Lerdwana S, Lamchiagdhase P, Siritanaratkul N, Webster HK (2004) Flow cytometric quantitation of red blood cell vesicles in thalassemia. Cytometry B Clin Cytom 57(1):23–31. doi:10.1002/cyto.b.10064
Westerman M, Pizzey A, Hirschman J, Cerino M, Weil-Weiner Y, Ramotar P, Eze A, Lawrie A, Purdy G, Mackie I, Porter J (2008) Microvesicles in haemoglobinopathies offer insights into mechanisms of hypercoagulability, haemolysis and the effects of therapy. Br J Haematol 142(1):126–135. doi:10.1111/j.1365-2141.2008.07155.x
Habib A, Kunzelmann C, Shamseddeen W, Zobairi F, Freyssinet JM, Taher A (2008) Elevated levels of circulating procoagulant microparticles in patients with beta-thalassemia intermedia. Haematologica 93(6):941–942. doi:10.3324/haematol.12460
Pattanapanyasat K, Gonwong S, Chaichompoo P, Noulsri E, Lerdwana S, Sukapirom K, Siritanaratkul N, Fucharoen S (2007) Activated platelet-derived microparticles in thalassaemia. Br J Haematol 136(3):462–471
Chaichompoo P, Kumya P, Khowawisetsut L, Chiangjong W, Chaiyarit S, Pongsakul N, Sirithanaratanakul N, Fucharoen S, Thongboonkerd V, Pattanapanyasat K (2012) Characterizations and proteome analysis of platelet-free plasma-derived microparticles in beta-thalassemia/hemoglobin E patients. J Proteome 76 :239–250. doi:10.1016/j.jprot.2012.06.004Spec No
Ruggeri ZM (2002) Platelets in atherothrombosis. Nat Med 8(11):1227–1234. doi:10.1038/nm1102-1227
Joseph M (1995) The generation of free radicals by blood platelets. Immunopharmacology of platelets. In. Academic Press, San Diego, pp. 209–223
Michelson AD (2003) How platelets work: platelet function and dysfunction. J Thromb Thrombolysis 16(1–2):7–12. doi:10.1023/b:thro.0000014586.77684.82
Furie B, Furie BC, Flaumenhaft R (2001) A journey with platelet P-selectin: the molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost 86(1):214–221
Furman MI, Barnard MR, Krueger LA, Fox ML, Shilale EA, Lessard DM, Marchese P, Frelinger AL 3rd, Goldberg RJ, Michelson AD (2001) Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol 38(4):1002–1006
Gawaz M, Neumann FJ, Ott I, Schiessler A, Schomig A (1996) Platelet function in acute myocardial infarction treated with direct angioplasty. Circulation 93(2):229–237
Marquardt L, Ruf A, Mansmann U, Winter R, Schuler M, Buggle F, Mayer H, Grau AJ (2002) Course of platelet activation markers after ischemic stroke. Stroke; a journal of cerebral circulation 33(11):2570–2574
Keawvichit R, Khowawisetsut L, Chaichompoo P, Polsrila K, Sukklad S, Sukapirom K, Khuhapinant A, Fucharoen S, Pattanapanyasat K (2012) Platelet activation and platelet-leukocyte interaction in beta-thalassemia/hemoglobin E patients with marked nucleated erythrocytosis. Ann Hematol 91(11):1685–1694. doi:10.1007/s00277-012-1522-2
Srihirun S, Tanjararak N, Chuncharunee S, Sritara P, Kaewvichit R, Fucharoen S, Pattanapanyasat K, Sibmooh N (2015) Platelet hyperactivity in thalassemia patients with elevated tricuspid regurgitant velocity and the association with hemolysis. Thromb Res 135(1):121–126. doi:10.1016/j.thromres.2014.10.010
Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, Saned M, Cesaretti C, Cappellini MD (2010) Splenectomy and thrombosis: the case of thalassemia intermedia. Journal of thrombosis and haemostasis : JTH 8(10):2152–2158. doi:10.1111/j.1538-7836.2010.03940.x
Mause SF, Weber C (2010) Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res 107(9):1047–1057. doi:10.1161/circresaha.110.226456
Montoro-Garcia S, Shantsila E, Marin F, Blann A, Lip GY (2011) Circulating microparticles: new insights into the biochemical basis of microparticle release and activity. Basic Res Cardiol 106(6):911–923. doi:10.1007/s00395-011-0198-4
Musallam KM, Taher AT, Karimi M, Rachmilewitz EA (2012) Cerebral infarction in beta-thalassemia intermedia: breaking the silence. Thromb Res 130(5):695–702. doi:10.1016/j.thromres.2012.07.013
Taher A, Isma'eel H, Mehio G, Bignamini D, Kattamis A, Rachmilewitz EA, Cappellini MD (2006) Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost 96(4):488–491
Eldor A, Rachmilewitz EA (2002) The hypercoagulable state in thalassemia. Blood 99(1):36–43
Atichartakarn V, Angchaisuksiri P, Aryurachai K, Onpun S, Chuncharunee S, Thakkinstian A, Atamasirikul K (2002) Relationship between hypercoagulable state and erythrocyte phosphatidylserine exposure in splenectomized haemoglobin E/beta-thalassaemic patients. Br J Haematol 118(3):893–898
Forlow SB, McEver RP, Nollert MU (2000) Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood 95(4):1317–1323
Gao Y, Lv L, Liu S, Ma G, Su Y (2013) Elevated levels of thrombin-generating microparticles in stored red blood cells. Vox Sang 105(1):11–17. doi:10.1111/vox.12014
Donadee C, Raat NJ, Kanias T, Tejero J, Lee JS, Kelley EE, Zhao X, Liu C, Reynolds H, Azarov I, Frizzell S, Meyer EM, Donnenberg AD, Qu L, Triulzi D, Kim-Shapiro DB, Gladwin MT (2011) Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation 124(4):465–476. doi:10.1161/CIRCULATIONAHA.110.008698
Acknowledgements
This work was supported by Faculty of Science, Mahidol University; Faculty of Science and Faculty of Medicine Ramathibodi Hospital, Mahidol University; Mahidol University Research Grants; Office of the Higher Education Commission and Mahidol University under the National Research University Initiative; Thailand Research Fund (TRF) (IRG5780009); The TRF Distinguished Research Professor Grant (DPG5980001); The Royal Golden Jubilee PhD Program of TRF; and Research Chair Grant, National Science and Technology Development Agency, Thailand. PK was supported under the Royal Golden Jubilee PhD Program of TRF.
Authors’ contributions
PK, KPh, and WK performed the experiments and analyzed the data. EN and NPM contributed to the concept of the study and interpretation. AK and SF contributed to the concept of the study and specimen collection. SS contributed to the concept of the study, design the experiments, the analysis of the data, and drafting the manuscript. KPa and PC were the principal investigator and take primary responsibility for the concept and design of the project, the analysis of the data, and drafting the manuscript. All authors reviewed and approved the final version to be published.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This work was supported by Faculty of Science, Mahidol University; Faculty of Science and Faculty of Medicine Ramathibodi Hospital, Mahidol University; Mahidol University Research Grants; Office of the Higher Education Commission and Mahidol University under the National Research University Initiative; Thailand Research Fund (TRF) (IRG5780009); The TRF Distinguished Research Professor Grant (DPG5980001); The Royal Golden Jubilee Program of TRF; and Research Chair Grant, National Science and Technology Development Agency, Thailand.
Conflict of interest
The authors declare that they have no conflict of interest.
Redundant publication
No substantial overlap with previous papers.
Additional information
Phatchanat Klaihmon and Kunwadee Phongpao contributed equally to this work.
Rights and permissions
About this article

Cite this article
Klaihmon, P., Phongpao, K., Kheansaard, W. et al. Microparticles from splenectomized β-thalassemia/HbE patients play roles on procoagulant activities with thrombotic potential. Ann Hematol 96, 189–198 (2017). https://doi.org/10.1007/s00277-016-2885-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-016-2885-6