
Abstract
The elastic behaviour and the structural evolution of microporous materials compressed hydrostatically in a pressure-transmitting fluid are drastically affected by the potential crystal–fluid interaction, with a penetration of new molecules through the zeolitic cavities in response to applied pressure. In this manuscript, the principal mechanisms that govern the P-behaviour of zeolites with and without crystal–fluid interaction are described, on the basis of previous experimental findings and computational modelling studies. When no crystal–fluid interaction occurs, the effects of pressure are mainly accommodated by tilting of (quasi-rigid) tetrahedra around O atoms that behave as hinges. Tilting of tetrahedra is the dominant mechanism at low-mid P-regime, whereas distortion and compression of tetrahedra represent the mechanisms which usually dominate the mid-high P regime. One of the most common deformation mechanisms in zeolitic framework is the increase of channels ellipticity. The deformation mechanisms are dictated by the topological configuration of the tetrahedral framework; however, the compressibility of the cavities is controlled by the nature and bonding configuration of the ionic and molecular content, resulting in different unit-cell volume compressibility in isotypic structures. The experimental results pertaining to compression in “penetrating” fluids, and thus with crystal–fluid interaction, showed that not all the zeolites experience a P-induced intrusion of new monoatomic species or molecules from the P-transmitting fluids. For example, zeolites with well-stuffed channels at room conditions (e.g. natural zeolites) tend to hinder the penetration of new species through the zeolitic cavities. Several variables govern the sorption phenomena at high pressure, among those: the “free diameters” of the framework cavities, the chemical nature and the configuration of the extra-framework population, the partial pressure of the penetrating molecule in the fluid (if mixed with other non-penetrating molecules), the rate of P-increase, the surface/volume ratio of the crystallites under investigations and the temperature at which the experiment is conducted. An overview of the intrusion phenomena of monoatomic species (e.g. He, Ar, Kr), small (e.g. H2O, CO2) and complex molecules, along with the P-induced polymerization phenomena (e.g. C2H2, C2H4, C2H6O, C2H6O2, BNH6, electrolytic MgCl2·21H2O solution) is provided, with a discussion of potential technological and geological implications of these experimental findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
“Microporous materials” are a class of compounds characterized by open-structures with cavities smaller than 20 Å in diameter. Cavities take the form of channels or cages. Materials with pores larger than 20 Å are the so-called “mesoporous materials”. Zeolites are the most common (in nature) and most used (in industrial processes) microporous materials. The structure of natural and synthetic zeolites is usually built up by a framework of SiO4–AlO4–PO4-tetrahedra. The extra-framework population consists of polar molecules (in particular H2O) and monovalent or divalent cations, which are commonly exchangeable. In hydrated zeolites, dehydration occurs at temperatures lower than 400 °C and is a usually reversible (and spontaneous) process. The tetrahedral framework may be interrupted by (OH, F) groups, which occupy tetrahedral apexes that are not shared with adjacent tetrahedra (Coombs et al. 1997). The general formula for common Si/Al zeolites is as follows: M + x L ++ y [Al(x+2y)Si n−(x+2y)O2n ]·mH2O (where M+ represents monovalent cations; L++ represents divalent cations; usually m < n).
The microporous nature of zeolites governs the four main properties of this class of materials: the selective (and spontaneous) cation-exchange capacity, the catalytic activity (mainly promoted by Brønsted acid sites), the T-induced reversible hydration/dehydration processes and the most-recently discovered P-induced sorption of molecules. These properties have made natural or synthetic zeolites an object of attention for a long series of applications, spanning from soil enhancement (e.g. zeolites are used as slow release fertilizers, in areas where environmental issues are of concern), environmental remediation, water treatment, animal feeding, biomedical and veterinary applications (e.g. drug delivery systems), cements and concretes production, gas separation, catalysis in the petroleum industry (e.g. almost all the world’s gasoline is produced using zeolites) and nuclear-waste processing (e.g. Komarneni 1985; Mumpton 1999; Kalló 2001; Maxwell and Stork 2001; Ming and Allen 2001; Bish et al. 2003; Ackley et al. 2003; Colella 2011; Gatta et al. 2016a). In this light, zeolites are nowadays considered as an important bulk commodity: the world production of natural zeolites in 2016 was about 2.8 million of tons (price: 100–230 dollar per ton) and the consumption of synthetic zeolites was approximately 1.6 million of tons (U.S. Geological Survey 2017). World reserves of natural zeolites have never been estimated. However, only in the U.S.A., resources are expected to approach 10 trillion tons for zeolite-rich deposits (U.S. Geological Survey 2017).
The peculiar behaviour of zeolites at non-ambient conditions, e.g. at high temperature (HT) or at high pressure (HP), led to a large number of in situ experiments over the last 60 years. More specifically, many experiments were devoted to the behaviour of zeolites in response to applied temperature: the mechanisms of T-induced dehydration, cation migration and rearrangement of extra-framework population have been investigated extensively, mainly by in situ single-crystal or powder X-ray/neutron-diffraction (e.g. Bish and Carey 2001; Pabalan and Bertetti 2001; Cruciani 2006 and references therein). In situ experiments on zeolites at high pressure have been done only in the last 15–20 years, mainly for technical difficulties, allowing the description of: (1) the compressional behaviour (and phase stability) in response to the applied pressure, along with the P-induced deformation mechanisms at the atomic scale (e.g. Gatta 2008, 2010a, b and references therein), (2) P-induced penetration of new molecules and its corresponding volume expansion (e.g. Lee et al. 2002a, b), (3) P-induced variation of the ionic conductivity of zeolites (e.g. Secco and Huang 1999; Rutter et al. 2000) and (4) P-induced amorphization processes (e.g. Gillet et al. 1996; Huang and Havenga 2001; Rutter et al. 2001; Greaves et al. 2003; Gulín-González and Suffritti 2004; Goryainov 2005).
The HP-behaviour of a zeolite compressed in a fluid is drastically dependent on the potential crystal–fluid interaction. Gatta (2008, 2010a) and Gatta and Lee (2014) described the relation between compressibility and microporosity, the framework flexibility (through deformation mechanisms at the atomistic level), and the different role played by framework (i.e. Si/Al-ordering, different cross-linking of the “building block unit”) and extra-framework configuration (i.e. nature of cations and absorbed molecules, ionic valence, ionic radii, coordination number) on the behaviour of zeolites when no crystal–fluid interaction occurs. The aim of this work is a comparative analysis, on the basis of previously published data, of the different behaviour of zeolites (natural or synthetic) when compressed in “penetrating” and in “non-penetrating” P-transmitting fluids (sensu Gatta 2008). More specifically, the effects of crystal–fluid interaction on the elastic behaviour and on the structure rearrangement of zeolites will be described, along with their potential geological and technological implications.
Experimental methods
The majority of the in situ high-pressure experiments on zeolites have been done by single-crystal and powder X-ray diffraction, as well as IR/Raman spectroscopy, using the so-called Merrill–Basset-type diamond anvil cell (DAC) (Merrill and Bassett 1974; Miletich et al. 2000); only a few experiments have been performed by in situ neutron powder diffraction, using the large-volume Paris–Edinburgh press (i.e. Besson et al. 1992; Colligan et al. 2005; Seryotkin et al. 2005).
HP-experiments with a DAC are usually conducted with the sample compressed hydrostatically in a P-transmitting fluid. P-transmitting media can be stable in the liquid or solid state at ambient conditions, or can be gaseous at ambient conditions and then liquefied at low-T before the loading of the DAC, or loaded into the P-chamber at high-P by means of a gas-loading device. The chemical nature and the behaviour of the P-transmitting fluid can play an important role in the HP-experiments on zeolites (Gatta 2008, 2010a; Angel et al. 2007; Gatta and Lee 2014). P-fluids can interact or not with the sample. Beside the potential crystal–fluid interaction, which will be extensively described below, it is highly desirable to ensure that the stress applied to the sample is homogeneous, i.e. without any differential stress or induced shear strain (Miletich et al. 2000; Angel et al. 2007). A hydrostatic medium cannot support shear stresses, simply because it has no shear strength. Non-hydrostatic stresses generate inhomogeneous strain in the crystal and, as a consequence, (sample) diffraction peaks broadening with a reduction of the signal-to-noise ratio. In addition, non-hydrostatic conditions can modify the compressional patterns of a given material (deduced on the basis of the unit-cell parameters variation with P) and can also suppress or promote phase transitions, including the promotion of P-induced amorphization phenomena (e.g. Decker et al. 1979; Kenichi 1999; Haines et al. 2001; Machon et al. 2003; Resel et al. 2004). Zeolites are relatively soft materials, and the occurrence of deviatoric stress usually has a dramatic impact on their behaviour at high pressure.
For experiments on zeolites at high pressure in a DAC, P-calibration has mainly been done by the so-called ruby fluorescence method (i.e. detecting the shift in the R1 emission line of ruby chips included in the compression chamber; P-uncertainty: ±0.05 GPa, Mao et al. 1986). A further method, especially used for single-crystal experiments, is based on the compressibility pattern of quartz (i.e. with quartz used as an internal standard in the compression chamber; P-uncertainty: <0.01 GPa, Angel et al. 1997). For the few experiments by in situ neutron diffraction with the large-volume press, lead as an internal standard (Colligan et al. 2005) or the calibration curve of the ILL hydraulic-press load vs. P (Seryotkin et al. 2005) have been used.
The behaviour of a given material at high pressure is usually described on the basis of its compression pattern (based on the evolution of the unit-cell parameters with P) and of its structural rearrangements at the atomic scale (by structure refinements based on the intensity data collected at high pressure). About zeolites, the compressional behaviour is usually well describable by in situ powder or single-crystal experiments. On the other hand, whereas the quality of the HP single-crystal data is usually good enough for structural refinements, the quality of powder data is often not sufficient, and this has led to an intensive use of computational modelling techniques—for example, “rigid unit mode” (RUM)/geometric approaches, classical force-field methodologies, ab initio structural optimizations and molecular dynamics simulations—as a complementary tool to unravel the structure evolution at the atomistic level, as described below. As a matter of fact, all the experiments in a DAC provided intensities, of the Bragg peaks, usually affected by a series of phenomena. Among those, the most critical are: the beam attenuation due to the absorption of the DAC components, parasitic diffraction of the crystalline components of the DAC (especially diamond anvils, their support plates and metal gasket), shadowing effects due to the DAC components (especially gasket and DAC steel body) (e.g. Miletich et al. 2005). For zeolites, which are usually materials with constituents with poor X-ray diffraction properties, the absorption phenomena are likely the most impacting on the data quality. In addition, in powder experiments an additional phenomenon usually occurs: the preferred orientation of crystallites, as common effect of uniaxial loads.
Computational modelling
The impressive evolution of high-performance computing (HPC) resources in the last decade, coupled with the concomitant development of theoretical chemistry, has fostered the application of computational techniques to a broad category of problems in zeolite science (Van Speybroeck et al. 2015). Theoretical tools are particularly precious in the study of events occurring at extreme conditions—e.g. high temperature or high pressure—i.e. in cases where obtaining experimental data at atomistic resolution or direct evidences of complex phenomena is often very difficult. In this contribution, we will focus on high-pressure investigations, underlining how modelling, in direct conjunction with experiment, can be used not only to aid structural determinations when refinement of crystallographic data is problematic, but also as a powerful predictive tool able to inspire the design and guide the fabrication of new zeolite materials through the use of high pressure. Nowadays, the scope of computational approaches in exploring pressure effects on open-framework silicates goes well beyond than that of providing the positions of the atoms in the unit cell starting from no empirically based information (Woodley and Catlow 2008). Nevertheless, structural elucidation performed “in silico” over the past two decades has fostered a substantial advance of our understanding about how these materials respond to compression, highlighting a key role of the guest species—molecules and cations—in guiding the deformation of the zeolite framework as a consequence of the applied pressure.
Computational approaches used in zeolite modelling may be categorized into three broad groups, based on the way in which the electronic structure and interatomic interactions of the system are represented and calculated: (1) “rigid unit mode” models and their extensions, namely template-based geometrical approaches; (2) classical force-field techniques—basically, Monte Carlo (MC) and molecular dynamics (MD) approaches; (3) quantum mechanical methodologies (a.k.a. “ab initio” or “first principles”).
The first category accounts solely for the strongest interactions in the system—covalent bonding and steric exclusion—and is constituted by the “flexibility-centred” methods. As they are based on simplified physical models, not only they are computationally fast and convenient, but they can also help interpreting experimental data and/or theoretical results at higher levels of theory. Application of these methods can provide valuable physical insight into the investigated phenomena, which is especially important in the high-pressure studies of zeolites (e.g. Sartbaeva et al. 2006, 2008; Wells et al. 2011). Specifically, the “rigid unit mode” (RUM) model (Giddy et al. 1993; Hammonds et al. 1994, Dove et al. 2000) treats the zeolite tetrahedra as fundamental, rigid, interacting units and analyses the flexibility of the structure in reciprocal space. Harmonic constraints are applied to the vertices of the tetrahedra, to penalize the separation of two adjacent units. RUMs, in which the polyhedra move as rigid bodies, without undergoing any distortion of their internal geometry, appear at zero frequency and are particularly useful to identify pressure-induced phase transitions. The template-based geometric analysis approach (Wells et al. 2002; Sartbaeva et al. 2006; Wells and Sartbaeva 2012, 2015), instead, works in the real space and considers both the atomic positions, and a set of geometrical templates reproducing the bonding pattern of groups of atoms—tetrahedra, in the case of zeolites. It is a more flexible approach, in the sense that there are still harmonic constraints—which connect atoms to the vertices of the tetrahedral templates—but, unlike in RUM, tetrahedra are not forced by construction to match exactly the input geometry. Rather, the templates and the atomic positions are iteratively relaxed to minimize mutual mismatches and avoid overlap between non-bonded atoms. Using this method, distortions from idealized geometry of the polyhedral templates can be readily visualized and quantified. This approach, implemented in the GASP code and recently reviewed (Wells and Sartbaeva 2012, 2015), has been successfully used for framework materials (Sartbaeva and Wells 2012). In particular, geometric simulations enabled to define an inherently geometrical feature of the zeolite framework: the “flexibility window” (Sartbaeva et al. 2006; Wells et al. 2015), i.e. the interval of unit-cell parameters in which the zeolite tetrahedral units can maintain their ideal shape. This important concept and its usefulness in analysing pressure-induced transformations will be discussed in the following section, together with some applications to high-pressure studies of zeolites.
Methods belonging to the second family are rooted in classical statistical mechanics and the interatomic interactions are modelled by effective potentials (Sanders et al. 1984; Demontis et al. 1987, 1988, 1990; De Boer et al. 1995). The parameters of the effective potentials are determined by fitting large sets of either experimental or higher level computational data (see, e.g. Gabrieli et al. 2014, 2016). In the study of zeolite materials, the most popular methods are Monte Carlo (MC) and molecular dynamics (MD) (Allen and Tildesley 1987; Demontis et al. 1990; Frenkel and Smit 2001). In the former one, the phase space of the system is sampled stochastically according to a given statistical ensemble, and physical quantities—such as energies, bond distances and cell parameters—are calculated by averaging over the sampled configurations. In MD, atoms are represented as classical particles obeying the Newton equations of motion, thus generating a trajectory; physical quantities are obtained by averaging over simulation time. Another widespread approach is grand-canonical MC, which allows to simulate systems with a variable number of particles. This technique was applied to the study of the pressure-induced H2O intrusion in hydrophobic silicalite-1 and LTA—as reported by Desbiens et al. (2005) and Coudert et al. (2009)—and also to the screening of zeolite structures for two important industrial processes: the purification of ethanol obtained from biomasses and isomerization of heavy alkanes in petroleum refining, both performed under moderate pressure conditions (Bai et al. 2015).
While MD can follow the time evolution of a system up to nanosecond scales, with MC it is possible to sample configurations not easily encountered along a standard trajectory. Nonetheless, both of them allow to simulate systems of larger size compared to quantum–mechanical calculations. They are normally applied to zeolites with large unit cells and to dynamic processes such as adsorption, intrusion and diffusion of extra-framework species (e.g. Smit and Maesen 2008; Combariza et al. 2013; Balestra et al. 2015; Gutiérrez-Sevillano et al. 2016; Viani et al. 2016). Although a few examples of first-principles MD applications are available—such as the site-to-site H2O diffusion in a partially dehydrated zeolite (Ceriani et al. 2004a)—classical MD still remains the method of choice for this kind of problems. To deal with temperature-induced or pressure-induced structural changes, special techniques are available—for example, constant-pressure molecular dynamics (MD) (Andersen 1980) or the Parrinello–Rahman approach (Parrinello and Rahman 1980, 1981), where the cell parameters are considered as additional dynamical variables, so that the simulation box is allowed to deform. However, very often the system must cross a significant energy barrier; in those cases, the phase transition becomes very unlikely in the accessible simulation time. To overcome this problem, free energy minimisation techniques were devised (Gale 1997); they were applied, e.g. to the monoclinic-to-orthorhombic phase transition in MFI zeolite at high temperature (Grau-Crespo et al. 2002). In another powerful approach, named metadynamics (Laio and Parrinello 2002; Martonák et al. 2003), penalty functions prevent the simulation from visiting again previously sampled configurations. Some examples of application to zeolites are the temperature-induced reconstructive phase transition from anhydrous Li-ABW to eucryptite (Ceriani et al. 2004b), and the simulation of pressure-induced amorphization in β-eucryptite at 3 GPa (Narayanan et al. 2013). In the latter case, simulations revealed that the amorphization mechanism, at the atomistic level, consisted in the tilting and distortion of tetrahedra centred at Al/Si sites, accompanied by changes in the Al coordination and disordering of Li cations.
In the third category of computational approaches, the electronic structure of the system is described quantum mechanically (“from first principles”) either via quantum chemistry methods—i.e. by solving the time-independent Schrödinger equation for the electronic wave function—or through density functional theory (DFT) approaches, where the energy is a unique functional of the electronic density. Since the exact form of the functional is unknown, approximated expressions are adopted (Parr and Yang 1989). In the simplest one, the local density approximation (LDA), the exchange correlation energy functional is calculated using the expression of a homogeneous gas of free electrons. To model zeolites, more accurate functional forms are used, which include dependency from the gradient of the density and are known as generalized gradient (GGA) approximations (e.g. Perdew 1986; Becke 1988; Lee et al. 1988; Perdew et al. 1996; Wu and Cohen 2006; Perdew et al. 2008). Hybrid functionals, which combine DFT with Hartree–Fock exchange (e.g. Becke 1993; Adamo and Barone 1999), can also be adopted; they are more accurate but more demanding than pure-DFT functionals. Also, an approximate treatment of dispersion effects—particularly crucial for modelling, e.g. the sorption of hydrocarbons in zeolites—may be introduced through the use of long-range corrections (e.g. Grimme 2006; Tkatchenko and Scheffler 2009; Grimme 2011).
Several functionals have been proposed over the years, and new ones are continuously developed. In this rapidly evolving scenario, benchmarking plays a crucial role to ensure that the choice of functional would be appropriate for the system under investigation. Benchmarking studies are generally designed to identify a functional (or a group of functionals) able to model a given set of physical quantities—e.g. cell parameters, bond distances and angles, vibrational and elastic properties—for a series of zeolite types by achieving a reasonable compromise between accuracy and cost. In this respect, dispersion-corrected GGA functionals perform particularly well, as it emerges from a series of recent benchmarks. Such detailed benchmark studies were performed both on hybrid functionals (Göltl and Hafner 2012; Göltl et al. 2012; Coudert 2013; Román-Román and Zicovich-Wilson 2015) and on GGA functionals using extensive databases of experimental data on aluminosilicate (Fischer 2015; Fischer et al. 2015; Bryukhanov et al. 2017) and neutral framework zeolites (Fischer et al. 2016; Fischer and Angel 2017).
First principles methodologies can properly describe the breaking and forming of chemical bonds occurring, e.g. in chemical reactions, or in reconstructive phase transitions, but imply a high computational overhead. Fortunately, the crystalline lattice of zeolites can be modelled using either periodic-DFT (e.g. Ballone et al. 2002; Otero Areán et al. 2007; Marx and Hutter 2009) or periodic ab initio methods (Pisani 1996, 1999; Dovesi et al. 2005; Larin et al. 2005; Demichelis et al. 2010), where a simulation box—characterized by a given atomic content, corresponding, e.g. to the crystallographic unit cell of the studied material—is replicated in the three dimensions forming an infinite lattice. Tensorial schemes for ab initio calculations of elastic properties (bulk modulus, shear modulus, Poisson ratio, etc.) at high pressure have been also implemented (e.g. Erba et al. 2017) and tested on garnets and other minerals (Erba et al. 2014a, b).
While standard (static) quantum chemical methods can accurately describe a limited number of atoms, classical approaches are more appropriate for an extended zeolite system. These methodologies can be combined giving rise to hybrid quantum mechanics/molecular mechanics (QM/MM) (see, e.g. Bludský et al. 2005; Morpurgo 2015); hybrid ab initio/DFT variants have also been proposed (see, e.g. Tuma and Sauer 2004, 2006; Piccini et al. 2016). Other approaches aimed at bridging the gap between first principles and classical descriptions are based on the partitioning of the total electron density among individual subsystems (Wesolowski and Warshel 1993; Tabacchi et al. 2005; Maerzke et al. 2009; De Silva and Wesolowski 2012), which maintains an accuracy level suitable for modelling, e.g. dye–zeolite hybrid composites (Zhou et al. 2013). There are, however, situations where both a quantum mechanical treatment of the electronic structure and a description of thermal effects via the atomic motion are needed. These requirements can be satisfied by the first principles molecular dynamics (FPMD) approach, originally proposed by Car and Parrinello (Car and Parrinello 1985; Remler and Madden 1990; Marx and Hutter 2009).
FPMD enables to study the time evolution of a system with first-principle accuracy because the forces on the atoms at each MD time step are obtained using a quantum mechanical description of the electrons. More commonly, the electronic structure is treated with DFT (Marx and Hutter 2009), even though wavefunction-based variants of this technique exist as well. The Car–Parrinello scheme for FPMD defines a fictitious dynamical system in which the potential energy surface depends on both the nuclear and the electronic degrees of freedom (Car and Parrinello 1985). The electronic wave function coefficients are propagated in time as classical degrees of freedom, and their dynamics generates at each time step the correct adiabatic electronic configuration corresponding to the new ionic positions. Hence, if at the beginning of the simulation the electronic orbitals correspond to the ground state, they will follow the motion of nuclei adiabatically and remain in that state as the nuclear configuration evolves in time. However, the dynamical parameters (fictitious electronic mass and time step) have to be chosen so that the transfer of energy between ions and electrons is kept very small during the simulation (Remler and Madden 1990). This condition is easily satisfied in systems with a large energy gap, such as zeolites.
FPMD has been implemented using different types of basis sets (Lippert et al. 1997; VandeVondele et al. 2005; Marx and Hutter 2009), namely plane waves (PW), projector augmented waves (PAW), and localized functions (VandeVondele et al. 2005). Periodic boundary conditions are generally adopted in all cases. In applications to zeolites, this implies the capability of describing the full crystal from first principles, thus properly reproducing the flexibility properties of the framework—which are especially important in governing the response of zeolites to an applied pressure. Whereas the direct simulation of a pressure-induced phase transition would require variable cell parameters, in many other cases it is convenient to hold the cell parameters fixed at the values experimentally determined at such pressure conditions (where available), which are known to be very accurate. Importantly, no constraints—either symmetry restraints or “frozen” nuclear positions—are normally imposed to the atoms in the simulation cell, which are thus left free to move according to their own potential energy surface determined by the interatomic interactions.
FPMD simulations, due to their high computational cost, are limited to elapsed times of the order of tens of ps, which are too short to observe activated events such as, e.g. chemical reactions, or reconstructive phase transitions. Such processes can be considered rare events on the FPMD time scale. To address these problems, rare-event sampling techniques have been developed, such as the above-mentioned metadynamics (Laio and Parrinello 2002; Barducci et al. 2011). In the case of zeolites, the FPMD extension of metadynamics (Iannuzzi et al. 2003) has been mainly applied to study activated processes occurring at high temperature, e.g. the methanol-to-olefin process catalysed by H-SAPO-34 (De Wispelaere et al. 2015), and the fabrication of zeolite hybrid functional materials (Calzaferri et al. 2003; Cao et al. 2016) through the intrusion of dye molecules inside the one-dimensional channels of zeolite L (Tabacchi et al. 2016). Other popular statistical sampling methods are the so-called “bluemoon” ensemble (Carter et al. 1989), the “transition path sampling” (Dellago et al. 1998) or the “nudged elastic band” (Jónsson et al. 1998; Sheppard et al. 2012). In all cases, a basic knowledge of the behaviour of the system at equilibrium conditions—which may generally be obtained via exploratory simulations—is needed. Considering the rapid and continuous increase in computing power, it may be foreseen that these methods will become useful tools in the prediction of the high-pressure behaviour of zeolite materials.
High-pressure behaviour without any crystal–fluid interaction
To describe the HP-behaviour of a zeolite without any interference of the P-fluid, the sample (polycrystalline or single crystal) is compressed in a “non-penetrating” P-transmitting medium (sensu Gatta 2008): a fluid made by molecules which cannot penetrate through the structure cavities in response to the applied pressure. Usually, the mix methanol:ethanol = 4:1, glycerol, isopropanol, perfluoroether, fluorinert, or various grades of silicone oils are used as nominally non-penetrating P-transmitting fluids (Angel et al. 2007; Gatta 2008; Klotz et al. 2009).
Simulation or structure refinements showed that open-framework materials accommodate the volume compression mainly by tilting, distortion and contraction of the primary building units: the tetrahedra. Tilting of tetrahedra occurs around the bridging oxygen atoms that act as “hinges”; as a consequence, this mechanism does not generate distortion of the tetrahedra, but changes the inter-tetrahedral T–O–T angles with P. Distortion of tetrahedra is reflected by changes in the intra-tetrahedral O–T–O angles, preserving the average T–O bond length. Contraction of tetrahedra is expressed by compression of the T–O bond distances. Tilting, distortion and contraction of tetrahedra act simultaneously at any pressure. However, tilting is an energetically less-costly mechanism if compared to distortion or contraction, and this corroborates the experimental evidence that tilting is the dominant mechanism at low-P regime, whereas distortion and then contraction becomes dominant at higher pressure, when tilting cannot accommodate the effect of pressure efficiently any further (Gatta 2010a; Gatta and Lee 2014). In other words, there is a hierarchy of the deformation mechanisms (Gatta 2010a), and this is independent of the nature of the cations in tetrahedral coordination (e.g. Si, Al, P, Be, B,…). As a matter of fact, at least up to 3–5 GPa, tetrahedra behave as “rigid-units” at least to a first approximation, e.g. the estimated fictive bulk modulus (defined as K P0,T0 = −V 0(∂P/∂V) P0,T0 = 1/β P0,T0, where β P0,T0 is the volume compressibility coefficient at ambient conditions) of SiO4 is 580(24) GPa (Zhang et al. 1998). Significantly, the rigid-unit behaviour is fully supported by available atomistic data obtained from FPMD simulations, and may also hold at above the 5 GPa threshold. For example, the Si and Al tetrahedral units of the zeolite gismondine undergo volume contractions of only 0.05 and 0.08%, respectively, at 7.6 GPa (Betti et al. 2007). Moreover, the above-mentioned hierarchy of deformation mechanisms is further corroborated by recent lattice-dynamic calculations on a low-silica zeolite with LTA framework (Gulín-González et al. 2016).
Tilting of tetrahedra can be observed in all the HP-experiments on zeolites, whose structure refinements or simulations are available. A common effect of tilting is the deformation of the secondary building units (SBU) of the zeolitic frameworks, which are represented by “open forms” (e.g. 4-, 5-, 6-, 8-, 10- or 12-membered rings of tetrahedra) or “3D closed forms” (e.g. double six-membered rings, T5O10 units of the “fibrous zeolites group”, T10O20 units of the “heulandite group”; Baerlocher et al. 2007). Usually, tilting of tetrahedra produces a continuous rearrangement of the structure, reflected by a monotonic unit-cell volume compression. It is common, for example, to observe that the ellipticity of the zeolitic channels tends to increase monotonically with P, via tilting of the tetrahedra that confine the channels, without any “inversion” (Gatta and Lee 2006); an “inversion” in ellipticity at high-P usually leads to a phase transition. More complex is the description of the response, at the atomic scale, of zeolites with secondary building units made by 3D closed forms, which behave as “rigid block-units” at a first approximation: at high pressure, their structures respond first by rotation of the SBUs, followed by SBU compression (but keeping the tetrahedra undeformed), and finally by deformation or compression of the tetrahedra themselves. The first two mechanisms are substantially based on tilting and are very efficient to accommodate the effect of pressure at low-medium P-regime. Two examples in this respect are those of the “fibrous zeolites group” (NAT, THO, and EDI framework type; SBU: T5O10 or 4 = 1 unit; Baerlocher et al. 2007) and of the “heulandite group” (HEU framework type; SBU: T10O20 or 4-4 = 1 unit; Baerlocher et al. 2007). All fibrous zeolites studied at high-P (i.e. natrolite, scolecite, thomsonite, edingtonite) show a similar deformation mechanism in response to applied pressure: cooperative rotation (anti-rotation) of the SBU about the axis of the SBU-chains, which in turn gives rise to the compression of the eight-membered rings channels (parallel to the SBU chain axis), with an increase of their ellipticity (Gatta 2005; Gatta and Wells 2004; Gatta et al. 2016b). In fibrous zeolites, the estimated fictive bulk modulus of the SBU is approximately twice those of the zeolites (i.e. ~115 GPa for the SBU vs. ~43 GPa for natrolite, ~55 GPa for scolecite, ~49 GPa for thomsonite and ~59 GPa for edingtonite) (Gatta 2005). The identical framework topology of natrolite and scolecite (i.e. NAT, Baerlocher et al. 2007) allowed also to speculate about the role played by the extra-framework population on the compressional behaviour of isotypic materials: the framework topology controls the main deformation mechanisms (in this case: cooperative anti-rotation of the SBU by tilting of tetrahedra), but the channel population (i.e. ionic radius of cations, valence, and their bonding configuration) controls the compressibility of the cavities, resulting in different compressibility of unit-cell volume. In addition, fibrous zeolites provide further experimental evidence: ordering of the Si/Al in the tetrahedral sites does not influence structure compressibility, which is virtually identical in orthorhombic (fully ordered Si/Al distribution) and tetragonal (highly disordered Si/Al distribution) edingtonite (Gatta et al. 2004a, b).
In heulandite, the corrugation (and thus the shortening) of the zig-zag chains of SBUs, parallel to [102], is the main deformation mechanism in response to applied pressure, which acts basically by tilting of tetrahedra. In addition, in this case, the estimated fictive bulk modulus of the SBU (i.e. ~63 GPa) is approx. twice that of the zeolite (i.e. ~28 GPa) (Comodi et al. 2001; Gatta et al. 2003). In general, the experimental findings on zeolites with closed-form SBUs show that the compressibility of the structure is the combined effect of soft channels and relatively stiff 3D SBUs compression.
Important quantitative insight into the tilting deformation mechanism may be obtained by investigating how the T–O–T angles evolve as a function of increasing pressure even in structures with “open” SBU. Interestingly, such angles were identified to be responsible for the pressure-induced framework modification up to 8–10 GPa on the basis of integrated experimental-computational data collected for a series of zeolites, e.g. bikitaite (BIK framework type, Ferro et al. 2002; Fois et al. 2002a; Baerlocher et al. 2007), yugawaralite (YUG framework type, Fois et al. 2005a, b; Baerlocher et al. 2007), and gismondine (GIS framework type, Betti et al. 2007; Baerlocher et al. 2007). For example, in gismondine, the average value of the T–O–T angle decreases significantly (from 142.8° to 137.8°) in passing to room pressure to 7.4 GPa (Betti et al. 2007). As a general trend, the decrease of the average T–O–T angle correlates with both applied pressure and volume contraction: namely, the higher the compression, the higher the variation of the angle. This analysis, therefore, brings further support to the intuitive argument that the framework responds to compression by modifying its most flexible element, that is, the junction between tetrahedral units.
A few representative examples can be considered to highlight that tilting can act as the main deformation effect even when the saturation of one mechanism is achieved at low-P. Gatta et al. (2005) and Gatta and Wells (2006) reported, by experiments and computational modelling, that the framework of levyne (LEV framework type, Baerlocher et al. 2007) reacts, under compression, by tilting of tetrahedra following two distinct deformation mechanisms: the first dominant at P < 1 GPa, through the cooperative rotation of tetrahedra belonging to the double six-membered rings; the second at P > 1 GPa, through compression of the four-membered ring (“joint-unit”) between the aforementioned six-membered rings, as the double six-membered rings have already reached a limit configuration at P ~ 1 GPa. These two mechanisms can explain the anomalous elastic behaviour of levyne, with an increase of the c-axis length between 0 and 1 GPa followed by a monotonic compression at P > 1 GPa. Up to 5 GPa, structure refinements and simulations show no distortion or compression of the tetrahedra, at a significant level, in levyne structure.
More recently, Comboni et al. (2017) reported the HP-behaviour of phillipsite (PHI framework type, Baerlocher et al. 2007) up to 9.4 GPa by an in situ single-crystal diffraction experiment. Despite using a hydrous P-transmitting fluid (i.e. methanol:ethanol:water = 16:3:1), no clear evidence of P-induced penetration of extra molecules through the zeolitic cavities was observed within the P-range investigated. However, two different compressional regimes occur. Between 0.0001 and 2.0 GPa, phillipsite behaves as an unusually stiff porous material: the refined bulk modulus is K P0,T0 = 89(8) GPa. Between 2.0 and 9.4 GPa, the material turns to be drastically softer and its bulk modulus decreases to K P0,T0 = 18.8(7) GPa. The structure refinements proved that, at P > 2 GPa, a P-induced change in the configuration of the H2O molecules, coupled with a change of the tilting mechanisms of the framework tetrahedra, gives rise to a completely different compressional behaviour. Accordingly, the evolution of the monoclinic β angle with pressure shows two distinct trends in the two compressional regimes: with a negative slope between 0.0001 and 2.0 GPa, and a positive slope between 2.0 and 9.4 GPa. The tilting of the tetrahedra, in response to the hydrostatic compression, causes the deformation of the eight-membered ring of tetrahedra which confines the [010] channel and, in turn, the inversion of β vs. P trend. In this specific case, the saturation of a tilting mechanism and the role played by the extra-framework population concur to a change of the compressional behaviour, without any P-induced phase transition.
Levyne and phillipsite are two representative examples of zeolites which do not undergo any P-induced phase transition, despite they experience a change of deformation mechanisms, reflected by a change of the unit-cell volume compressional pattern. The list can be extended, for example, to gobbinsite (GIS framework type, Baerlocher et al. 2007; Gatta et al. 2010, 2012; Gatta and Lotti 2011), cancrinite (CAN framework type, Baerlocher et al. 2007; Lotti et al. 2012) or nepheline (a feldspathoid, Gatta and Angel 2007). On the other hand, some zeolites with high “framework density” (FD, defined as the number of T atoms in a volume of 1000 Å3, Baerlocher et al. 2007) or hydrophobic zeolites (i.e. zeolites nominally without any extra-framework population) tend to react to applied pressure with phase transitions, which are usually displacive in character, and the high-pressure polymorphs can have distorted tetrahedra. A representative example, in this respect, is that of zeolites of the analcime group (i.e. analcime, leucite, wairakite and pollucite; ANA framework type, Baerlocher et al. 2007): all these zeolites experience a first-order phase transition from the high-symmetry low-P polymorph (cubic analcime and pollucite, tetragonal leucite and monoclinic wairakite) to a triclinic high-P polymorph, at relatively low-P (~1 GPa for analcime, ~2.4 GPa for leucite, ~2.5 GPa for wairakite, ~0.7 GPa for pollucite; Gatta et al. 2006, 2008, 2009a; Ori et al. 2008a). The phase transitions are fully reversible upon decompression. For analcime, in particular, structure refinements (based on single-crystal intensity data) revealed that the main deformation mechanisms of the HP-polymorph act through deformation of the four- and six-membered rings of tetrahedra by tilting of the polyhedra, along with their significant distortion. A spectacular HP-behaviour with two P-induced phase transitions in all-silica ferrierite (FER framework type, Baerlocher et al. 2007) was recently reported by Lotti et al. (2015a), by in situ single crystal and powder X-ray diffraction experiments, using penetrating and non-penetrating fluids. Using silicone oil, as a polymeric non-penetrating P-fluid, the experimental data show a remarkable flexibility of the ferrierite framework at high pressure with two displacive phase transitions, following the path Pmnn-to-P121/n1-to-P21/n11, the first at ~0.7 GPa and the second at ~1.24 GPa (Fig. 1). The transitions are fully reversible. As reported by Lotti et al. (2015a), the two monoclinic space groups do not share a group–subgroup relationship: the multistage phase transition requires an intermediate structure with P-1 symmetry, as common subgroup of both P121/n1 and P21/n11. The three polymorphs share a virtually identical bulk compressibility, but with a different anisotropic compressional pattern. Also in this case, the structure evolution in response to applied pressure is mainly governed by tilting, and the phase transitions are the effect of deformation mechanisms saturation, followed by the promotion of new ones in a structure configuration which is energetically more favourable.

Structure evolution of the all-silica ferrierite compressed in a non-penetrating fluid (see text for further details)
Phase transitions can drive to “new materials” which are industrially important: a new zeolite ITQ-50 was obtained by compressing a synthetic all-silica zeolite ITQ-29 in a DAC with a non-penetrating P-transmitting fluid (Jordá et al. 2013). As from the crystallographic data it was possible to obtain only the cell parameters of the new material, the structure was obtained with the aid of classical force-field calculation. Remarkably, the pressure-synthesized microporous material showed better performances than the precursor zeolite in the propene/propane separation—an important industrial process.
Besides the modification induced to the framework, it is useful to highlight how the organization of the extra-framework species is modified by compression. For example, the natural zeolite bikitaite hosts in its one-dimensional channels hydrogen bonded chains of H2O molecules, which at room conditions display a behaviour typical of a solid phase of water at low dimensionality—hence, the name “one-dimensional ice” (Fois et al. 1999, 2001b)—and an impressive resistance towards dehydration despite not being hydrogen bonded to the framework (Ceriani et al. 2004a). Indeed, upon compression, the one-dimensional ice structure persisted up to 5 GPa, but new hydrogen bonds were formed between the H2O chains and the framework oxygen atoms (Ferro et al. 2002). Even more intriguingly, a similar behaviour was found when bikitaite was compressed in penetrating (water-containing) medium up to 4 GPa: the H2O chains were preserved up to the highest pressure with no intercalation of additional H2O molecules (Seryotkin 2016), highlighting thus the exceptional stability of this confined H2O nanostructure. Actually, also the synthetic zeolite Li-ABW hosts in its monodimensional channels the same peculiar H2O wires (Fois et al. 2001a, b); in this case, however, the supramolecular H2O architecture is easily disrupted when moderate pressures are applied (Fois et al. 2008a). Theoretical calculations revealed that the reason why these two similar systems display such a different response to compression is the electric polarization of the zeolite framework, which is considerably higher in bikitaite compared to Li-ABW, thus providing a greater electrostatic stabilization to the H2O chain (Fois et al. 2001a, 2008a). Porous materials with monodimensional channel systems can host other interesting examples of confined H2O nanostructures—such as the H2O-triple helix in VPI-5 aluminophosphate (Fois et al. 2002b), or the pressure-induced H2O nanotube in zeolite LTL (Lee et al. 2007). The interest in the behaviour of one-dimensional channel materials—at both standard and high-P conditions—stems from the possibility of filling them, upon H2O evacuation, with guest species of suitable size, for example, drugs (Delle Piane et al. 2014) or luminescent molecules, giving rise to low-dimensionality systems endowed by technologically appealing properties (see, e.g. Calzaferri et al. 2003; Fois et al. 2005c; Manzano et al. 2013; Gigli et al. 2014; Cucinotta et al. 2014, Calzaferri 2017; Gartzia-Rivero et al. 2017).
The key question is: why some zeolites experience P-induced phase transitions (with no crystal–fluid interaction) and others not? In addition, which are the limit conditions above which an open-framework structure undergoes a phase transition? Is it possible to predict a transition pressure? The state of the art of the knowledge does not allow to provide an unambiguous answer to these questions. However, the experimental findings about P-induced phase transitions in zeolites (with no crystal–fluid interaction) appear to be well predictable by the “flexibility window” theory. The “flexibility window” (Sartbaeva et al. 2006) is a pervasive property of zeolitic frameworks and it defines a range of densities over which the corner-sharing TO4 units, making up the framework, can in principle be made perfectly tetrahedral (i.e. a range of densities over which the tetrahedra retain their holosymmetric shape). This window is limited at high density by contacts between oxygen atoms on neighbouring TO4 units, and at low density by extension of the intra-tetrahedral T–O bonds (though not, in general, by linear T–O–T angles). Gatta et al. (2009b) reported that the ANA cubic framework, for example, displays a narrow flexibility window: the framework could be compressed by only 3% in volume before oxygen atoms came into contact. The geometric simulations on the ANA framework (using a tetrahedral geometry appropriate to the Si:Al ratio of natural cubic analcime) (Sartbaeva et al. 2006, 2008; Wells et al. 2011), with the experimental cubic unit-cell parameters measured at high pressure (Gatta et al. 2006), showed that the structures are perfectible over the whole observed range of cubic analcime, from ambient conditions up to around 1 GPa. The theoretical upper and lower limits of the flexibility window in cubic analcime were obtained by simulations using the unit-cell parameters outside the range observed in the experiment. The low-density edge of the window was found to be near the density observed for the analcime structure at ambient conditions (i.e. the ambient structure is close to maximal extension), whereas the high-density edge of the window on compression lies near the last observed cubic structure before the transition. Overall, the P-induced phase transition in analcime, observed at about 1 GPa, is well predictable on the basis of the flexibility window of this structure.
High-pressure behaviour with crystal–fluid interaction
“Penetrating” P-transmitting media are a class of fluids which contain molecules able to penetrate, through selective sorption, into the zeolitic cavities in response to applied pressure. This phenomenon implies a drastic effect on the compressional pattern and on the P-induced deformation mechanisms at the atomic level. Elemental gaseous media at ambient conditions (e.g. helium, nitrogen, neon, argon, xenon, kripton) and small molecules (e.g. H2O, CO2) are potentially penetrating media. Even some larger molecules, usually used as P-transmitting media (e.g. methanol, ethanol, ethylene, ethylene glycol, acetylene) could be considered as potentially penetrating media. Penetrability of external molecules (or atoms) at high pressure is governed by several variables, among those the most important are: the “free diameters” of the framework cavities, the chemical nature and the configuration of the extra-framework population, the partial pressure of the penetrating molecule in the fluid (if mixed with other non-penetrating molecules, e.g. P(H2O) in a mixture of alcohol–H2O), the temperature at which the experiment is conducted. A given molecule can, therefore, act as a penetrating medium for some zeolites and as a non-penetrating medium for other zeolites.
The effects of the P-induced penetration depend on the nature of the penetrating atoms or molecules through the zeolitic cavities, and, for a comparative analysis, it is convenient to distinguish among monoatomic species, small molecules and larger molecules.
P-induced penetration of monoatomic species
Lee et al. (2010) showed that, when compressed in liquid Ar, natural natrolite (of ideal composition Na16Al16Si24O80·16H2O, NAT framework type, Baerlocher et al. 2007) can incorporate a significant amount of Ar under moderate pressure and temperature conditions (i.e. 60 °C for 10 h). The P-induced penetration of Ar gives rise to a spectacular P-induced expansion of the unit-cell volume (i.e. ~6.5% larger unit-cell volume than the starting zeolite) at P ~ 3.0–3.5 GPa, with a new Ar-bearing form of natrolite described as Na16Al16Si24O80·16H2O·6Ar. The expansion of the unit-cell volume is governed by the expansion of the eight-membered ring channels along [001], where the extra-framework population lies. The structure refinement of Na16Al16Si24O80·16H2O·6Ar showed that Na+ remains six-coordinated, with the Ar–Na distances ranging between 2.90 and 3.22 Å and the Ar–O distances at ~3.24 Å. In other words, Ar interacts via short-range van der Waals forces. Natrolite served as an efficient host system for further experiments with monoatomic penetrating species: Xe and Kr. Seoung et al. (2014) reported how Ag-natrolite, ideally Ag16Al16Si24O80·16H2O, adsorbs xenon into its eight-membered ring channels at 1.7 GPa and 250 °C, while Ag+ is reduced to metallic Ag0 and possibly oxidized to Ag2+. The sorption of Xe gives rise to an expansion by 3.2% of the unit-cell volume, with a weak interaction of Xe and Ag (i.e. Ag–Xe ~3.1 Å). Surprisingly, the sorption of xenon is irreversible after pressure release and requires heat to desorb. Using liquid Kr as P-transmitting fluid, Ag-natrolite adsorbs Kr at 2.1 GPa and after annealing at 250 °C, with a partial reduction of Ag+ to metallic Ag0. The penetration phenomenon leads to a moderate unit-cell volume expansion (only ∼1.2%) and appears to be fully reversible under decompression.
In zeolites with larger cavities than those in natrolite, the P-induced penetration of monoatomic species does not imply a unit-cell volume expansion, but rather a different compressional behaviour if compared to that obtained in a non-penetrating fluid. For example, Niwa et al. (2013) reported how the synthetic zeolite Linde-type A (LTA framework type, Baerlocher et al. 2007) compressed in liquid He and Ar shows different compressional patterns, which are, in turn, different if compared to those reported for compression in liquid water (penetrating) or in silicone oil (non-penetrating polymeric fluid). Structure refinements were not performed, leaving open questions about the mechanisms at the atomic scale. However, the evolution of unit-cell volume vs. P is sufficient to prove that the P-induced penetration of He, Ar and H2O occurs, with a reduction of the compressibility, if compared to that with a non-penetrating fluid, by 50, 15 and 700%, respectively. The sorption of extra H2O molecules affects dramatically the compressibility of Linde-type A. The reduction of compressibility reflects the “pillar effect” that the new intruded atoms or molecules generate, making the zeolitic cavities less compressible (not collapsible).
P-induced penetration of small molecules
Experiments devoted to the P-induced penetration of small molecules are probably the most common. The P-induced penetration phenomenon was first discovered using water, or hydrous mixtures, by Hazen (1983) and Hazen and Finger (1984), on the basis of the significantly different compressional pattern of a synthetic zeolite (i.e. Linde Na-A; LTA framework type, Baerlocher et al. 2007) in different fluids. Lee et al. (2002a, b) reported the very first structure refinements proving the P-induced sorption of extra-H2O molecules, from the P-transmitting fluid through the zeolitic cavities (the so-called P-induced “superhydration”), in a natrolite and its synthetic counterpart K16Ga16Si24O80·12H2O. Natrolite, ideally Na16Al16Si24O80·16H2O (NAT framework type, Baerlocher et al. 2007), transforms to Na16Al16Si24O80·32H2O at ~1.2 GPa (via the intermediate ordered-paranatrolite phase with 24 H2O molecules per formula unit, stable between 0.9 and 1.2 GPa, Lee et al. 2005), doubling the number of molecules p.f.u. of H2O with a consequent unit-cell volume expansion by ~2.5%, if compared to natrolite stable at ambient P. The extra H2O molecules intruded in the eight-membered ring channel are bonded to Na, which increases its coordination number. The P-induced superhydration effect is completely reversible in natrolite but not in its synthetic counterpart K16Ga16Si24O80·12H2O, suggesting that the chemical nature of extra-framework (and likely framework) population plays an important role in governing the penetration reversibility. The P-induced penetration of H2O in natrolite was described with different experimental methods (Colligan et al. 2005; Seryotkin et al. 2005), and it was observed in almost all the fibrous zeolites with spectacular volume expansion (e.g. natrolite, scolecite and thomsonite, e.g. Lee et al. 2002a, b, 2005; Colligan et al. 2005; Gatta 2005; Likhacheva et al. 2006, 2007; Seoung et al. 2013, 2015; Seryotkin et al. 2017), with the exception of edingtonite (EDI framework type, Baerlocher et al. 2007; Gatta et al. 2004a, b): the large Ba-polyhedron fills very efficiently the eight-membered ring channel along [001], which is in an already expanded configuration if compared to the other fibrous zeolites, hindering further penetration of H2O molecules.
The role of the cations in natrolite under superhydration conditions was recently explored theoretically by investigating the P-induced behaviour of Na-, Rb-, and Cs-natrolite (Kremleva et al. 2013) and of the K-substituted counterpart (Kremleva et al. 2014). With calculations based on density functional theory (DFT), these studies were able to reproduce approximately the critical pressure values at which the corresponding transformations were found to occur in experiments, providing, therefore, valuable insight into their microscopic details. Based on the modelling results, these authors also predicted the possible formation, at high-pressure conditions, of two isomers of superhydrated K-NAT, with either positive or negative chain rotation angles (Kremleva et al. 2014).
An additional P-induced expansion phenomenon was observed in laumontite, ideally Ca4Al8Si16O48·nH2O with n ≤ 18 (LAU framework type, Baerlocher et al. 2007). Using a partially dry sample with 12 H2O m.p.f.u., Lee et al. (2004) showed, using a hydrous P-transmitting fluid and a powder sample, that laumontite experiences a phase transition at P = 0.2(1) GPa, with a spectacular unit-cell volume increase of ~2.6%. The structure refinements proved that the expansion reflects the transition to a fully hydrated form (with 18 H2O m.p.f.u.), by selective sorption of extra H2O, which leads to the expansion of the eight-membered ring channels along [001]. Significantly, P-induced hydration in laumontite was first investigated by a computer modelling study performed with a classical force field by White et al. (2004). Such a study predicted the occurrence of full hydration at moderate pressures, which was then confirmed by the experiments, evidencing that, when pressure is applied, the structural stability of the laumontite-type framework increases because of the fully occupied H2O network.
Fibrous zeolites and (partially dehydrated) laumontite represent rare examples of P-induced insertion of H2O molecules with spectacular expansion of the unit-cell volume. Usually, the penetration of extra H2O molecules from the P-fluid is not accompanied by unit-cell volume expansion, as shown, e.g. for: the synthetic Linde-type A (ideally Na12Al12Si12O48·26 H2O, LTA framework type; Hazen 1983; Hazen and Finger 1984; Arletti et al. 2003; Likhacheva et al. 2009; Niwa et al. 2013); synthetic Li-, (Na,Cs)- and Cd-RHO zeolites [(Li,Na,Cs)12(Al12Si36O96)·44H2O, RHO framework type; Lee et al. 2001]; synthetic all-silica zeolite Y (FAU framework type, Colligan et al. 2004); gismondine (ideally Ca4Al8Si8O32·16H2O, GIS framework type; Ori et al. 2008b) and its synthetic K-gallosilicate counterpart (K-GaSi-GIS, Lee et al. 2008); boggsite (ideally Ca8Na3Al19Si77O192·70H2O, BOG framework type; Arletti et al. 2010); synthetic Na-ZSM-5 (i.e. (Na4.58K0.02)(Ca0.18Mg0.03Ba0.01Fe0.05Sr0.01)(Al4.48Si91.35)O192 ·28.39H2O, MFI framework type, Arletti et al. 2011) and H-ZSM-5 (i.e. (H6.8Na1.1)(Al7.9Si89.8)O192 ·36H2O, MFI framework type, Quartieri et al. 2011); all-silica ferrierite (FER framework type; Lotti et al. 2015a); paulingite (ideally (K,Na,Ca0.5,Ba0.5)10(Al10Si32O84)· nH2O, with n = 27–44, PAU framework type; Gatta et al. 2015); synthetic AlPO4-5 (AFI-framework type; Lotti et al. 2016).
The experiment of Colligan et al. (2004), on a purely siliceous zeolite Y (FAU framework type, Baerlocher et al. 2007), deserves a particular attention. Colligan et al. (2004) compressed a synthetic all-silica zeolite Y (FAU framework type) in silicone oil and in methanol:ethanol:water = 16:3:1 mix using a DAC, and described its HP-behaviour on the basis of in situ synchrotron X-ray powder diffraction data, with Rietveld structure refinements and computational modelling. This was probably the first experiment in which a neutral and hydrophobic zeolitic framework was used to describe P-induced penetration phenomena, giving the opportunity to examine the effect of pressure on a porous silicate without interferences due to extra-framework charge-balancing cations and their interactions with framework oxygen atoms. Compressed in the mixture of alcohol–water, this zeolite shows a drastically lower compressibility than that observed in (non-penetrating) silicone oil. In addition, in alcohol–water mix, two distinct compressional patterns occurred, with a changeover at 4 GPa. The Rietveld structure refinements proved that new extra-framework sites occur at high pressure, modelled as partially or fully occupied by oxygen atoms of H2O molecules. The sum of the extra-framework site occupancies increases with pressure, and the pore filling saturation is achieved at about 4.0 GPa. This last experimental finding allowed the authors to explain the change of the compressional behaviour of this zeolite compressed in methanol:ethanol:water = 16:3:1 mix: (1) P-induced intrusion of H2O molecules is the principal process that occurs between 0.0001 and 4 GPa, leading to a drastically lower compressibility if compared to that observed in silicone oil; (2) when the filling of the pores is completed, at P > 4 GPa a new compressional pattern, with higher compressibility, is observed and the effect of hydrostatic compression is predominantly accommodated by framework deformation (mainly via tetrahedral tilting). Unfortunately, no structure refinements were performed in decompression, leaving open questions about the reversibility of the intrusion process. With this experiment, Colligan et al. (2004) showed how polar molecules (i.e. H2O molecules) can be intruded in a neutral framework in response to applied pressure.
Some experiments have been performed using liquid CO2 as penetrating P-transmitting fluid. Natrolite (Na16Al16Si24O80·16H2O), for example, transforms to Na16Al16Si24O80·16H2O·8CO2 at 1.5 GPa (Lee et al. 2011). The penetration of extra CO2 molecules through the eight-membered ring channels, running along [001], leads to an expansion of the channels and, in turn, to a spectacular unit-cell volume increase by ~6.8%. The CO2-bearing natrolite stable at high pressure contains ~12 wt % of CO2, and its symmetry decreases from orthorhombic (Sp. Gr. Fdd2) to monoclinic (Sp. Gr. Cc). The intruded CO2 molecules give rises to a rearrangement of the extra-framework population, with a migration of the H2O molecules toward one side of the channel. CO2 molecules interact with both Na and H2O, and lie in a plane almost perpendicular to the channel direction. The structure refinements after decompression showed that Na16Al16Si24O80·16H2O·8CO2 is meta-stable after P-release (even after an equilibration time of 1 h).
Haines et al. (2010) showed that the P-induced intrusion of extra molecules of CO2 in silicalite (MFI framework type) hinders its P-induced amorphization (at least up to 20–23 GPa), whereas in a non-penetrating fluid this zeolite showed effects of amorphization at 4–5 GPa.
P-induced penetration of complex molecules and polymerization phenomena
Santoro et al. (2013) reported the first evidence of P-induced photo-polymerized ethylene in the channels of silicalite (MFI framework type, Baerlocher et al. 2007), using single crystal and powder of SiO2–silicalite compressed in supercritical fluid C2H4. The penetration of C2H4 molecules occurs at 0.5–1.5 GPa, and the polymerization is promoted under ultraviolet (351–364 nm) irradiation. Experimental evidence, based on optical spectroscopy and X-ray diffraction, confirmed that the structure of C2H4-bearing silicalite, recovered at ambient pressure, contains single polyethylene chains confined by the zeolitic channels. The C2H4-bearing silicalite shows a significant increase of its bulk modulus and density if compared to the parental silicalite, in response to the P-induced pore-filling effect.
On the same zeolite, a further experiment was reported by Scelta et al. (2014) devoted to the P-induced polymerization of acetylene molecules (to poly-acetylene chains) through the zeolitic cavities. The authors used a multi-methodological approach, based on in situ and ex situ measurements (by IR spectroscopy, Raman spectroscopy and X-ray diffraction), to describe how with a DAC and using only high pressure (∼4 GPa; no temperature or ultraviolet irradiation) it is possible to promote the penetration and re-organization (via polymerization) of C2H2 molecules through the zeolite channels.
Silicalite was also selected as the zeolitic host for a further experiment aimed to investigate the P-induced polymerization in all-silica zeolites: Santoro et al. (2015) reported the P-induced synthesis of all-transoid polycarbonyl [–(C=O)–] n in a zeolite, starting from mixtures of (solid) CO and powder or single crystal of silicalite, compressed in a DAC. Using a multi-methodological approach (based on IR, Raman, single-crystal X-ray diffraction, and ab initio computational methods for calculating the vibrational spectrum of polymerised CO), the authors reported how compressing the zeolite in solid CO, evidence of CO penetration through the zeolitic cavities with a re-organization in a polymeric configuration were found. The experimental findings indicated that the average interaction between confined polymerised CO and the host silicalite is of the van der Waals type, and the resulting IR spectra are compatible with the all-transoid polycarbonyl [–(C=O)–] n chains predicted by DFT studies. The ex situ measurements proved that the P-induced penetration and polymerization of CO is an irreversible process. To the best of our knowledge, this is one of the very rare examples in which the P-induced penetration of external molecules occurs in a solid host compressed in a solid medium.
The same group of researchers (Santoro et al. 2016) extended their experiments on a different zeolite with a monodimensional system of channels: the all-silica ZSM-22 (TON framework type, Baerlocher et al. 2007). Polycrystalline ZSM-22 compressed (up to 5–10 GPa) in (liquid) acetylene and (liquid) CO, loaded cryogenically in a DAC, experiences similar phenomena as those previously described for silicalite: the intrusion and the subsequent irreversible polymerization of C2H2 (to poly-acetylene) and CO (to polycarbonyl [–(C=O)–] n ).
Arletti et al. (2015) investigated the behaviour of a synthetic high-silica mordenite (MOR framework type, Baerlocher et al. 2007) compressed in a non-penetrating fluid (i.e. silicone oil) and a series of potentially penetrating fluids: the mix methanol:ethanol:water = 16:3:1, water:ethanol = 3:1, and ethylene glycol, by in situ (i.e. synchrotron X-ray powder diffraction, Raman spectroscopy) and ex situ measurements (i.e. synchrotron X-ray powder diffraction, IR spectroscopy). The experimental findings showed that: (1) the elastic behaviour with a non-penetrating fluid is consistent with that previously reported (with a P-induced phase transition from a C-centred to a primitive space group at ~1 GPa, Lotti et al. 2015b) and (2) the P-induced intrusion of guest molecules, from the P-fluids through the cavities of this zeolite, occurs for all the (nominally) penetrating fluids and at low pressure. For example, evidence of the ethylene glycol penetration was reported already at 0.1 GPa and this phenomenon appears to be only partially reversible upon decompression. Whereas methanol or ethanol cannot be intruded at high pressure in Na-mordenite (i.e. Na6Al6.02Si42.02O96 ·19H2O), as reported by Gatta and Lee (2006) and Lotti et al. (2015b), the absence of the extra-framework population promotes the penetration of these molecules through the empty channels of high-silica mordenite in response to applied pressure.
More recently, Richard et al. (2016) used a combination of in situ Raman spectroscopy and X-ray diffraction to investigate the P-induced insertion of BNH6 (ammonia borane, solid at ambient conditions) in the cavities of the hydrophobic silicalite-1F (MFI framework type, Baerlocher et al. 2007). A single crystal of silicalite was compressed in a powder of BNH6, using a DAC. The experimental findings showed how BNH6 molecules penetrate through the cavities of the silicalite-1F structure at very low P (~0.1 GPa), and the insertion leads to the appearance of new Raman modes. Raman spectra collected at high-P showed how orientational disorder of the –BH3 and –NH3 groups, pertaining to the intruded molecules, occur within the P-range investigated, if compared to the bulk ammonia borane used as P-transmitting medium. In situ X-ray powder diffraction experiments showed that the compressibility of the BNH6-bearing silicalite-1F is three times lower than that of the parental silicalite-1F (with empty cavities), in response to the P-induced pore-filling effect.
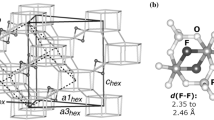
Very recently, Arletti et al. (2017) showed how the hydrophobic all-silica ferrierite (FER framework type, Baerlocher et al. 2007) compressed in the ethanol:water = 1:3 mixture experiences a transition between 0.8 and 1.3 GPa from the orthorhombic (Sp. Gr. Pmnn) to monoclinic symmetry (Sp. Gr. P21/n), coupled with a penetration of the P-fluid molecules. The X-ray powder diffraction data and modelling (using a dispersion-corrected density functional approximation) showed that the (H2O, ethanol)-bearing ferrierite, stable at high pressure, is able to separate the ethanol–water mixture into ethanol dimer wires and H2O tetramer squares (Fig. 2). The specific zeolite type—ferrierite—is pivotal for achieving the H2O–ethanol organization in such a peculiar two-dimensional arrangement. Ferrierite has two parallel channel systems of different diameter—six- and ten-membered rings—which are perfectly tailored to host, respectively, H2O tetramers and ethanol dimers. Surprisingly, the confined supramolecular organization remains stable even upon complete pressure release, which is a key requirement in view of potential technological applications. Indeed, the intrusion of pure H2O in all-silica FER was observed by Cailliez et al. (2008) at considerable lower pressures (below 0.30 GPa) using a porosimeter; such an experiment achieved complete filling of the zeolite framework, but H2O was reversibly extruded upon pressure release (Cailliez et al. 2008). Notably, powder X-ray diffraction data indicated intrusion of H2O molecules at comparable pressure values (0.2 GPa) when ferrierite was compressed in a DAC with a methanol:ethanol:H2O = 16:3:1 mixture—in this case, the maximum pressure reached was 1.5 GPa, and H2O was retained also at room conditions (Arletti et al. 2014). This suggests that pressures higher than 0.3 GPa are instrumental for the irreversible encapsulation of H2O and ethanol molecules in ferrierite, ensuring thus the stability of the supramolecular architecture of dimers and tetramers at room conditions (Arletti et al. 2017).

Supramolecular organization of the H2O and ethanol molecules inside the two-dimensional channel system of hydrophobic all-silica ferrierite. The guest molecules are in ball-and-stick representation. Hydrogen bonds are shown as dashed lines. Atom colours: H white, C cyan, O(ethanol) red; O(H2O) blue. The structure is projected in the ab plane
Previous experiments on the same zeolites were performed by Lotti et al. (2015a) on single crystal and polycrystalline samples, using silicone oil (as a polymeric non-penetrating P-fluid) and methanol:ethanol:H2O = 16:3:1 mixture, ethylene glycol and 2methyl-2propen-1ol (as potentially penetrating fluids). The experiments with the potentially penetrating media enhanced the occurrence of P-induced intrusion of fluid molecules, with different phase-transition paths and compressibility patterns. However, the disordered distribution of the maxima in the calculated residual electron density map, obtained by single-crystal X-ray structure refinements, did not allow to define a unique scenario of the configuration of the intruded molecules (Lotti et al. 2015a). The starting orthorhombic polymorph was always restored upon decompression with all the used P-fluids. The authors highlighted the different HP-behaviour observed for single crystal and powder sample using the same P-fluid, along with the different P-induced phase-transition paths in response to different process kinetics (Lotti et al. 2015a).
The aforementioned experiments on all-silica silicalite (MFI framework type), ZSM-22 (TON framework type), mordenite (MOR framework type) and ferrierite (FER framework type) were all devoted to describe the P-induced penetration of molecular guest systems in a zeolitic host. Recently, Arletti et al. (2016) reported the behaviour of an all-silica ferrierite compressed in an electrolytic MgCl2·21H2O solution. The P-induced intrusion was found to occur at very low P (about 0.19 GPa); the phenomenon is reversible (as proved by the X-ray diffraction before and after the P-induced intrusion experiment) and it is affected by a moderate hysteresis. The Rietveld refinement, based on the synchrotron diffraction data collected at P ~ 0.28 GPa (above the intrusion pressure), showed that both ions and H2O molecules of the aqueous solution (used as P-transmitting fluid) were intruded in the cavities, with an ordered distribution: (1) the Mg2+ site lies at the centre of the FER cage and it is coordinated by four H2O molecules (with partial site occupancy) in a square planar configuration, (2) the Cl− site is located in the ten-membered ring channels parallel to [001], coordinated by two H2O molecules. Mg2+ and Cl− are partially solvated. The idealized composition of the electrolyte guest solution in the zeolitic cavities is: MgCl2·10H2O. At P ~ 0.68 GPa, a phase transition from orthorhombic (Sp. Gr. Pmnn) to monoclinic (Sp. Gr. P21/n) symmetry was also observed.
Discussion and conclusions
If we consider the behaviour of zeolites when compressed in penetrating and non-penetrating P-fluids, the first general consideration we can make is that the P-induced intrusion of new molecules through the zeolitic cavities has a drastic impact of the elastic behaviour and on the structure evolution of a given zeolite. The experimental findings pertaining to compression in non-penetrating fluids showed that:
-
1.
The flexibility observed in this class of open-framework materials, in response to applied P or T, is based mainly on tilting of (quasi-rigid) tetrahedra around O atoms that behave as hinges. Tilting of tetrahedra is the dominant mechanisms at low-mid P-regime, whereas distortion and compression of tetrahedra represent the mechanism which usually dominate the mid-high P-regime. One of the most common deformation mechanisms in zeolitic framework, able to accommodate the effect of pressure, is the increase, though without inversion, of channels ellipticity. As we can learn from the experiments on isotypic zeolites, the deformation mechanisms are dictated by the topological configuration of the tetrahedral framework and are not influenced by the Si/Al/P distribution or by the extra-framework population. However, the compressibility of the cavities is controlled by the nature and bonding configuration of ionic and molecular content, resulting in different unit-cell volume compressibility in isotypic structures.
-
2.
The range of compressibility of zeolites is significantly large, with bulk moduli ~10 < K P0,T0 < ~ 90 GPa. The microporosity does not necessarily imply high compressibility, as several zeolites are stiffer than non-zeolitic rock-forming minerals (e.g. quartz, feldspars, feldspathoids, scapolites, micas; Gatta and Lee 2014). In general, zeolites with stuffed channels (i.e. with an extra-framework population) are stiffer than zeolites with empty channels. If the microporosity is represented by the framework density, then the compressibility of zeolites is not directly related to microporosity.
The experimental findings pertaining to compression in penetrating fluids, and thus with crystal–fluid interaction, showed that not all the zeolites experience a P-induced intrusion of new monoatomic species or molecules from the P-transmitting fluids. For example, zeolites with well-stuffed channels at ambient conditions (e.g. natural zeolites) tend to hinder the penetration of new species through the channels. There must be several variables that govern the sorption phenomena at high pressure beyond the “free diameters” of the framework cavities, among those: the chemical nature and the configuration of the extra-framework population, the partial pressure of the penetrating molecule in the fluid (if mixed with other non-penetrating molecules, e.g. P(H2O) in a mixture of alcohol–H2O), likely the rate of the P-increase, the surface/volume ratio of the crystallites under investigations, the temperature at which the experiment is conducted. As shown by Lotti et al. (2015a), the rate of the P-increase and the surface/volume ratio of the crystallites can play an important role in governing the crystal–fluid interaction, through P-induced penetration phenomena, and deserve further investigations.
The re-organization of the intruded molecules in zeolitic hosts, promoted by applied pressure and by other variables (e.g. ultraviolet irradiation, moderate temperature), is one of the most fascinating discoveries in material science over the last decade, with potential technological and geological implications. The aforementioned experiments of Santoro et al. (2013, 2015, 2016), Scelta et al. (2014), Richard et al. (2016) and Arletti et al. (2015, 2017) indicate new routes for creating hybrid host–guest composite materials, where an inorganic framework drives the formation of organic polymer with low dimensionality, acting as a stable host for it. The new hybrid inorganic-host/organic-guest materials display completely different physicochemical properties if compared to the parental zeolites, in which the interesting properties imparted by pressure would be retained also at standard conditions, and could be exploited in applications. In this fascinating perspective, a crucial question to be addressed is whether moderate pressures, e.g. below 1 GPa, might be sufficient to induce “technologically appealing” irreversible processes, such as the supramolecular organization of simple species in complex patterns, or the formation of one-dimensional polymer chains. Avoiding the use of extreme pressures would be highly desirable for practical applications; for this reason, future investigations along this route should be aimed not only to obtain new composites, but also to determine, or predict, the pressure value at which the transformation would become quantitative and irreversible. Whether supramolecular organization of the included species appears at a certain pressure onset, or if it gradually evolves with increasing pressure, is just one of the questions to be addressed to gather molecular-level knowledge, and hence control, of these processes. A further issue to be investigated is the influence of the composition of the P-transmitting media on the intrusion, organization, and transformation processes inside the framework. Additionally, unravelling the molecular details of the pressure-driven penetration of guest species inside the zeolite pores would be a highly challenging task, which would involve a thorough study of the external surfaces of the zeolite material (Hendriks et al. 2017) under non-standard conditions. In this respect, modelling could play a crucial role: the behaviour of surfaces and interfaces under high-temperature conditions has already enabled to capture new phenomena that were not directly accessible to experimental observation (Ceriani et al. 2004a; Fois et al. 2000, 2010b; Tabacchi et al. 2015b)—for example, that the opening of zeolite pores may be enlarged by concerted rotations of the tetrahedral units induced by the intrusion of bulky molecules (Tabacchi et al. 2016). With this premises, we envisage that a combined experimental and modelling endeavour to explore the interfaces of open-framework materials at high-pressure conditions might reveal the occurrence of new, unexpected processes of key relevance for future advances in zeolite science and technology.
The confinement of molecular species inside zeolite channels at normal pressure is already a successful strategy to build supramolecular architectures: materials with innovative properties have been fabricated by constraining molecules in an ordered arrangement within these nanosized spaces. The encapsulation of dyes into the monodimensional system of channels of zeolite L has led to functional composites suitable for a variety of applications—from solar energy technology to nanomedicine (Popovic et al. 2007; Calzaferri 2012; Fois et al. 2012; Cucinotta et al. 2014; Insuwan et al. 2016; Gartzia-Rivero et al. 2017). Host–guest assemblies working as light-harvesting antenna systems are promising materials for the field of molecule-based devices (Calzaferri et al. 2003; Fois et al. 2010a; Martínez-Martínez et al. 2014). These systems usually consist of chromophores hosted in the one-dimensional pores of zeolite L. Like in the antenna systems of natural photosynthetic organisms, upon photon absorption, a confined dye molecule can transfer its excitation energy to a neighbouring dye molecule, and the energy transfer continues up to the end of the supramolecular chain, where energy could be collected (Calzaferri et al. 2003). To interface the artificial antenna with other components in a working device, the zeolite termination should be functionalized with suitable molecules named “stopcocks” (Maas and Calzaferri 2002), which can also be used to avoid leaking of dye molecules from zeolite channels (Calzaferri 2012; Tabacchi et al. 2015a; Cao et al. 2016; Gartzia-Rivero et al. 2017). Based on this principle, highly versatile composites to be used as tailor-made devices for multiple applications have been created, but their behaviour under non-standard conditions is still to be explored. The study of high-pressure effects on dye–zeolite hybrids would be indispensable for future progress of this technology from fundamental bases. Moreover, by exploiting high pressure, the intrusion of different kinds of dyes might be obtained, with the possibility of organizing these molecules in a complex pattern determined by the topology of the framework and by the related intermolecular interactions, which could ultimately lead to hybrid composites with new, unexpected opto-electronic properties.
High-pressure effects might probably be exploited to improve catalytic processes of relevance for industrial and sustainable applications. These processes may either involve zeolites with strong Lewis acid sites—such as the titanium silicalite TS1 catalyst (Taramasso et al. 1983) adopted in olefin epoxidation processes (Bellussi et al. 1992)—or, more commonly, moderately hydrophilic synthetic zeolites (i.e. with high Si/Al ratio) having protons as extra-framework cations (see, e.g. Fois et al. 2008b). These Brønsted acid sites may activate the intruded molecules. These zeolites are used, for example, in the production of biofuels, as catalysts for cracking and isomerization reactions. Thanks to its framework topology, exhibiting relatively small cavities, hydrophilic ferrierite shows an impressive transition state selectivity for such reactions (see, e.g. Martínez and Corma 2011). However, just because of the small pore diameters, the reaction normally takes place only on the external surface of the zeolite, at its pore opening (Wiedemann et al. 2016). As a consequence of this “pore-mouth catalysis”, only a small proportion of the catalytic sites is actually exploited. Reasonably, moderate pressures could favour the intrusion of the reactant molecules inside the zeolite pores, thus enhancing the performances of the industrial process. Concerning Lewis-acid catalysts, as the olefin epoxidation cycle involves the direct participation of the zeolite framework as active oxygen mediator (Spano et al. 2006), it can be argued that the use of pressure may help to lower the associated free energy barrier, with beneficial effects on catalysis. Besides improving catalytic processes, high-pressure effects might be also effectively exploited to create new zeolite structures starting from a common, economically convenient parent material. Importantly, the new phases might display better performances than the starting material, as demonstrated by Jordá et al. (2013).
In this broad scenario, modelling studies play a key role. Simulations have become an extremely versatile tool for addressing the complex behaviour of zeolites at high-pressure conditions, and for connecting the experimentally measured response of the system to its features at the atomistic-detail level. Nowadays, the scope of computational investigations is no longer limited to the interpretation of experimental results: they should aim at opening new routes in the various aspects of zeolite technology. Integrated theoretical–experimental approaches would be instrumental in this perspective: they are currently widely adopted and are going to gain further momentum from the growing increase of computing power and the continuous development of faster numerical algorithms.
Concerning the geological implications of the experimental findings on the P-induced crystal–fluid interaction in zeolites, it is not difficult to consider the potential role played by zeolites (especially Ca-bearing zeolites, like laumontite) found as among the main mineralogical components of the oceanic basalts and their transformation products, as we know by the ODP—ocean drilling project (e.g. Alt et al. 1986; Sevigny et al. 1992; Yasukawa et al. 2014; http://iodp.americangeosciences.org/vufind/). For example, these zeolites may act as potential carrier of H2O or CO2 (or even H2S, CH4, Ar, Xe, or Kr), sorbed under the combined effect of pressure and temperature during genetic and post-genetic conditions, which can be later released when the oceanic crust is subducted. In addition, whereas it is well known the utilization of synthetic zeolites in petrochemistry (e.g. Vermeiren and Gilson 2009 and references therein), the role played by natural zeolites on generation, migration and accumulation of hydrocarbons (processes that occur at moderate P/T conditions) is still unknown. Some zeolite-like minerals, for example, melanophlogite (a polymorph of SiO2 with an open-framework structure, also called chlatrasil; Tribaudino et al. 2008, 2010; Gatta et al. 2014), contain CH4, CO2 or N2 in their structural cavities. Natural methane clathrates—solids condensed at moderate pressure in which a large amount of methane is trapped within a framework of H2O—share with zeolites the framework topologies and the host–guest structures. The experimental findings pertaining to the HP-behaviour of zeolites can be potentially extended to clathrates. For example, methane clathrates are stable at a higher temperature than liquefied natural gas (i.e. 110–250 K), and this promotes some interest in converting natural gas into clathrates instead of using the conventional liquefaction technology for transportation. The production of methane clathrates from natural gas requires less energy and a smaller refrigeration plant if compared to liquefied natural gas. Can the application of moderate pressure improve the methane clathrates condensation with a more efficient insertion of CH4 in the H2O framework?
References
Ackley MW, Rege SU, Saxena H (2003) Application of natural zeolites in the purification and separation of gases. Micropor Mesopor Mater 61:25–42
Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110:6158–6170
Allen M, Tildesley D (1987) Computer simulation of liquids. Clarendon Press, Oxford
Alt JC, Honnorez J, Laverne C, Emmermann R (1986) Hydrothermal alteration of a 1 km section through the upper oceanic crust, deep sea drilling project hole 504B: mineralogy, chemistry and evolution of seawater–basalt interactions. J Geophys Res 91:10309–10335
Andersen HC (1980) Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys 72:2384–2393
Angel RJ, Allan DR, Miletich R, Finger LW (1997) The use of quartz as an internal pressure standard in high-pressure crystallography. J Appl Crystallogr 30:461–466
Angel RJ, Bujak M, Zhao J, Gatta GD, Jacobsen SJ (2007) Effective hydrostatic limits of pressure media for high-pressure crystallographic studies. J Appl Crystallogr 40:26–32
Arletti R, Ferro O, Quartieri S, Sani A, Tabacchi G, Vezzalini G (2003) Structural deformation mechanisms of zeolites under pressure. Am Mineral 88:1416–1422
Arletti R, Fois E, Gigli L, Vezzalini G, Quartieri S, Tabacchi G (2017) Irreversible conversion of a water–ethanol solution into an organized two-dimensional network of alternating supramolecular units in a hydrophobic zeolite under pressure. Angew Chem Int Ed 56:2105–2109
Arletti R, Leardini L, Vezzalini G, Quartieri S, Gigli L, Santoro M, Haines J, Rouquette J, Konczewicz L (2015) Pressure-induced penetration of guest molecules in high-silica zeolites, the case of mordenite. Phys Chem Chem Phys 17:24262–24274
Arletti R, Quartieri S, Vezzalini G (2010) Elastic behaviour of zeolite boggsite in silicone oil and aqueous medium: a case of high-pressure-induced over-hydration. Am Mineral 95:1247–1256
Arletti R, Ronchi L, Quartieri S, Vezzalini G, Ryzhikov A, Nouali H, Daou TJ, Patarin J (2016) Intrusion–extrusion experiments of MgCl2 aqueous solution in pure silica ferrierite: Evidence of the nature of intruded liquid by in situ high pressure synchrotron X-ray powder diffraction. Micropor Mesopor Mater 235:253–260
Arletti R, Vezzalini G, Morsli A, Di Renzo F, Dmitriev V, Quartieri S (2011) Elastic behaviour of MFI-type zeolites: 1- compressibility of Na-ZSM-5 in penetrating and non-penetrating media. Micropor Mesopor Mater 142:696–707
Arletti R, Vezzalini G, Quartieri S, Di Renzo F, Dmitriev V (2014) Pressure-induced water intrusion in FER-type zeolites and the influence of extraframework species on structural deformations. Microp Mesop Mater 191:27–37
Baerlocher C, McCusker LB, Olson DH (2007) Atlas of zeolite framework types, 6th edn. Elsevier, Amsterdam
Bai P, Jeon MY, Ren L, Knight C, Deem MW, Tsapatsis M, Siepmann JI (2015) Discovery of optimal zeolites for challenging separations and chemical transformations using predictive materials modelling. Nat Commun 6:5912
Balestra SRG, Hamad S, Ruiz-Salvador AR, Domínguez-García V, Merkling PJ, Dubbeldam D, Calero S (2015) Understanding nanopore window distortions in the reversible molecular valve zeolite RHO. Chem Mater 27:5657–5667
Ballone P, Quartieri S, Sani A, Vezzalini G (2002) High-pressure deformation mechanism in scolecite: a combined computational-experimental study. Am Mineral 87:1194–1206
Barducci A, Bonomi M, Parrinello M (2011) Metadynamics. WIREs Comput Mol Sci 1:826–843
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys Rev A 38:3098–3100
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Bellussi G, Carati A, Clerici MG, Maddinelli G, Millini R (1992) Reactions of titanium silicalite with protic molecules and hydrogen peroxide. J Catal 133:220–230
Besson JM, Nelmes RJ, Hamel G, Loveday JS, Weill G, Hull S (1992) Neutron powder diffraction above 10 GPa. Phys B 180:907–910
Betti C, Fois E, Mazzucato E, Medici C, Quartieri S, Tabacchi G, Vezzalini G, Dmitriev V (2007) Gismondine under HP: deformation mechanism and re-organization of the extra-framework species. Microporous Mesoporous Mater 103:190–209
Bish DL, Carey JW (2001) Thermal behaviour of natural zeolites. Rev Mineral Geochem 45:403–452
Bish DL, Vaniman DT, Chipera SJ, Carey JW (2003) The distribution of zeolites and their effects on the performance of a nuclear waste repository at Yucca Mountain, Nevada, U.S.A. Am Mineral 88:1889–1902
Bludský O, Silhan M, Nachtigall P, Bucko T, Benco L, Hafner J (2005) Theoretical investigation of CO interaction with copper sites in zeolites: periodic DFT and hybrid quantum mechanical/interatomic potential function study. J Phys Chem B 109:9631–9638
Bryukhanov IA, Rybakov AA, Larin AV, Trubnikov DN, Vercauteren DP (2017) The role of water in the elastic properties of aluminosilicate zeolites: DFT investigation. J Mol Model 23:68
Cailliez F, Trzpit M, Soulard M, Demachy I, Boutin A, Patarin J, Fuchs AH (2008) Thermodynamics of water intrusion in nanoporous hydrophobic solids. Phys Chem Chem Phys 10:4817–4826
Calzaferri G (2012) Nanochannels: hosts for the supramolecular organization of molecules and complexes. Langmuir 28:6216–6231
Calzaferri G (2017) Entropy in multiple equilibria, theory and applications. Phys Chem Chem Phys 19:10611–10621
Calzaferri G, Huber S, Maas H, Minkowski C (2003) Host–guest antenna materials. Angew Chem Int Ed 42:3732–3758
Cao P, Khorev O, Devaux A, Sägesser L, Kunzmann A, Ecker A, Häner R, Brühwiler D, Calzaferri G, Belser P (2016) Supramolecular organization of dye molecules in zeolite L channels: synthesis, properties, and composite materials. Chem Eur J 22:4046–4060
Car R, Parrinello M (1985) Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett 55:2471–2474
Carter EA, Ciccotti G, Hynes JT, Kapral R (1989) Constrained reaction coordinate dynamics for the simulation of rare events. Chem Phys Lett 156:472–477
Ceriani C, Fois E, Gamba A, Tabacchi G, Ferro O, Quartieri S, Vezzalini G (2004a) Dehydration dynamics of bikitaite: Part II. Ab initio molecular dynamics study. Am Mineral 89:102–109
Ceriani C, Laio A, Fois E, Gamba A, Martoňák R, Parrinello M (2004b) Molecular dynamics simulation of reconstructive phase transitions on an anhydrous zeolite. Phys Rev B 70:113403
Colella C (2011) A critical reconsideration of biomedical and veterinary applications of natural zeolites. Clay Miner 46:295–309
Colligan M, Forster PM, Cheetham AK, Lee Y, Vogt T, Hriljac JA (2004) Synchrotron X-ray powder diffraction and computational investigation of purely siliceous zeolite Y under pressure. J Am Chem Soc 126:12015–12022
Colligan M, Lee Y, Vogt T, Celestian AJ, Parise JB, Marshall WG, Hriljac JA (2005) High pressure neutron powder diffraction study of superhydrated natrolite. J Phys Chem B 109:18223–18225
Combariza AF, Gomez DA, Sastre G (2013) Simulating the properties of small pore silica zeolites using interatomic potentials. Chem Soc Rev 42:114–127
Comboni D, Gatta GD, Lotti P, Merlini M, Liermann H-P (2017) On the P-induced behaviour of the zeolite phillipsite: an in situ single-crystal synchrotron X-ray diffraction study. Phys Chem Miner 44:1–20
Comodi P, Gatta GD, Zanazzi PF (2001) High-pressure structural behaviour of heulandite. Eur J Miner 13:497–505
Coombs DS, Alberti A, Armbruster T, Artioli G, Colella C, Galli E, Grice JD, Liebau F, Mandarino JA, Minato H, Nickel EH, Passaglia E, Peacor DR, Quartieri S, Rinaldi R, Ross M, Sheppard RA, Tillmanns E, Vezzalini G (1997) Recommended nomenclature for zeolite minerals: report of the Subcommittee on Zeolites of International Mineralogical Association, Commission on new minerals and minerals names. Can Miner 35:1571–1606
Coudert FX (2013) Systematic investigation of the mechanical properties of pure silica zeolites: stiffness, anisotropy, and negative linear compressibility. Phys Chem Chem Phys 15:16012–16018
Coudert FX, Cailliez F, Vuilleumier R, Fuchs AH, Boutin A (2009) Water nanodroplets confined in zeolite pores. Faraday Discuss 141:377–398
Cruciani G (2006) Zeolites upon heating: Factors governing their thermal stability and structural changes. J Phys Chem Solids 67:1973–1994
Cucinotta F, Guenet A, Bizzarri C, Mróz W, Botta C, Milián-Medina B, Gierschner J, De Cola L (2014) Energy transfer at the zeolite L boundaries: towards photo- and electroresponsive materials. ChemPlusChem 79:45–57
De Boer K, Jansen APJ, Van Santen RA (1995) Structure–stability relationships for all-silica structures. Phys Rev B 52:12579–12590
De Wispelaere K, Ensing B, Ghysels A, Meijer EJ, Van Speybroeck V (2015) Complex reaction environments and competing reaction mechanisms in zeolite catalysis: insights from advanced molecular dynamics. Chem Eur J 21:9385–9396
Decker DL, Petersen S, Debray D, Lambert M (1979) Pressure-induced ferroelastic phase transition in Pb3(PO4)2: a neutron-diffraction study. Phys Rev B 19:3552–3555
Dellago C, Bolhuis PG, Chandler D (1998) Efficient transition path sampling: Application to Lennard-Jones cluster rearrangements. J Chem Phys 108:9236–9245
Delle Piane M, Corno M, Pedone A, Dovesi R, Ugliengo P (2014) Large-scale B3LYP simulations of ibuprofen adsorbed in MCM-41 mesoporous silica as drug delivery system. J Phys Chem C 119:26737–26749
Demichelis R, Civalleri B, Ferrabone M, Dovesi R (2010) On the performance of eleven DFT functionals in the description of the vibrational properties of aluminosilicates. Int J Quant Chem 110:406–415
Demontis P, Suffritti GB, Quartieri S, Fois ES, Gamba A (1987) Molecular dynamics studies on zeolites. II: a simple model for silicates applied to anhydrous natrolite. Zeolites 7:122–127
Demontis P, Suffritti GB, Quartieri S, Fois ES, Gamba A (1988) Molecular dynamics studies on zeolites. 3. Dehydrated zeolite A. J Phys Chem 92(4):867–871
Demontis P, Suffritti GB, Quartieri S, Fois ES, Gamba A (1990) Molecular dynamics studies on zeolites. 4. Diffusion of methane in silicalite. J Phys Chem 94(10):4329–4334
Desbiens N, Demachy I, Fuchs AH, Kirsch-Rodeschini H, Soulard M, Patarin J (2005) Water condensation in hydrophobic nanopores. Angew Chem Int Ed 44:5310–5313
Dove MT, Trachenko KO, Tucker MG, Keen DA (2000) Rigid Unit Modes in framework structures: theory, experiment and applications. Rev Mineral Geochem 39:1–33
Dovesi R, Orlando R, Civalleri B, Roetti C, Saunders VR, Zicovich-Wilson CM (2005) CRYSTAL: a computational tool for the ab initio study of the electronic properties of crystals. Z Kristallogr 220:571–573
Erba A, Caglioti D, Zicovich-Wilson CM, Dovesi R (2017) Nuclear-relaxed elastic and piezoelectric constants of materials: Computational aspects of two quantum-mechanical approaches. J Comput Chem 38:257–264
Erba A, Mahmoud A, Orlando R, Dovesi R (2014a) Elastic properties of six silicate garnet end members from accurate ab initio simulations. Phys Chem Minerals 41:151–160
Erba A, Mahmoud A, Orlando R, Dovesi R (2014b) Erratum to: elastic properties of six silicate garnet end-members from accurate ab initio simulations. Phys Chem Miner 41:161–162
Ferro O, Quartieri S, Vezzalini G, Fois E, Gamba A, Tabacchi G (2002) High-pressure behaviour of bikitaite: an integrated theoretical and experimental approach. Am Mineral 87:1415–1425
Fischer M (2015) Structure and bonding of water molecules in zeolite hosts: benchmarking plane-wave DFT against crystal structure data. Z Kristallogr 230:325–336
Fischer M, Angel R (2017) Accurate structures and energetics of neutral-framework zeotypes from dispersion-corrected DFT calculations. J Chem Phys 146:174111
Fischer M, Delgado MR, Areán CO, Duran CO (2015) CO adsorption complexes in zeolites: how does the inclusion of dispersion interactions affect predictions made from DFT calculations? The case of Na-CHA. Theor Chem Acc 134:91
Fischer M, Evers FO, Formalik F, Olejniczak A (2016) Benchmarking DFT-GGA calculations for the structure optimisation of neutral-framework zeotypes. Theor Chem Acc 135:257
Fois E, Gamba A, Medici C, Tabacchi G (2005c) Intermolecular electronic excitation transfer in a confined space: a first-principles study. ChemPhysChem 6:1917–1922
Fois E, Gamba A, Medici C, Tabacchi G, Quartieri S, Mazzucato E, Arletti R, Vezzalini G, Dmitriev V (2008a) High pressure deformation mechanism of Li-ABW: Synchrotron XRPD study and ab initio molecular dynamics simulations. Microporous Mesopor Mater 115:267–280
Fois E, Gamba A, Tabacchi G (2000) First-principles simulation of the intracage oxidation of nitrite to nitrate sodalite. Chem Phys Lett 329:1–6
Fois E, Gamba A, Tabacchi G, Arletti R, Quartieri S, Vezzalini G (2005a) The “template” effect of the extra-framework content on zeolite compression: The case of yugawaralite. Am Mineral 90:28–35
Fois E, Gamba A, Tabacchi G, Ferro O, Quartieri S, Vezzalini G (2002a) A theoretical investigation on pressure-induced changes in the vibrational spectrum of zeolite bikitaite. Stud Surf Sci Catal 142:1877–1884
Fois E, Gamba A, Tabacchi G, Quartieri S, Arletti R, Vezzalini G (2005b) High-pressure behaviour of yugawaralite at different water content: an ab initio study. Stud Surf Sci Catal 155:271–280
Fois E, Gamba A, Tabacchi G, Quartieri S, Vezzalini G (2001a) Water molecules in single file: first-principles studies of one-dimensional water chains in zeolites. J Phys Chem B 105:3012–3016
Fois E, Gamba A, Tabacchi G, Quartieri S, Vezzalini G (2001b) On the collective properties of water molecules in one-dimensional zeolitic channels. Phys Chem Chem Phys 3:4158–4163
Fois E, Gamba A, Tabacchi G, Trudu F (2008b) First principles studies on boron sites. Stud Surf Sci Catal 174:751–754
Fois E, Gamba A, Tilocca A (2002b) Structure and dynamics of the flexible triple helix of water inside VPI-5 molecular sieves. J Phys Chem B 106:4806–4812
Fois E, Tabacchi G, Barreca D, Gasparotto A, Tondello E (2010b) “Hot” surface activation of molecular complexes: insight from modelling studies. Angew Chem Int Ed 49:1944–1948
Fois E, Tabacchi G, Calzaferri G (2010a) Interactions, behaviour and stability of fluorenone inside zeolite nanochannels. J Phys Chem C 114:10572–10579
Fois E, Tabacchi G, Calzaferri G (2012) Orientation and order of xanthene dyes in the one-dimensional channels of zeolite L: bridging the gap between experimental data and molecular behaviour. J Phys Chem C 116:16784–16799
Fois E, Tabacchi G, Quartieri S, Vezzalini G (1999) Dipolar host/guest interactions and geometrical confinement at the basis of the stability of one-dimensional ice in zeolite bikitaite. J Chem Phys 111:355–359
Frenkel D, Smit B (2001) Understanding molecular simulation. Academic Press, San Diego
Gabrieli A, Sant M, Demontis P, Suffritti GB (2014) Fast and efficient optimization of molecular dynamics force fields for microporous materials: bonded interactions via force matching. Microporous Mesoporous Mater 197:339–347
Gabrieli A, Sant M, Demontis P, Suffritti GB (2016) A combined energy-force fitting procedure to develop DFT-based force fields. J Phys Chem C 120:26309–26319
Gale JD (1997) GULP: a computer program for the symmetry-adapted simulation of solids. J Chem Soc Faraday Trans 93:629–637
Gartzia-Rivero L, Bañuelos J, López-Arbeloa I (2017) Photoactive nanomaterials inspired by nature: LTL zeolite doped with laser dyes as artificial light harvesting systems. Materials 10:495
Gatta GD (2005) A comparative study of fibrous zeolites under pressure. Eur J Miner 17:411–422
Gatta GD (2008) Does porous mean soft? On the elastic behaviour and structural evolution of zeolites under pressure. Z Kristallogr 223:160–170
Gatta GD (2010a) Extreme deformation mechanisms in open-framework silicates at high-pressure: evidence of anomalous inter-tetrahedral angles. Micropor Mesopor Mater 128:78–84
Gatta GD, Angel RJ (2007) Elastic behaviour and pressure-induced structural evolution of nepheline: implications for the nature of the modulated superstructure. Am Mineral 92:1446–1455
Gatta GD, Bersani D, Lottici PP, Tribaudino M (2014) High-pressure Raman study of CH4 in melanophlogite (type I clathrate). Miner Mag 78:1661–1669
Gatta GD, Birch DW, Rotiroti N (2010) Reivestigation of the crystal structure of the zeolite gobbinsite: a single-crystal X-ray diffraction study. Am Miner 95:481–486
Gatta GD, Boffa Ballaran T, Comodi P, Zanazzi PF (2004a) Isothermal equation of state and compressional behaviour of tetragonal edingtonite. Am Miner 89:633–639
Gatta GD, Boffa Ballaran T, Comodi P, Zanazzi PF (2004b) Comparative compressibility and equation of state of orthorhombic and tetragonal edingtonite. Phys Chem Miner 31:288–298
Gatta GD, Brundu A, Cappelletti P, Cerri G, de’Gennaro B, Farina M, Fumagalli P, Guaschino L, Lotti P, Mercurio M (2016a) New insights on pressure, temperature, and chemical stability of CsAlSi5O12, a potential host for nuclear waste. Phys Chem Minerals 43:639–647
Gatta GD, Comodi P, Zanazzi PF (2003) New insights on high-pressure behaviour of microporous materials from X-ray single-crystal data. Micropor Mesopor Mater 61:105–115
Gatta GD, Comodi P, Zanazzi PF, Boffa Ballaran T (2005) Anomalous elastic behaviour and high-pressure structural evolution of zeolite levyne. Am Miner 90:645–652
Gatta GD, Lee Y (2006) On the elastic behaviour of zeolite mordenite: a synchrotron powder diffraction study. Phys Chem Miner 32:726–732
Gatta GD, Lee Y (2014) Zeolites at high pressure: A review. Miner Mag 78:267–291
Gatta GD, Lotti P (2011) On the low-temperature behaviour of the zeolite gobbinsite: a single-crystal X-ray diffraction study. Micropor Mesopor Mater 143:467–476
Gatta GD, Lotti P, Nestola F, Pasqual D (2012) On the high-pressure behaviour of gobbinsite, the natural counterpart of the synthetic zeolite Na–P2. Micropor Mesopor Mater 163:259–269
Gatta GD, Nestola F, Boffa Ballaran T (2006) Elastic behaviour, phase transition and pressure-induced structural evolution of analcime. Am Miner 91:568–578
Gatta GD, Rotiroti N, Boffa Ballaran T, Pavese A (2008) Leucite at high pressure: elastic behaviour, phase stability and petrological implications. Am Miner 93:1588–1596
Gatta GD, Rotiroti N, Boffa Ballaran T, Sanchez-Valle C, Pavese A (2009a) Elastic behaviour and phase-stability of pollucite, a potential host for nuclear waste. Am Miner 94:1137–1143
Gatta GD, Sartbaeva A, Wells AS (2009b) Compression behaviour and flexibility window of the analcime-like feldspathoids: experimental and theoretical findings. Eur J Miner 21:571–580
Gatta GD, Scheidl KS, Pippinger T, Skála R, Lee Y, Miletich R (2015) High-pressure behaviour and crystal–fluid interaction under extreme conditions in paulingite [PAU-topology]. Micropor Mesopor Mater 206:34–41
Gatta GD, Tabacchi G, Fois E, Lee Y (2016b) Behaviour at high pressure of Rb7NaGa8Si12O40·3H2O (a zeolite with EDI topology): a combined experimental–computational study. Phys Chem Miner 43:209–216
Gatta GD, Wells SA (2004) Rigid unit modes at high pressure: an explorative study of a fibrous zeolite-like framework with EDI topology. Phys Chem Minerals 31:465–474
Gatta GD, Wells SA (2006) Structural evolution of zeolite levyne under hydrostatic and non-hydrostatic pressure: geometric modelling. Phys Chem Miner 33:243–255
Gatta GD (2010b) Microporous materials at high pressure: Are they really soft? In: Boldyreva E, Dera P (eds) High-pressure crystallography: from fundamental phenomena to technological applications. NATO science for peace and security—series b (physics and biophysics). Springer Science, Berlin, pp 481–491
Giddy AP, Dove MT, Pawley GS, Heine V (1993) The determination of rigid unit modes as potential soft modes for displacive phase transitions in framework crystal structures. Acta Crystallogr A 4:697–703
Gigli L, Arletti R, Tabacchi G, Fois E, Vitillo JG, Martra G, Agostini G, Quartieri S, Vezzalini G (2014) Close-packed dye molecules in zeolite channels self-assemble into supramolecular nanoladders. J Phys Chem C 118:15732–15743
Gillet P, Malézieux JM, Itié JP (1996) Phase changes and amorphization of zeolites at high pressure: the case of scolecite and mesolite. Am Miner 81:651–657
Goryainov SV (2005) Pressure-induced amorphization of Na2Al2Si3O10·2H2O and KAlSi2O6 zeolites. Phys Status Solidi 202:R25–R27
Grau-Crespo R, Acuay E, Ruiz-Salvador, AR (2002) A free energy minimisation study of the monoclinic–orthorhombic transition in MFI zeolite. Chem Commun 2544–2545. doi:10.1039/B208064H
Greaves GN, Meneau F, Sapelkin A, Colyer LM, Gwynn IA, Wade S, Sankar G (2003) The rheology of collapsing zeolites amorphized by temperature and pressure. Nature Mat 2:622–629
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem 27:1787–1799
Grimme S (2011) Density functional theory with London dispersion corrections. Wiley Interdiscip Rev Comput Mol Sci 1:211–228
Gulín-González J, Pupo CT, Conyedo EN, Ruiz-Puentes A, Demontis P, Suffritti GB (2016) A lattice dynamics study of ZK-4 microporous material under different temperature and pressure conditions. Micropor Mesopor Mater 226:191–200
Gulín-González J, Suffritti GB (2004) Amorphization of calcined LTA zeolites at high pressure: a computational study. Microporous Mesoporous Mater 69:127–134
Gutiérrez-Sevillano JJ, Calero S, Hamad S, Grau-Crespo R, Rey F, Valencia S, Palomino M, Balestra SRG, Ruiz-Salvador AR (2016) Critical role of dynamic flexibility in Ge-containing zeolites: impact on diffusion. Chem Eur J 22:10036–10043
Göltl F, Grüneis A, Bučko T, Hafner J (2012) Van der Waals interactions between hydrocarbon molecules and zeolites: periodic calculations at different levels of theory, from density functional theory to the random phase approximation and Møller–Plesset perturbation theory. J Chem Phys 137:114111
Göltl F, Hafner J (2012) Structure and properties of metal-exchanged zeolites studied using gradient-corrected and hybrid functionals. I. Structure and energetics. J Chem Phys 136:064501
Haines J, Cambon O, Levelut C, Santoro M, Gorelli F, Garbarino G (2010) Deactivation of Pressure-induced amorphization in silicalite SiO2 by insertion of guest species. J Am Chem Soc 132:8860–8861
Haines J, Léger JM, Gorelli F, Hanfland M (2001) Crystalline post-quartz phase in silica at high pressure. Phys Rev Lett 87:15503
Hammonds KD, Dove MT, Giddy AP, Heine V (1994) Crush: a Fortran program for the analysis of the rigid-unit mode spectrum of a framework structure. Am Miner 79:1207–1209
Hazen RM (1983) Zeolite molecular sieve 4A: anomalous compressibility and volume discontinuities at high pressure. Science 219:1065–1067
Hazen RM, Finger LW (1984) Compressibility of zeolite 4A is dependent on the molecular size of the hydrostatic pressure medium. J Appl Phys 56:1838–1840
Hendriks FC, Schmidt JE, Rombouts JA, Lammertsma K, Bruijnincx PC, Weckhuysen BM (2017) Probing zeolite crystal architecture and structural imperfections using differently sized fluorescent organic probe molecules. Chem Eur J 23:6305–6314
Huang Y, Havenga EA (2001) Why do zeolites with LTA structure undergo reversible amorphization under pressure? Chem Phys Lett 345:65–71
Iannuzzi M, Laio A, Parrinello M (2003) Efficient exploration of reactive potential energy surfaces using Car–Parrinello molecular dynamics. Phys Rev Lett 90:238302
Insuwan W, Rangsriwatananon K, Meeprasert J, Namuangruk S, Surakhot Y, Kungwan N, Jungsuttiwong S (2016) Combined experimental and theoretical investigation on fluorescence resonance energy transfer of dye loaded on LTL zeolite. Microp Mesop Mater 241:372–382
Jordá JL, Rey F, Sastre G, Valencia S, Palomino M, Corma A, Segura A, Errandonea D, Lacomba R, Manjón FJ, Gomis Ó, Kleppe AK, Jephcoat AP, Amboage M, Rodríguez-Velamazán JA (2013) Synthesis of a novel zeolite through a pressure-induced reconstructive phase transition process. Angew Chem Int Ed 52:10458–10462
Jónsson H, Mills G, Jacobsen KW (1998) Nudged elastic band method for finding minimum energy paths of transitions. In: Berne BJ, Ciccotti G, Coker DF (eds) Classical and quantum dynamics in condensed phase simulations. World Scientific, Singapore, pp 51–66
Kalló D (2001) Applications of natural zeolites in water and wastewater treatment. Rev Miner Geochem 45:519–550
Kenichi T (1999) Absence of the c/a anomaly in Zn under high pressure with a helium-pressure medium. Phys Rev B 60:6171–6174
Klotz S, Chervin J-C, Munsch P, Le Marchand G (2009) Hydrostatic limits of 11 pressure transmitting media. J Phys D Appl Phys 42:075413
Komarneni S (1985) Phillipsite in Cs decontamination and immobilization. Clays Clay Min 33:145–151
Kremleva A, Vogt T, Rösch N (2013) Monovalent cation-exchanged natrolites and their behaviour under pressure. A computational study. J Phys Chem C 117:19020–19030
Kremleva A, Vogt T, Rösch N (2014) Potassium-exchanged natrolite under pressure. Computational study vs experiment. J Phys Chem C 118:22030–22039
Laio A, Parrinello M (2002) Escaping free-energy minima. Proc Natl Acad Sci USA 99:12562–12566
Larin AV, Trubnikov DN, Vercauteren DP (2005) Improvement of X-ray diffraction geometries of water physisorbed in zeolites on the basis of periodic Hartree-Fock calculations. Int J Quantum Chem 102:971–979
Lee Y, Hriljac JA, Parise JB, Vogt T (2005) Pressure-induced stabilization of ordered paranatrolite: a solution to the paranatrolite controversy. Am Mineral 90:252–257
Lee Y, Hriljac JA, Vogt T (2004) Pressure-induced migration of zeolitic water in laumontite. Phys Chem Minerals 31:421–428
Lee Y, Hriljac JA, Vogt T (2010) Pressure-induced argon insertion into an auxetic small pore zeolite. J Phys Chem C 114:6922–6927
Lee Y, Hriljac JA, Vogt T, Parise JB, Edmondson M, Anderson P, Corbin D, Nagai T (2001) Phase transition of zeolite RHO at high-pressure. J Am Chem Soc 123:8418–8419
Lee Y, Kao CC, Kim SJ, Lee HH, Lee DR, Shin TJ, Choi JY (2007) Water nanostructures confined inside the quasi-one-dimensional channels of LTL zeolite. Chem Mater 19:6252–6257
Lee Y, Kim SJ, Kao CC, Vogt T (2008) Pressure-induced hydration and order-disorder transition in a synthetic potassium gallosilicate zeolite with gismondine topology. J Am Chem Soc 130:2842–2850
Lee Y, Liu D, Seoung D, Liu Z, Kao CC, Vogt T (2011) Pressure- and heat-induced insertion of CO2 into an auxetic small-pore zeolite. J Am Chem Soc 133:1674–1677
Lee Y, Vogt T, Hriljac JA, Parise JB, Artioli G (2002a) Pressure-induced volume expansion of zeolites in the natrolite family. J Am Chem Soc 124:5466–5475
Lee Y, Vogt T, Hriljac JA, Parise JB, Hanson JC, Kim SJ (2002b) Non-framework cation migration and irreversible pressure-induced hydration in a zeolite. Nature 420:485–489
Lee C, Yang W, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Likhacheva AY, Malyshev ME, Manakov AY, Goryainov SV, Ancharov AI (2009) Non-hydrostatic compression of zeolite NaA in water medium: connection to anomalous conductivity. Z Kristallogr 224:137–143
Likhacheva AY, Seryotkin YV, Manakov AY, Goryainov SV, Ancharov AI, Sheromov MA (2006) Anomalous compression of scolecite and thomsonite in aqueous medium to 2 GPa. High Pres Res 26:449–453
Likhacheva AY, Seryotkin YV, Manakov AY, Goryainov SV, Ancharov AI, Sheromov MA (2007) Pressure-induced over-hydration of thomsonite: a synchrotron powder diffraction study. Am Mineral 92:1610–1615
Lippert G, Hutter J, Parrinello M (1997) A hybrid Gaussian and plane wave density functional scheme. Mol Phys 92:477–488
Lotti P, Arletti R, Gatta GD, Quartieri S, Vezzalini G, Merlini M, Dmitriev V, Hanfland M (2015a) Compressibility and crystal–fluid interactions in all-silica ferrierite at high pressure. Micropor Mesopor Mater 218:42–54
Lotti P, Gatta GD, Comboni D, Merlini M, Pastero L, Hanfland M (2016) AlPO4-5 zeolite at high pressure: crystal-fluid interaction and elastic behaviour. Micropor Mesopor Mat 228:158–167
Lotti P, Gatta GD, Merlini M, Liermann H-P (2015b) High-pressure behaviour of synthetic mordenite–Na: an in situ single-crystal synchrotron X-ray diffraction study. Z Kristallogr 230:201–211
Lotti P, Gatta GD, Rotiroti N, Cámara F (2012) High-pressure study of a natural cancrinite. Am Mineral 97:872–882
Maas H, Calzaferri G (2002) Trapping energy from and injecting energy into dye-zeolite nanoantennae. Angew Chem Int Ed 41:2284–2288
Machon D, Dmitriev VP, Bouvier P, Timonin PN, Shirokov VB, Weber H-P (2003) Pseudoamorphization of Cs2HgBr4. Phys Rev B 68:144104
Maerzke KA, McGrath MJ, Kuo IFW, Tabacchi G, Siepmann JI, Mundy CJ (2009) Vapor–liquid phase equilibria of water modelled by a Kim-Gordon potential. Chem Phys Lett 479:60–64
Manzano H, Gartzia-Rivero L, Bañuelos J, López-Arbeloa I (2013) Ultraviolet–visible dual absorption by single BODIPY dye confined in LTL zeolite nanochannels. J Phys Chem C 117:13331–13336
Mao HK, Xu J, Bell PM (1986) Calibration of the ruby pressure gauge to 800 kbar under quasi-hydrostatic conditions. J Geophys Res 91:4673–4676
Martoňák R, Laio A, Parrinello M (2003) Predicting crystal structures: the Parrinello–Rahman method revisited. Phys Rev Lett 90:75503
Martínez C, Corma A (2011) Inorganic molecular sieves: Preparation, modification and industrial application in catalytic processes. Coord Chem Rev 255:1558–1580
Martínez-Martínez V, García R, Gómez-Hortigüela L, Sola Llano R, Pérez-Pariente J, López-Arbeloa I (2014) Highly luminescent and optically switchable hybrid material by one-pot encapsulation of dyes into MgAPO-11 unidirectional nanopores. ACS Photonics 1:205–211
Marx D, Hutter J (2009) Ab initio molecular dynamics: basic theory and advanced methods. Cambridge University Press, Cambridge
Maxwell IE, Stork WHJ (2001) Hydrocarbon processing with zeolites. Stud Surf Sc Catal 137:747–819
Merrill L, Bassett WA (1974) Miniature diamond anvil pressure cell for single-crystal X-ray diffraction studies. Rev Sci Instr 45:290–294
Miletich R, Allan DR, Kuhs WF (2000) High-pressure single-crystal techniques. Rev Miner Geochem 41:445–519
Miletich R, Hejny C, Krauss G, Ullrich A (2005) Diffraction techniques: Shedding light on structural changes at extreme conditions. In: Miletich R (ed) Mineral behaviour at extreme conditions. European Mineralogical Union Notes in Mineralogy, vol 7, pp 281–338
Ming DW, Allen ER (2001) Use of natural zeolites in agronomy, horticulture, and environmental soil remediation. Rev Miner Geochem 45:619–654
Morpurgo S (2015) A DFT study on Cu(I) coordination in Cu-ZSM-5: effects of the functional choice and tuning of the ONIOM approach. J Comput Chem 36:660–669
Mumpton FA (1999) La roca magica: Uses of natural zeolites in agriculture and industry. Proc Natl Acad Sci USA 96:3463–3470
Narayanan B, Reimanis IE, Ciobanu CV (2013) Atomic-scale mechanism for pressure-induced amorphization of β-eucryptite. J Appl Phys 114:083520
Niwa K, Tanaka T, Hasegawa M, Okada T, Yagi T, Kikegawa T (2013) Pressure-induced noble gas insertion into Linde-type A zeolite and its incompressible behaviours at high pressure. Micropor Mesopor Mater 182:191–197
Ori S, Quartieri S, Vezzalini G, Dmitriev V (2008a) Pressure-induced structural deformation and elastic behaviour of wairakite. Am Miner 93:53–62
Ori S, Quartieri S, Vezzalini G, Dmitriev V (2008b) Pressure-induced over-hydration and water ordering in gismondine: a synchrotron powder diffraction study. Am Miner 93:1393–1403
Otero Areán C, Nachtigallova D, Nachtigall P, Garrone E, Delgado MR (2007) Thermodynamics of reversible gas adsorption on alkali-metal exchanged zeolites—the interplay of infrared spectroscopy and theoretical calculations. Phys Chem Chem Phys 9:1421–1437
Pabalan RT, Bertetti FP (2001) Cation-exchange properties of natural zeolites. Rev Miner Geochem 45:453–518
Parr RG, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, New York, Oxford
Parrinello M, Rahman A (1980) Crystal structure and pair potentials: a molecular-dynamics study. Phys Rev Lett 45:1196–1199
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7185
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822–8824
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Perdew J, Ruzsinszky A, Csonka G, Vydrov O, Scuseria G, Constantin L, Zhou X, Burke K (2008) Restoring the density-gradient expansion for exchange in solids and surfaces. Phys Rev Lett 100:136406
Piccini GM, Alessio M, Sauer J (2016) Ab initio calculation of rate constants for molecule–surface reactions with chemical accuracy. Angew Chem Int Ed 55:5235–5237
Pisani C (1999) Software for the quantum-mechanical simulation of the properties of crystalline materials: state of the art and prospects. J Mol Struct (Theochem) 463:125–137
Pisani C (1996) Quantum mechanical ab initio calculation of the properties of crystalline materials. Lect Notes Chem 67:47–75
Popovic Z, Otter M, Calzaferri G, De Cola L (2007) Self-assembling living systems with functional nanomaterials. Angew Chem Int Ed 46:6188–6191
Quartieri S, Montagna G, Arletti R, Vezzalini G (2011) Elastic behaviour of MFI-type zeolites: compressibility of H-ZSM-5 in pentrating and non-penetrating media. J Solid State Chem 184:1505–1516
Remler D, Madden P (1990) Molecular dynamics without effective potentials via the Car–Parrinello approach. Mol Phys 70:921–966
Resel R, Oehzelt M, Shimizu K, Nakayama A, Takemura K (2004) On the phase-transition in anthracene induced by high pressure. Solid State Commun 129:103–106
Richard J, León Cid S, Rouquette J, van der Lee A, Bernard S, Haines J (2016) Pressure-induced insertion of ammonia borane in the siliceous zeolite, silicalite-1F. J Phys Chem C 120:9334–9340
Román-Román EI, Zicovich-Wilson CM (2015) The role of long-range van der Waals forces in the relative stability of SiO2–zeolites. Chem Phys Lett 619:109–114
Rutter MD, Secco RA, Huang Y (2000) Ionic conduction in hydrated zeolite Li-, Na- and K-A at high pressures. Chem Phys Lett 331:189–195
Rutter MD, Uchida T, Secco RA, Huang Y, Wang Y (2001) Investigation of pressure-induced amorphization in hydrated zeolite Li-A and Na-A using synchrotron X-ray diffraction. J Phys Chem Solids 62:599–606
Sanders MJ, Leslie M, Catlow CRA (1984) Interatomic potentials for SiO2. J Chem Soc Chem Commun 19:1271–1273
Santoro M, Dziubek K, Scelta D, Ceppatelli M, Gorelli FA, Bini R, Thibaud J-M, Di Renzo F, Cambon O, Rouquette J, Hermet P, van der Lee A, Haines J (2015) High pressure synthesis of all-transoid polycarbonyl [–(C=O)–] n in a zeolite. Chem Mater 27:6486–6489
Santoro M, Gorelli FA, Bini R, Haines J, van der Lee A (2013) High-pressure synthesis of a polyethylene/zeolite nano-composite material. Nat Commun 4:1557
Santoro M, Scelta D, Dziubek K, Ceppatelli M, Gorelli FA, Bini R, Garbarino G, Thibaud J-M, Di Renzo F, Cambon O, Hermet P, Rouquette J, van der Lee A, Haines J (2016) Synthesis of 1D polymer/zeolite nanocomposites under high pressure. Chem Mater 28:4065–4071
Sartbaeva A, Gatta GD, Wells SA (2008) Flexibility window controls pressure-induced phase transition in analcime. Europhys Lett 83:26002
Sartbaeva A, Wells SA (2012) Framework flexibility and rational design of new zeolites for catalysis. Appl Petrochem Res 2:69–72
Sartbaeva A, Wells SA, Treacy MMJ, Thorpe MF (2006) The flexibility window in zeolites. Nature Mat 5:962–965
Scelta D, Ceppatelli M, Santoro M, Bini R, Gorelli FA, Perucchi A, Mezouar M, van der Lee A, Haines J (2014) High pressure polymerization in a confined space: conjugated chain/zeolite nanocomposites. Chem Mater 26:2249–2255
Secco RA, Huang Y (1999) Pressure-induced disorder in hydrated Na-A zeolite. J Phys Chem Solids 60:999–1002
Seoung D, Lee Y, Cynn H, Park C, Choi KY, Blom DA, Evans WJ, Kao CC, Vogt T, Lee Y (2014) Irreversible xenon insertion into a small pore zeolite at moderate pressures and temperatures. Nature Chem 6:835–839
Seoung D, Lee Y, Kao CC, Vogt T, Lee Y (2013) Super-hydrated zeolites: pressure-induced hydration in natrolites. Chem Eur J 33:11100
Seoung D, Lee Y, Kao CC, Vogt T, Lee Y (2015) Two-step pressure-induced superhydration in small pore natrolite with divalent extra-framework cations. Chem Mat 27:3874–3880
Seryotkin YV (2016) Evolution of the bikitaite structure at high pressure: a single-crystal X-ray diffraction study. Micropor Mesopor Mater 226:415–423
Seryotkin YV, Bakakin VV, Fursenko BA, Belitsky IA, Joswig W, Radaelli PG (2005) Structural evolution of natrolite during over-hydration: a high-pressure neutron diffraction study. Eur J Miner 17:305–313
Seryotkin YV, Bakakin VV, Likhacheva AY, Dementiev SN, Rashchenko SV (2017) Structural behaviour of Tl-exchanged natrolite at high pressure depending on the composition of pressure-transmitting medium. Phys Chem Miner. doi:10.1007/s00269-017-0887-0) (in press)
Sevigny JH, Whitechurch H, Storey M, Salters VJM (1992) Zeolite-facies metamorphism of central Kerguelen Plateau basalts. Proc Ocean Drilling Program, Scientific Results 120:63–69
Sheppard D, Xiao P, Chemelewski W, Johnson DD, Henkelman G (2012) A generalized solid-state nudged elastic band method. J Chem Phys 136:074103
De Silva P, Wesolowski TA (2012) Exact non-additive kinetic potentials in realistic chemical systems. J Chem Phys 137:094110
Smit B, Maesen TL (2008) Molecular simulations of zeolites: adsorption, diffusion, and shape selectivity. Chem Rev 108:4125–4184
Spano E, Tabacchi G, Gamba A, Fois E (2006) On the role of Ti(IV) as a Lewis acid in the chemistry of titanium zeolites: formation, structure, reactivity, and aging of Ti—peroxo oxidizing intermediates. A first principles study. J Phys Chem B 110:21651–21661
Tabacchi G, Calzaferri G, Fois E (2016) One-dimensional self-assembly of perylene-diimide dyes by unidirectional transit of zeolite channel openings. Chem Commun 52:11195–11198
Tabacchi G, Fois E, Calzaferri G (2015a) Structure of nanochannel entrances in stopcock-functionalized zeolite L composites. Angew Chem Int Ed 54:11112–11116
Tabacchi G, Hutter J, Mundy CJ (2005) A density-functional approach to polarizable models: a Kim–Gordon response density interaction potential for molecular simulations. J Chem Phys 123:074108
Tabacchi G, Fois E, Barreca D, Carraro G, Gasparotto A, Maccato C (2015b) Modelling the first activation stages of the Fe(hfa)2TMEDA CVD precursor on a heated growth surface. In: Advanced processing and manufacturing technologies for nanostructured and multifunctional materials ii: a collection of papers presented at the 39th international conference on advanced ceramics and composites. Wiley, Hoboken, pp 83–90
Taramasso M, Perego G, Notari B (1983) U.S. Patent 441051
Tkatchenko A, Scheffler M (2009) Accurate molecular Van Der Waals interactions from ground-state electron density and free-atom reference data. Phys Rev Lett 102:073005
Tribaudino M, Artoni A, Mavris C, Bersani D, Lottici PP, Belletti D (2008) Single-crystal X-ray and Raman investigation on melanophlogite from Varano Marchesi (Parma, Italy). Am Mineral 93:88–94
Tribaudino M, Gatta GD, Lee Y (2010) A high-pressure cubic-to-tetragonal phase-transition in melanophlogite, a SiO2 clathrate phase. Micropor Mesopor Mater 129:267–273
Tuma C, Sauer J (2004) A hybrid MP2/planewave-DFT scheme for large chemical systems: proton jumps in zeolites. Chem Phys Lett 387:388–394
Tuma C, Sauer J (2006) Treating dispersion effects in extended systems by hybrid MP2:DFT calculations—protonation of isobutene in zeolite ferrierite. Phys Chem Chem Phys 8:3955–3965
U.S. Geological Survey (2017) Mineral commodity summaries 2016. U.S. Geological Survey, Reston, Virginia
Van Speybroeck V, Hemelsoet K, Joos L, Waroquier M, Bell RG, Catlow CRA (2015) Advances in theory and their application within the field of zeolite chemistry. Chem Soc Rev 44:7044–7111
VandeVondele J, Krack M, Mohamed F, Parrinello M, Chassaing T, Hutter J (2005) Quickstep: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput Phys Commun 167:103–112
Vermeiren W, Gilson JP (2009) Impact of zeolites on the petroleum and petrochemical industry. Top Catal 52:1131–1161
Viani L, Minoia A, Cornil J, Beljonne D, Egelhaaf HJ, Gierschner J (2016) Resonant energy transport in dye-filled monolithic crystals of zeolite L: modelling of inhomogeneity. J Phys Chem C 120:27192–27199
Wells SA, Dove MT, Tucker MG (2002) Real-space rigid-unit-mode analysis of dynamic disorder in quartz, cristobalite and amorphous silica. J Phys Condens Matter 14:4567–4584
Wells SA, Leung KM, Edwards PP, Sartbaeva A (2015) A flexibility windows in faujasite with explicit water and methanol extra-framework content. Dalton Trans 44:5978–5984
Wells SA, Sartbaeva A (2012) Template-based geometric simulation of flexible frameworks. Materials 5:415–431
Wells SA, Sartbaeva A (2015) GASP: software for geometric simulations of flexibility in polyhedral and molecular framework structures. Mol Simul 41:1409–1421
Wells SA, Sartbaeva A, Gatta GD (2011) Flexibility windows and phase transitions of ordered and disordered ANA framework zeolites. Europhys Lett 94:56001
Wesolowski TA, Warshel A (1993) Frozen density functional approach for ab initio calculations of solvated molecules. J Phys Chem 97:8050–8053
White CLIM, Ruiz-Salvador AR, Lewis DW (2004) Pressure-induced hydration effects in the zeolite laumontite. Angew Chem Int Ed 54:469–472
Wiedemann SCC, Ristanović Z, Whiting GT, Reddy Marthala VR, Kärger J, Weitkamp J, Wels B, Bruijnincx PCA, Weckhuysen BM (2016) Large ferrierite crystals as models for catalyst deactivation during skeletal isomerisation of oleic acid: evidence for pore mouth catalysis. Chem Eur J 22:199–210
Woodley SM, Catlow R (2008) Crystal structure prediction from first principles. Nat Mat 7:937–946
Wu Z, Cohen R (2006) More accurate generalized gradient approximation for solids. Phys Rev B 73:235116
Yasukawa K, Liu H, Fujinaga K, Machida S, Haraguchi S, Ishii T, Nakamura K, Kato Y (2014) Geochemistry and mineralogy of REY-rich mud in the eastern Indian Ocean. J Asian Earth Sci 93:25–36
Zhang L, Ahsbahs H, Kutoglu A (1998) Hydrostatic compression and crystal structure of pyrope to 33 GPa. Phys Chem Miner 25:301–307
Zhou X, Wesolowski TA, Tabacchi G, Fois E, Calzaferri G, Devaux A (2013) First-principles simulation of the absorption bands of fluorenone in zeolite L. Phys Chem Chem Phys 15:159–167
Acknowledgements
The authors thank the Italian Ministry of Education, MIUR-Project: “Futuro in Ricerca 2012-ImPACT- RBFR12CLQD”. G. Tabacchi thanks Prof. E. Fois for useful discussions on the role of computational modelling in the investigation of high-pressure phenomena in open frameworks. Two anonymous reviewers are thanked for the revision of the manuscript. The Editor, M. Rieder, is warmly thanked for this invited paper to celebrate the 40th of Physics and Chemistry of Minerals.
Author information
Authors and Affiliations
Corresponding author
Additional information
Invited review article to commemorate the 40th anniversary of the journal.
Rights and permissions
About this article

Cite this article
Gatta, G.D., Lotti, P. & Tabacchi, G. The effect of pressure on open-framework silicates: elastic behaviour and crystal–fluid interaction. Phys Chem Minerals 45, 115–138 (2018). https://doi.org/10.1007/s00269-017-0916-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-017-0916-z