
Abstract
A study of the effect of substitution of Mg and Ca in garnet solid solution (Grtss) was carried out using Raman spectroscopy to probe changes to the crystal lattice. The garnet solid solutions with composition changing along pyrope (Py; Mg3Al2Si3O12) and grossular (Gr; Ca3Al2Si3O12) binary were synthesized from glass at 6 GPa and 1400 °C and a second series of Grtss with composition Py40Gr60 were synthesized at 6 GPa but different temperatures from 1000 to 1400 °C. Raman mode assignments were made based on a comparison with the two end members pyrope and grossular, which show consistent result with literature study on single crystals data. The correlation between the Raman mode frequencies and compositional changes along the pyrope–grossular binary suggests a two-mode behavior for Mg and Ca cations in the garnet structure. The full widths at half-maximum of selected Raman modes increase on moving away from the end members and are about double the end-member values in the mid-position, where the frequencies closely linearly change with composition. The frequencies of the translational modes of the SiO4 tetrahedron (T(SiO4)) show large deviations from linearity indicating a strong kinematic coupling with the translational modes of the Ca and Mg cations. The anomalies in T(SiO4) are linked to mixing unit cell volume, suggesting that the nonlinear mixing volume behavior along the pyrope–grossular binary is related to the resistance of the Si–O bond to expansion and compression, which is caused by substitution of Mg and Ca cations in the dodecahedral sites. Annealing temperature also shows effect on Raman mode frequencies, but the main factor controlling the changes in mode frequencies along pyrope–grossular binary is composition.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Garnets are common minerals found in many metamorphic rocks. They also make up an important constituent of the mantle transition zone (e.g., Irifune and Ringwood 1987, 1993). The general garnet formula X3Y2(SiO4)3 can accommodate a range of divalent cations in the X site (mainly Ca, Mg, Fe and Mn) and trivalent cations in the Y site (mainly Al, Fe, Cr) resulting in large number of possible minerals. In order to simplify our characterization of the relationships between the chemical composition and the physical properties of the garnet minerals we often think of them as pure end members, for example grossular garnet (Gr: Ca3Al2Si3O12) or pyrope (Py: (Mg3Al2Si3O12), with intermediate compositions described in terms of solid solutions of the two (for example: (Ca0.5Mg0.5)3Al2Si3O12 is Gr50Py50). A first-order approach to relating end-member properties to those of intermediate compositions involves using a linear interpolation such as Vegard’s law (Vegard 1921). Many structural and chemical properties of complex garnet solid solutions can be obtained through careful characterization of the corresponding chemically simpler end members. However, deviations from ideal mixing are well-known for many mixing properties particularly for those binary systems in which there is a significant difference in the size of the cation being substituted, for example the pyrope–grossular solid solution series (e.g., Newton et al. 1977; Geiger et al. 1987; Ganguly et al. 1993; Geiger and Feenstra 1997; Dachs and Geiger 2006; Du et al. 2015, 2016). Understanding these deviations is important since they can lead to local structural heterogeneities caused by substitutions which, for example, control element partitioning behaviors (e.g., van Westrenen et al. 1999). This has resulted in many recent studies that focused on the local structural environment changes with Mg/Ca substitution in pyrope–grossular system, including theoretical calculations (e.g., Bosenick et al. 2000, 2001; Freeman et al. 2006; Kelsey et al. 2008), extended X-ray absorption fine structure (EXAFS) analysis (Oberti et al. 2006; Quartieri et al. 2008), and a nuclear magnetic resonance (NMR) study (Kelsey et al. 2008), showed qualitatively consistent results that the X–O bond lengths vary with composition. However, one would expect that compositional changes should also affect the rotation angle of the SiO4 tetrahedra; therefore, mechanistic details on the effect of substitution on the crystal structure are still unclear. For example, X-ray absorption spectroscopy studies of pyrope–grossular garnets suggest that the geometries of the local coordination of Ca and Mg cations do not change continuously along the pyrope–grossular binary (Oberti et al. 2006; Quartieri et al. 2008) which might explain the two-mode behavior of the translational Raman mode of X-cation (Ca, Mg) during substitution (Hofmeister and Chopelas 1991a; Kolesov and Geiger 1998) and the bimodal distribution of microstrain peaks along pyrope–grossular binary reported by Du et al. (2016).
Complete and accurate knowledge of how the crystal structure changes with composition along the pyrope–grossular binary is essential for correctly computing their thermodynamic and physical properties. Because of the large available calorimetric, volumetric, spectroscopic, and phase equilibrium constraints from experimental studies of the pyrope–grossular garnet solid solutions, many computational studies have attempted atomistic simulations to elucidate mixing properties in order to predict their thermodynamic behavior over a wide range of temperatures and pressures (e.g., Dove 2001; Bosenick et al. 2000, 2001; Vinograd et al. 2004; Baima et al. 2015). However, none of these studies was able to demonstrate quantitative agreement with the available experimental constraints. The most recent Monte Carlo simulation on excess free energy of pyrope–grossular garnets predicated a significant short-range ordering of Mg and Ca cations for garnet solid solution with composition (Mg0.5Ca0.5)3Al2Si3O12 (Vinograd and Sluiter 2006). Although there are no experimental results showing phase transition from cubic Ia-3d to I4122 tetragonal symmetry at 600K as predicted by Vinograd and Sluiter (2006), the description that the two dodecahedral sites become structurally distinct through substitution was consistent with other theoretical and experimental studies (e.g., Bosenick et al. 2000, 2001; Quartieri et al. 2008). In addition, Monte Carlo simulations suggest that the configurational entropy of the pyrope–grossular solid solutions with intermediate compositions might be significantly decreased from the ideal mixing values due to the development of short-range ordering. Therefore, the effect of cation arrangements during substitution in the pyrope–grossular solid solutions should be taken into account for future thermodynamic calculations (Vinograd and Sluiter 2006).
Vibrational spectroscopy provides an excellent probe of the microscopic as well as the macroscopic behavior of minerals. Local effects, including effects of compression, heating, or compositional alteration on specific sites or atomic motions inside the structure, can be examined. Both Raman and infrared spectra of five end-member garnets have been reported (Hofmeister and Chopelas 1991b; Kolesov and Geiger 1998; Mernagh and Liu 1990), but accurate prediction of the mode frequencies of solid solutions by extrapolation from the end members is inadequate due to the nonlinear mixing behavior among garnet systems. The only one reported Raman spectra along the pyrope–grossular binary by Kolesov and Geiger (1998) showed deviation from ideal mixing between the two end members. However, the reported Raman spectra of the two end members also differed largely from the single-crystal Raman spectra (Gillet et al. 1992), leaving some uncertainties with regard to their reported Raman spectra for the pyrope–grossular solid solutions. More work is needed to clear up these uncertainties and to test the nonlinear compositional dependence of the Raman modes along the pyrope–grossular garnet binary.
There has been a long-standing discussion of the behavior of the Mg cation located in the dodecahedral site of pyrope. It could be either dynamically disordered (e.g., Armbruster et al. 1992; Kolsesov and Geiger 1998, 2000) or statically disordered over subsites (e.g., Hofmeister and Chopelas 1991b; Nakatsuka et al. 2011). The large temperature dependence of the line width of the band at ~135 cm−1 has been interpreted as evidence for thermally disordered of Mg in pyrope (Kolsesov and Geiger 2000). Peak shift and line width variations of phonon spectra are related to local structural changes associated with mixing (Boffa Ballaran and Woodland 2006). It has been shown that the substitution of Mg/Ca inside pyrope–grossular solid solutions will cause different degrees of cation ordering (e.g., Bosenick et al. 2000, 2001; Du et al. 2016). Whether this static ordering of Mg/Ca cations results in changes in the Raman mode frequencies of the pyrope–grossular solid solutions is still unknown.
To address these issues we have collected a new set of Raman data from the synthetic end members pyrope and grossular and a series of solid solutions synthesized under the same pressure and temperature condition. We have also collected spectra of a series of garnets of composition Py40Gr60 synthesized in the same manner, but annealed at different temperatures to test the effect of annealing temperature on microstructure and any possible correlations with the Raman mode frequencies and excess volumes. Systematic line broadening of Raman spectra of solid solutions is quantitative analysis to characterize the local structural changes due to substitution. Specifically, nonlinearity and unexpected trends in the vibrational modes will be closely examined to provide structural information, which brings insights on their volumetric properties.
Experimental methods
Samples synthesis
Pyrope–grossular solid solutions, with eight different compositions Py100-xGrx (x = 0, 10, 20, 40, 60, 80, 90, 100) were synthesized from anhydrous glasses of stoichiometric composition. As previously reported in Du et al. (2015) the glasses were made by quenching molten mixtures of finely ground CaCO3, MgO, Al2O3, and SiO2 powders of appropriate stoichiometry. Microprobe analysis confirmed that the compositions of the glasses were the same as the starting proportions of the oxides. As previously reported in Du et al. (2015, 2016, 2017), the eight crystalline samples were synthesized at the Lamont-Doherty Earth Observatory (LDEO) in a multi-anvil (MA) device by pressurizing to 6 GPa and holding at 1400 °C for 0.5 h. Four additional samples with composition Py40Gr60 were synthesized at 6GPa but by holding at 1000, 1100, 1200, and 1400 °C for 48 h in order to explore the effect of annealing temperature on microstructure. We chose the Py40Gr60 composition since this has been shown to exhibit the largest deviation from ideal mixing volume when synthesized at 6 GPa and 1400 °C (Du et al. 2016). The quenched samples were checked by electron microprobe and scanning electron microscopy to confirm that they were of single phase and of the same composition as the starting glasses (Du et al. 2015). The grain size of these garnet samples were found to be about 5–10 µm. The unit cell volume of these garnet crystals were calculated from X-ray diffraction data collected at high-resolution synchrotron light sources (Du et al. 2016, 2017). The synthesis or annealing conditions and unit cell volume of all Py40Gr60 garnets are summarized in Table 1.
Raman spectroscopy
Raman spectra were obtained with a Renishaw InVia Reflex laser Raman microprobe at the School of Earth and Space Sciences, Peking University. The system is built around a Leica DM LM microscope combined with an f/1.8 holographic imaging spectrograph and a diode pumped solid-state laser which emits a 50 mW beam at 532 nm (18,797 cm−1). Samples were placed beneath the microscope objective and excited by the laser directed through the microscope optics. The scattered light was collected along the same optical path as the incoming laser in 180° backscattering geometry. The laser beam spot was ~1 µm in diameter which is much smaller than the size of the grains in our sample. Ten spectra were collected from each garnet sample from different points on the sample grain giving Raman data from 100 to 1100 cm−1. The peak-fitting routines in Origin pro 2016 were used to obtain peak parameters, Raman mode frequencies, and peak widths [full width at half-maximum (FWHM)] from these spectra. Lorentz peak shape functions were used in a single peak-fitting procedure in line with previous studies (Hofmeister and Chopelas 1991a). Occasionally extra peaks were included in the fitting procedure to account for small shoulders in order to ensure a satisfactory fit to the main peak.
Results
Raman mode frequencies change with composition
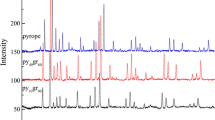
Figure 1 shows the eight Raman spectra collected from our pyrope–grossular solid solution series. Values of the mode frequencies for peaks in these spectra determined using our peak-fitting procedure are contained in Table 2. The Raman spectra of all eight garnets show the same overall appearance containing sharp peaks grouped in three regions: low-frequency peaks (180–420 cm−1), medium-frequency peaks (450–700 cm−1), and high-frequency peaks (850–1100 cm−1). The peak positions of the end members pyrope and grossular agree well with literature data from both synthetic polycrystalline samples (Gillet et al. 1992) and single crystals with same compositions (Kolesov and Geiger 1998) (Table 3). The Raman modes of the end members are well assigned to the vibrational modes allowed by crystal symmetry after Hofmeister and Chopelas (1991a) (Table 2). The observed vibrational modes are grouped into three main regions: the highest frequency modes (A, B, C, D) are dominated by stretching motions of Si–O ((Si–O)str), the modes at around 550 cm−1 (E, F, G, H, I, J) are dominated by bending motions of Si–O ((Si–O)bend), and the modes at around 350 cm−1 (K, L, M, N, O) are dominated by rotation motions of SiO4 (R(SiO4)). In addition, the mode at around 200 cm−1 (S) is due to translational motions of SiO4 (T(SiO4)) (Kolesov and Geiger 1998). Garnet solid solutions with compositions along this binary were similarly assigned as the end members. Several bands between 240 and 350 cm−1 (P, Q, R) are distinct in the grossular-rich samples, but weak in the pyrope-rich samples. They decrease in intensity towards the intermediate solid solution compositions until they are no longer observable in the Py60Gr40 spectrum (Fig. 1). They have previously been identified as modes associated with X-site cations (Ca in this case) in the dodecahedral site with the decrease in intensity being due to substitution of Mg for Ca and the two-mode behavior caused by the large size difference between the mixing cations Mg and Ca (Hofmeister and Chopelas 1991a). In the pyrope-rich samples, we see peaks due to Mg-translational modes (~133 cm−1). The frequencies associated with the Mg and Ca-translational modes do not change linearly with composition between the two end-member garnets as was previously observed (Kolesov and Geiger 1998).

Raman spectra of garnets along pyrope–grossular binary. The marked P, Q, R peaks represent the newly appearing Raman modes (translational modes of X-cations) for grossular-rich garnets. E and F peaks mark the Raman modes associated with bending motion of Si–O bond, showing discontinued changes with composition. And these discontinuities represent the two-mode behavior of Raman spectra of pyrope–grossular garnets
Figure 2 shows the compositional dependencies of the internal mode frequencies of SiO4, (Si–O)str and (Si–O)bend along the pyrope–grossular binary. The stretching motions of Si–O bonds show a strong dependence on garnet composition (Fig. 2a), and the bending motions of Si–O bonds show a relatively weak dependence on garnet composition (Fig. 2b). The internal mode frequencies of SiO4 in pyrope-rich garnets are higher than the grossular-rich garnets. This trend has been found for garnets in general (Hofmeister and Chopelas 1991a) that the internal modes of SiO4 tetrahedra show simple dependencies on lattice parameters. Two (Si–O)bend bands (E and F) appear in two different ranges for pyrope-rich garnets (646–686 cm−1) and grossular-rich garnets (593–646 cm−1) (Table 2; Fig. 2b) indicating a discontinuous change with composition.

Compositional dependence of frequencies of Raman mode corresponding with internal motions of SiO4-tetrahedra: a Si–O stretching and b Si–O bending. The Raman modes marked as E and F in Fig. 1 (600–700 cm−1) show discontinuity along pyrope–grossular binary
Selected internal vibrational mode frequencies and widths as a function of composition along pyrope–grossular binary are shown with details in Fig. 3. The frequencies for internal modes are seen to decrease linearly with Ca content in garnet composition (Fig. 3a1 and b1), as previously observed (Kolesov and Geiger 1998). The stretching motions show stronger dependence on composition and hence unit cell volume than the bending modes as expected (Hofmeister and Chopelas 1991a) with (Si–O)str varying at an average rate of ~130 ± 20 cm−1/Å compared to an average garnet value of about 120 cm−1/Å and the (Si–O)bend varying at an average rate of 63 cm−1/Å compared to an average garnet value of about ~90 cm−1/Å. The higher rate of change for the (Si–O)str modes is due to the fact that the force constants for stretching modes are much smaller than those bending modes thus amplifying the effect. The Raman mode frequency (L) associated with the rotation of the SiO4-tetrahedron (R(SiO4)) is roughly independent of composition (Fig. 3c1), and the translational modes of the SiO4-tetrahedron (T(SiO4)) (S) changes nonlinearly with composition (Fig. 3d1), indicating the coupling of this vibration with translational mode of the X-site cations.

Selected Raman mode frequencies and their peak widths change with composition along pyrope–grossular binary. a (Si–O) stretching, b (Si–O) bending, c SiO4 rotation, and d SiO4 translation. The empty circles are data from Kolesov and Geiger (1998), and the gray circles are Py40Gr60 garnets synthesized at different temperatures
Raman mode frequencies vs. synthesis temperatures
Figure 4 shows the Raman spectra from the Py40Gr60 garnet solid solutions synthesized at 6 GPa and a range of temperatures from 1000 to 1400 °C. The Raman spectra of these five Py40Gr60 garnets have the same overall appearance. The selected Raman mode frequencies determined from these spectra using out peak-fitting method are shown in Fig. 5. The Raman mode associated with translational motions of the SiO4-tetrahedra and its peak width (FWHM) for all of these five garnet samples are summarized in Table 1 and Fig. 6. We can see that there is a weak-positive correlation between translational mode frequency and unit cell parameter (Fig. 6a) and that the Raman mode lines become broader with increasing annealing temperature (Fig. 6b).

Raman spectra of Py40Gr60 garnets synthesized at different temperatures from 1000 to 1400 °C, showing same overall appearance

Selected Raman mode frequencies of Py40Gr60 garnets annealed at different temperatures

a The translational mode frequencies of SiO4-tetrahedron of Py40Gr60 annealed at different temperatures. b Peak width of Raman mode corresponding to translational motion of SiO4-tetrahedron decreases with increasing on unit cell dimension and decreasing on annealing temperature
Discussion
Similar compositional dependence of selected Raman modes for the pyrope–grossular garnets was reported by Kolesov and Geiger (1998), although their Raman mode frequencies associated with bending of Si–O bond and rotation of SiO4-tetrahedron are both smaller than that we report in this study (Fig. 3b1, c1). Kolesov and Geiger (1998) reported Raman mode frequencies of end-member garnets from both single-crystal and polycrystalline samples and that the single-crystal values are larger than those from the polycrystalline samples and agree well with our data and other literature studies (Table 3). The Raman laser spot size (~1 µm) that we used was smaller than the grain size (5–10 µm) of our samples. This could be the reason why the Raman data presented here is closer to the data collected from single crystals and Raman frequency values from pyrope–grossular garnet solid solutions are larger than those from polycrystalline samples as reported by Kolesov and Geiger (1998).
As shown by Bosenick et al. (1995), the grossular-rich garnets that were used by Kolesov and Geiger (1998) were synthesized from seeded glass, where the garnet seed was hydrothermally synthesized from garnet glass with 7 wt% distilled water. There was no report of the water content of these garnets in their paper and whether there may be an effect on the vibrational spectra from any OH included in their material. The garnet solid solutions used in the previous study were synthesized at temperatures from 1050 to 1400 °C (Bosenick et al. 1995). As mentioned above, the synthesis temperature has a slight effect on the Raman mode frequencies. On the other hand, the garnet samples used in this study were all synthesized anhydrously from dry glass at the same temperature (1400 °C). Therefore, the Raman mode frequencies from our samples should be more consistent and provide a better data set for calculation of thermodynamic parameters via lattice dynamic models.
The low-frequency mode at around 210 cm−1 in pyrope that shifts gradually to 180 cm−1 in grossular is related to the translational motions of the SiO4-tetrahedra rather than the translational motions of the X-cation (Mg, Ca) (Kolesov and Geiger 1998). Our new observation shows that the translational motions of the SiO4 tetrahedra deviate strongly from linearity along the solid solution series (Fig. 3d1). Since these Raman mode frequencies are close to those associated with the translational motions of the Mg and Ca cations (T (Mg, Ca)), the large deviation suggests a strong kinematic coupling between the T(SiO4) and T (Mg, Ca) modes. This implies that the frequency of the T(SiO4) mode depends on the bond strengths of surrounding polyhedral rather than on the internal SiO4 bonds. Therefore, the decrease in frequency of the T(SiO4) and T (Mg, Ca) modes from pyrope to grossular is caused by expansion of the lattice due to substitution of the smaller Mg2+ cation with the much larger Ca2+ cation. The negative deviation from linearly changing with composition of the Raman modes of T(SiO4) indicates an increased volume for intermediate compositions. We have observed similar mixing volume deviations previously from unit cell volume measurements the behavior of which as a function of composition we compare with the Raman mode frequency data in Fig. 7. The large excess volume change with composition is caused by large size mismatch between Ca (1.12 Å) and Mg (0.89 Å) (Shannon 1976), which then causes changes to the rotational orientation of the SiO4 tetrahedra and changes in the Si–O bond lengths.

Deviation of translational mode frequencies of SiO4 tetrahedra from linearity along pyrope–grossular binary (empty symbols). The solid symbols are excess volume along this binary reported by Du et al. (2016). The corresponding changes on rotation of SiO4 tetrahedra caused by substitution of Mg and Ca cations show good correlation with unit cell volume changes along pyrope–grossular binary. The dashed line is polynomial fitting to the excess volume data
Figure 3 shows plots of the peak width (FWHM) of Raman modes as a function of composition (Table 4). The Raman modes of the synthetic end members, pyrope and grossular, are very sharp indicating that the cations are well ordered. The Raman modes of garnet solid solutions show peak broadening, reaching a maximum near the Py50Gr50 composition (Fig. 3a2, b2, and c2). For solid solution garnets line broadening in the vibrational spectra can be caused by defects or disorder (Boffa Ballaran et al. 1999). Similar Raman mode peak broadening has been observed for the pyrope–majorite solid solutions, which was interpreted as cation disorder due to the Mg2+ + Si4+ = 2Al3+ substitution. This mode broadening was interpreted as a characteristic precursor effect for structural phase transitions, which was confirmed in the pyrope–majorite system by X-ray diffraction as well as vibrational spectroscopy (Liu et al. 2017). We did not observe any structural phase transition for the pyrope–grossular system during our previous X-ray diffraction studies (Du et al. 2016, 2017). We did, however, observe similar X-ray diffraction peak broadening which was found to be related to the arrangement of Ca and Mg cations in the dodecahedral sites (Du et al. 2016, 2017). When one atom (Mg or Ca) is replaced by the other on the dodecahedral site, the local structure relaxes around it resulting in structural variations on the same length scale as phonons. The line broadening that we present here for Si–O stretching, bending, and rotation modes of SiO4 may be due to the effect of these substitutions on the Raman mode frequencies. However, the line width of the Raman mode associated with the translational motion of SiO4 (T(SiO4)) shows a large difference compared to the other three modes (Fig. 3d2). Garnet solid solutions with compositions close to 50:50 (Py40Gr60 and Py60Gr40) show narrower peaks than those with compositions close to the two end members. As discussed above, the Raman modes of T(SiO4) show strong coupling with cation (Mg and Ca) substitutions, therefore we might expect a much broader peak width for T(SiO4). However, we observed the opposite. The peak widths of garnets with intermediate compositions are narrower than those with compositions close to the end members, indicating that the physical origins of the peak broadening for T(SiO4) is different comparing with R(SiO4). If we took line broadening as indicative of the range of local distortions, the local structural heterogeneities in a crystal will cause strain gradients. The microstrain calculated from X-ray diffraction data varies in a similar double-peak pattern as the peak width of the Raman mode T(SiO4) (Fig. 8), suggesting that they both probe the same distortions of the crystal structure. As mentioned above, the frequency of the T(SiO4) mode is largely affected by the surrounding X-cations, because it is dependent on whether it is strongly compressed by the larger cation (Ca) in the neighboring dodecahedron or weekly compressed by the smaller cation (Mg). Along this line, we would expect that the arrangement of Mg and Ca cation in the dodecahedral site will affect the Raman mode frequency of T(SiO4). If we took the peak broadening of the T(SiO4) mode as a sign of disordering then the much narrower peaks for Py60Gr40 and Py40Gr60 indicate that these two garnets have a more ordered structure, which is consistent with previous studies using NMR spectroscopy and synchrotron X-ray diffraction (e.g., Bosenick et al. 1995, 1999; Du et al. 2016). As proposed by Bosenick et al. (1995), garnet solid solutions with intermediate composition along the pyrope–grossular binary show larger degree of short-range ordering than those with compositions close to the end members. It was also highlighted through an ab initio calculation that the energies of particular atomic configurations are closely related to the third nearest neighbor (3NN) arrangement of the Mg and Ca cations, which are edge sharing with the same SiO4 tetrahedron resulting in the pairing of the same cations (Mg–Mg and Ca–Ca) in the S3NN site being unfavorable from an energy point of view due to a higher configurational energy (Freeman et al. 2006). If we assume that the broadening of the X-ray diffraction peaks and the Raman peaks associated with the T(SiO4) mode both indicate more microstrain, then this would indicate that garnets with composition close to the end members have more cation pairings (Mg–Mg and Ca–Ca) in the third nearest neighbor position than garnets with intermediate compositions (e.g., Py60Gr40 and Py40Gr60) which thus have less possibility of randomly generated Mg–Mg or Ca–Ca (S3NN) pairings giving an intrinsic degree of short-range ordering (e.g., Bosenick et al. 1995, 2000, 2001; Dove et al. 2000; Vinograd et al. 2004; Lavrentiev et al. 2006; Vinograd and Sluiter 2006). This less “strained” structure may have relatively large excess volume because Ca–Mg pairs pack less efficiently than strain-causing Mg–Mg and Ca–Ca pairs.

Peak width of Raman mode corresponding to the translational motion of SiO4 tetrahedra. The open symbols are microstrain data calculated from synchrotron X-ray diffraction pattern (Du et al. 2017) with equation \(B_{\text{garnet}} \cos \theta\) = \(\frac{K\lambda}{\left\langle L \right\rangle > \cos \theta}\cos \theta + 4\eta \tan \theta \cos \theta\) = \(\frac{K\lambda}{\left\langle L \right\rangle} + 4\eta \sin \theta\), where λ is the wavelength of the X-ray, and θ is the Bragg angle, K is a constant of value approximately 0.9 (Langford and Wilson 1978), L is the crystal size (diffraction domain size), \(\frac{K\lambda}{\left\langle L \right\rangle \cos \theta}\) is the size effect on XRD peak broadening (Scherrer 1918), 4ηtanθ is the width of the peak due to microstrain (Stokes and Wilson 1944), and η is the microstrain. Similar two-peaked pattern indicates the local distortional strain by Mg–Ca size misfit affects the translational mode of SiO4 tetrahedra. The dashed line is polynomial fitting to the microstrain data
The short-range ordering of Mg and Ca in the pyrope–grossular garnets was reported to decrease with increasing synthesis temperature (Bosenick et al. 1999). Garnets synthesized at higher temperatures might be expected to exhibit larger microstrain as more strain-causing S3NN pairs are formed resulting in broader Raman peaks as high synthesis temperature corresponds to a more disordered cation distribution. The Raman spectra of Py40Gr60 synthesized at different temperatures show that the peak width of the T(SiO4) Raman mode increases with increasing synthesis temperature, which corresponds to a more disordered structure (Fig. 6). Therefore, unlike internal modes of SiO4, which change linearly with composition, the Raman mode of T(SiO4) deviates largely from linearity and shows strong correlation with the microstrain inside the garnet structure and so may be a good indicator of the degree of cation ordering in solid solutions.
Conclusions
An analysis of Raman mode frequencies of garnets along the pyrope–grossular solid solution series shows that the vibrational spectra change with composition and annealing temperatures. The Raman modes associated with the stretching and bending motions of Si–O bonds and the rotational motions of the SiO4 tetrahedra show simple linear dependences on composition, but the translational modes of SiO4 tetrahedra show a nonlinear trend due to stronger coupling with translational modes of the X (Ca and Mg) cations.
The peak widths of the selected Raman modes (A1g) show a nonlinear dependence on composition, and reach a maximum near the intermediate garnets. The arrangement of Mg and Ca has a large effect on the translational motion of the SiO4 tetrahedra. The peak widths of translational modes of T(SiO4) are slightly “M” shaped vs. composition and may result from different short-range ordering status of Mg and Ca cations during substitution.
References
Armbruster T, Geiger CA, Lager GA (1992) Single crystal X-ray refinement of almandine-pyrope garnets at 298 and 100 K. Am Min 77:512–523
Baima J, Ferrabone M, Orlando R, Erba A, Dovesi R (2015) Thermodynamics and phonon dispersion of pyrope and grossular silicate garnets from ab initio simulations. Phys Chem Min 43:137–149
Boffa Ballaran T, Woodland AB (2006) Local structure of ferric iron-bearing garnets deduced by IR-spectroscopy. Chem Geol 225:360–372
Boffa Ballaran T, Carpenter MA, Geiger CA, Koziol AM (1999) Local structural heterogeneity in garnet solid solutions. Phys Chem Mineral 26:554–569
Bosenick A, Geiger CA, Schaller T, Sebald A (1995) A 29Si MAS NMR and IR spectroscopic investigation of synthetic pyrope-grossular garnet solid solutions. Am Min 80:691–704
Bosenick A, Geiger CA, Phillips BL (1999) Local Ca-Mg distribution of Mg-rich pyrope-grossular garnets synthesized at different temperatures revealed by 29Si MAS NMR spectroscopy. Am Min 84:1422–1432
Bosenick A, Dove MT, Geiger CA (2000) Simulation studies on the pyrope-grossular garnet solid solution. Phys Chem Min 27:398–418
Bosenick A, Dove MT, Heine V, Geiger CA (2001) Scaling of thermodynamic mixing properties in garnet solid solutions. Phys Chem Min 28:177–187
Dachs E, Geiger CA (2006) Heat capacities and entropies of mixing of pyrope-grossular (Mg3Al2Si3O12–Ca3Al2Si3O12) garnet solid solutions: a low-temperature calorimetric and a thermodynamic investigation. Am Min 91:894–906
Dove MT (2001) Computer simulations of solid solutions. In: Geiger CA (ed) Solid solutions in silicate and oxide systems of geological importance. EMU notes in mineralogy, vol 3. Eötvös University Press, Budapest, pp 225–250
Dove MT, Bosenick A, Myers ER, Warren MC, Redfer SAT (2000) Modeling in relation to cation ordering. Phase Trans 71:205–226
Du W, Clark SM, Walker D (2015) Thermo-compression of pyrope-grossular garnet solid solution: non-linear compositional dependence. Am Min 100:215–222
Du W, Clark SM, Walker D (2016) Excess mixing volume, microstrain, and stability of pyrope-grossular garnets. Am Min 101:193–204
Du W, Li X, Li B (2017) Microstrain in pyrope-grossular garnet solid solution at high pressure: a case study of Py90Gr10 and Py10Gr90 up to 15 GPa. Phys Chem Min 44(6):377–388
Freeman CL, Allan NL, van Westrenen W (2006) Local cation environments in the pyrope-grossular Mg3Al2Si3O12–Ca3Al2Si3O12 garnet solid solution. Phys Rev B 74:134203
Ganguly J, Cheng W, O’Neill HC (1993) Syntheses, volume, and structural changes of garnets in the pyrope-grossular join; implications for stability and mixing properties. Am Min 78:583–593
Geiger CA, Feenstra A (1997) Molar volumes of mixing of almandine-pyrope and almandine-spessartine garnets and the crystal chemistry and thermodynamic-mixing properties of the aluminosilicate garnets. Am Min 82:571–581
Geiger CA, Newton RC, Kleppa OJ (1987) Enthalpy of mixing of synthetic almandine-grossular and almandine-pyrope garnets from high-temperature solution calorimetry. Geochim Cosmochim Acta 51:1755–1763
Gillet P, Fiquet G, Malezieux JM, Geiger CA (1992) High-pressure and high-temperature Raman spectroscopy of end-member garnets: pyrope, grossular and andradite. Eur J Min 4:651–664
Hofmeister AM, Chopelas A (1991a) Vibrational spectroscopy of end-member silicate garnets. Phys Chem Min 17:503–526
Hofmeister AM, Chopelas A (1991b) Thermodynamic properties of pyrope and grossular from vibrational spectroscopy. Am Min 76:880–891
Irifune T, Ringwood AE (1987) Phase transformation in primitive MORB and pyrolite compositions to 25 GPa and some geophysical implications. In: Manghnani MH, Syono Y (eds) High pressure research in mineral physics, vol 39. TERRAPUB Tokyo/American Geophysical Union, Washington, DC, pp 235–246
Irifune T, Ringwood AE (1993) Phase transformations in subducted oceanic crust and buoyancy relationships at depths of 600–800 km in the mantle. Earth Planet Sci Lett 117(1–2):101–110
Kelsey KE, Stebbins JF, Du L, Mosenfelder JL, Asimow PD, Geiger CA (2008) Cation order/disorder behavior and crystal chemistry of pyrope-grossular garnets: an 17O 3QMAS and 27Al MAS NMR spectroscopic study. Am Min 93:134–143
Kolesov BA, Geiger CA (1998) Raman spectra of silicate garnets. Phys Chem Min 25:142–151
Kolesov BA, Geiger CA (2000) Low-temperature single-crystal Raman spectrum of pyrope. Phys Chem Min 27:645–649
Langford JI, Wilson AJC (1978) Scherrer after sixty years: a survey and some new results in the determination of crystallite size. J Appl Crystallogr 11:102–113
Lavrentiev MY, van Westrenen W, Allan NL, Freeman CL, Purton JA (2006) Simulation of thermodynamic mixing properties of garnet solid solutions at high temperatures and pressures. Chem Geol 225:336–346
Liu Z, Du W, Shinme T, Gréaux S, Zhou C, Arimoto T, Kunimoto T, Irifune T (2017) Garnets in the majorite-pyrope system: symmetry, lattice microstrain, and order-disorder of cations. Phys Chem Min 44:237–245
Mernagh TP, Liu L (1990) Pressure dependence of Raman spectra from the garnet end-members pyrope, grossularite and almandine. J Raman Spec 21:305–309
Nakatsuka A, Shimokawa M, Nakayama N, Ohtaka O, Arima H, Okube M, Yoshiasa A (2011) Static disorders of atoms and experimental determination of Debye temperature in pyrope: low- and high-temperature single-crystal X-ray diffraction study. Am Min 96:1593–1605
Newton RC, Charlu TV, Kleppa OJ (1977) Thermochemistry of high pressure garnets and clinopyroxenes in the system CaO-MgO-Al2O3-SiO2. Geochim Cosmochim Acta 41:369–377
Oberti R, Quartieri S, Dalconi MC, Boscherini F, Iezzi G, Boiocchi M, Eeckhout SG (2006) Site preference and local geometry of Sc in garnets: part I. Multifarious mechanisms in the pyrope-grossular join. Am Min 91:1230–1239
Quartieri S, Boscherini F, Dalconi C, Iezzi G, Meneghini C, Oberti R (2008) Magnesium K-edge EXAFS study of bond-length behavior in synthetic pyrope-grossular garnet solid solutions. Am Min 93:495–498
Scherrer P (1918) Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachrichten der Kgl Gesellschaft der Wissenschaftern zu Göttingen 26:98–100
Shannon RD (1976) Revised effective ionic radii. Acta Crystallogr A32:751–767
Stokes AR, Wilson AC (1944) The diffraction of X rays by distorted crystal aggregates. Proc Phys Soc 56:174–181
van Westrenen W, Blundy J, Wood B (1999) Crystal-chemical controls on trace element partitioning between garnet and anhydrous silicate melt. Am Min 84:838–847
Vegard L (1921) Die Konstitution der Mischkristalle und die Raumfüllung der Atome. Zeitschrift für Physik 5:17–26
Vinograd VL, Sluiter MHF (2006) Thermodynamics of mixing in pyrope-grossular, Mg3Al2Si3O12-Ca3Al2Si3O12, solid solution from lattice dynamics calculations and Monte Carlo simulations. Am Min 91:1815–1830
Vinograd VL, Sluiter M, Winkler B, Putnis A, Hålenius U, Gale JD, Becker U (2004) Thermodynamics of mixing and ordering in pyrope-grossular solid solution. Min Mag 68:101–121
Acknowledgements
We thank two anonymous reviewers for their constructive comments on our manuscript and Dr. Larissa Dobrzhinetskaya for processing this paper. The authors thank Hongrui Ding from School of Earth and Space Sciences in Peking University for help with Raman Spectroscopy measurements. This work was supported by the Strategic Priority Research Program (B) of Chinese Academy of Sciences (Grant No. XDB18000000), and by the DREAM project of MOST, China (Grant No. 2016YFC0600408) to Xi Liu.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Du, W., Han, B., Clark, S.M. et al. Raman spectroscopic study of synthetic pyrope–grossular garnets: structural implications. Phys Chem Minerals 45, 197–209 (2018). https://doi.org/10.1007/s00269-017-0908-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-017-0908-z