
Abstract
We present a systematic experimental study on the phase transition, lattice microstrain, and order–disorder of cations for garnets in the majorite–pyrope system. Polycrystalline gem-quality garnets were synthesized at high pressure and high temperature using a Kawai-type multi-anvil apparatus. A phase transition from a cubic to tetragonal structure is clearly observed for garnets with the majorite content of more than 74 mol % through X-ray diffraction (XRD) and Raman scattering studies. Microstrain of garnets, evaluated with the Williamson–Hall plot on XRD profiles, shows a nonlinear dependence of the garnet compositions. The variation of the XRD peak broadening suggests the lattice microstrain of these garnets may be associated with the local structural heterogeneities due to the substitution of different cations via the coupled substitution (Mg2+ + Si4+ = 2Al3+) in the garnet structure. The width variation of Raman scattering peaks indicates that cation disorder occurs in the garnet structure for intermediate compositions. It is found that intermediate garnets and end-members have a minimum of microstrain, while those between end-members and intermediate compositions possess a larger microstrain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Garnet is one of the most abundant rock-forming minerals in the Earth’s crust, upper mantle, and mantle transition zone (Novak and Gibbs 1971; Ringwood and Major 1971; Akaogi and Akimoto 1977; Irifune and Ringwood 1987). Garnet in the Earth’s mantle usually exists as the solid solution and contains considerable amounts of sodium (Na), aluminum (Al), calcium (Ca), and iron (Fe). Among these garnet solid solutions, the majorite (MgSiO3)–pyrope (Mg3Al2Si3O12) solid system is the most dominant and related garnet solutions in the Earth’s mantle transition zone (Irifune and Ringwood 1987; Liu et al. 2015). Therefore, accurate knowledge of crystal structures, lattice strains, thermodynamics, and other physical properties of garnets in the majorite–pyrope system is indispensable for understanding the physical dynamics of the mantle transition zone.
Garnet, such as pyrope, usually has a cubic structure with an \(\text{Ia}\overline{\text{3}} \text{d}\) space group and consists of ZO4 tetrahedra and YO6 octahedra, giving rise to a three-dimensional framework (Novak and Gibbs 1971). However, for the majorite–pyrope system, the synthetic MgSiO3 majorite has a tetragonal structure with a I41/a space group at ambient conditions, which is formed through a symmetry reduction from the space group \(\text{Ia}\overline{\text{3}} \text{d}\) (cubic) to the space group I41/a (tetragonal) due to Mg–Si ordering in the octahedral sites upon cooling (Angel et al. 1989). Pervasive twinning textures in the synthetic MgSiO3 majorite provide the evidence for the symmetry reduction during cooling in their syntheses, when majorite went across the cubic–tetragonal phase boundary (Wang et al. 1993; Heinemann et al. 1997). Furthermore, the phase transition from the cubic (\(\text{Ia}\overline{\text{3}} \text{d}\)) to tetragonal (I41/a) symmetry is also found for garnets with the majorite content of more than 75–80 mol % (Mj75Py25, Mj: majorite; Py: pyrope) (Parise et al. 1996; Heinemann et al. 1997). This phase transition, an improper ferroelastic transition (Heinemann et al. 1997), may have important effects on the elasticity and relative strength (Sinogeikin et al. 1997; Hunt et al. 2010; Liu et al. 2015). However, Mj75Py25 in the work by Heinemann et al. (1997) was observed as a cubic garnet, whereas it was regarded as a tetragonal structure due to some newly appearing tetragonal peaks found by Parise et al. (1996). Furthermore, some studies by Sinogeikin et al. (1997) and Gwanmesia et al. (2000) did not find this phase transition. Crystallographic refinements suggested that tetragonal garnets of Mj64Py26–Mj38Py62 had a space group of I41/acd (Nakatsuka et al. 1999). Orthorhombic symmetry was also proposed for the Mj93Py7 garnet through Raman spectroscopy study (Rauch et al. 1996). Hence, this phase transition on the dependence of chemical compositions along the majorite–pyrope binary is still ambiguous due to the different synthetic conditions and analytical methods.
It is interesting to understand the macroscopic physical properties of garnets through microscopic views. For example, the thermodynamic mixing behavior of pyrope–grossular garnets can be interpreted by their microscopic structural strain (Boffa Ballaran et al. 1999; Boffa Ballaran and Carpenter 2003; Bosenick et al. 2001; Dapiaggi et al. 2005; Freeman et al. 2006; Du et al. 2016), which is caused by the substitution of different cations into their lattice sites due to the mismatch of cation size. The majorite–pyrope system provides an excellent substitution solid system for investigating microscopic–macroscopic relationships and the physical nature behind thermodynamic behaviors. Because of the different ionic radii of cations in this system (Mg2+: 0.69 Å, Al3+: 0.54 Å, Si4+: 0.43 Å) (Shannon 1976), Al3+ would substitute Mg2+ and Si4+ in octahedral sites in the garnet structure via the coupled substitution (Mg2+ + Si4+ = 2Al3+) (Parise et al. 1996; Heinemann et al. 1997), which should theoretically result in the local structure heterogeneity and lattice microstrain (Boffa Ballaran et al. 1999; Bosenick et al. 2001; Dapiaggi et al. 2005; Freeman et al. 2006; Du et al. 2016). Nevertheless, there have been no studies on the lattice microstrain in this system.
Because of the high symmetry of the crystallographic structure for garnets, vibrational properties of garnets in the majorite–pyrope system have been widely studied by Raman spectroscopy (McMillan et al. 1989; Kolesov and Geiger. 1998; Giesting and Hofmeister 2002; Giesting et al. 2004; Hofmeister et al. 2004). In particular, the Raman vibrational spectrum of garnets is quite sensitive to the local structure distortion such as SiO4-tetrahedra and AlO6-octahedra and cation order–disorder effects. Some earlier studies found that substitution of Mg and Si by Al resulted in cation disorder in the majorite–pyrope system, which significantly affected the thermal conductivity (Giesting and Hofmeister 2002), and was also associated with the phase transition and thermodynamics (Giesting et al. 2004; Li et al. 2007). This site-substitution disorder in garnet solid solutions can be well reflected in Raman spectroscopy. However, Raman spectroscopy study of garnets in the majorite–pyrope system is still very limited, especially for the majorite-rich garnets.
Here, we synthesized a series of high-quality garnets with different compositions along the majorite–pyrope binary at high pressure and high temperature using a Kawai-type multi-anvil apparatus combined with tungsten carbide anvils. Additionally, we reported the chemical compositional and site-substitutional dependence of their physical properties related to symmetry, microstrain, order–disorder of cations for garnets.
Experimental methods
A series of single-phase polycrystalline Mj x Py1−x garnets (x mol % majorite and (1−x) mol % pyrope) in the majorite–pyrope system were synthesized from the starting material of glass, which was quenched from the fused high-purity oxide mixture consisting of MgO, Al2O3, and SiO2 at 1600–1800 °C. The compositions of synthetic garnets are consistent with those of the glass starting material within analytical errors. The fine-ground glass powder was put in an oven at 110 °C for 24 h to get rid of the water effect and then densely packed into Au capsules using a hand press on the hot-plate at ~150 °C. The sealed gold capsules were placed into an MgO sleeve, which insulates the capsule from the rhenium (Re) heater in 11/5 (OEL/TEL = Octahedral Edge Length of pressure medium/Truncated Edge Length of the anvil) cell assembly (Fig. 1). High-pressure and high-temperature experiments were performed at 18–19 GPa under 1700 °C for 2 h using a Kawai-type multi-anvil apparatus (Orange-3000) at the Geodynamics Research Center, Ehime University, Japan. After that, the pressure was decompressed slowly for about 10–12 h at 500 K to remove the stress in the sample. Pressure was calibrated against the press load on the basis of phase transition of ZnS (15.5 GPa), GaAs (18.7 GPa), and GaP (23 GPa) at room temperature (Irifune et al. 1996; Liu et al. 2016). Pressure calibration at high temperatures was calibrated by the phase transformation of MgSiO3 and 95MgSiO3·5Al2O3 (x mol %) from ilmenite/majorite to bridgmanite at 2000 K (Irifune et al. 1996; Liu et al. 2016). Temperature was monitored by W97Re3–W75Re25 thermocouple located in the center of the Re heater without correction for the pressure effect on the thermocouple EMF.

Schematic cross section of the high-pressure 11/5 cell assemblage for hot-pressing polycrystalline garnets
The microstructure and chemical composition (Supplementary Table S1) of the recovered samples were checked by a field-emission scanning electron microprobe (FE-SEM; JEOL-7000F) and scanning electron microprobe (SEM; JEOL, JSR-1000) with an energy-dispersive spectrometer (EDS) at the Geodynamics Research Center, Ehime University, Japan. The chemical composition was calibrated with MgSiO3 enstatite, MgSi2O4 forsterite, and Al2O3 corundum as standards. The recovered samples were polished using 1-μm diamond paste and examined by a microfocus X-ray diffraction (XRD) with a rotating Cu anode at 40 kV, 30 mA. Some selected synthetic samples (pyrope and Mj74Py26 garnet) are well grounded into powder and checked by powder XRD with a rotating Co anode at 40 kV, 40 mA in Bayerisches Geoinstitut, University of Bayreuth, Germany. High-purity Si was used as an external standard to calibrate 2θ for XRD, respectively. High-quality XRD profiles of these garnets were collected by extending the exposure time to 2 h. Raman spectroscopy was performed using a confocal micro-Raman system (Renishaw, RS-SYS1000) equipped with an Ar ion laser (λ = 514.5 nm). Raman shift was calibrated by a silicon standard, and its uncertainty is less than ±0.5 cm−1.
Results and discussions
X-ray diffraction: symmetry study
Chemical compositions, pressure and temperature conditions for synthesis, lattice parameters, and unit-cell volumes are summarized in Table 1. Selected representative photomicrographs, second and back-scattered electron images of the quenched samples are displayed in Fig. 2. The synthetic polycrystalline garnets with grain size of 2–5 μm are well sintered, nearly cylindrical (diameter ~2 mm and height ~1 mm) in shape, free of microcracks, and translucent or transparent in color. The well-sintered garnets without microcracks suggest that there is almost no stress in our synthetic samples. These synthetic garnets are homogeneous in their compositions from the back-scattered electron observation combined with their chemical composition analysis (Supplementary Table S1).

Representative second and back-scattered electron images with photomicrographs (inside) showing the textures of a Mj59Py41 and b Mj80Py20, respectively
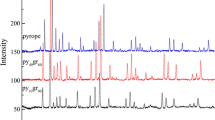
Figure 3 shows some representative high-quality XRD patterns of garnet solid solutions at room conditions. Although the distinguishability between the cubic and tetragonal structure for garnets is subtle and difficult to observe, we can clearly see the differences between them through a long exposure time. In all the XRD patterns, reflections of garnets between pyrope and Mj59Py41 garnet are well assigned to a cubic structure (\(\text{Ia}\overline{\text{3}} \text{d}\)), while those with the majorite content higher than 74 mol % can be resolved into a tetragonal structure (I41/a) (Fig. 3a). In Fig. 3b, the new peaks of (031)/(013), (222), (053)/(035), (424)/(244), and (626), which are absent in cubic garnets (Mj59Py41, Mj41Py59, Mj20Py80, and pyrope), are first observed in the XRD pattern of Mj74Py26 and exist in the other majorite-rich garnets with the majorite content higher than 74 mol %. It is found that the reflections of (031)/(013)/(053)/(035) and (222)/((442)/(662) belong to the classes of 0kl: k, l = 2n and hhl: 2 h + l = 4n for the tetragonal symmetry with the space group I41/a (Angel et al. 1989; Parise et al. 1996), which are absent for the cubic (\(\text{Ia}\overline{\text{3}} \text{d}\)) symmetry, suggesting that garnets with majorite contents higher than 74 mol % are probably tetragonal.

a Representative XRD patterns of Mj x Py1−x (x majorite mol %) garnets. The marked asterisks indicate the reflections are from the tetragonal garnets, but absent in the cubic garnets. The vertical blue and red lines represent the diffraction indices of cubic (Novak and Gibbs 1971) and tetragonal garnet (Angel et al. 1989), respectively. b The enlarged image of XRD patterns of garnets from Mj39Py61 to Mj80Py20
We also plotted the (400) and (800) reflections for these garnets (Fig. 4a). The (400) reflection becomes broader at Mj74Py26 and gradually splits into two peaks toward majorite. In the case of the (800) reflection, it visibly broadens with increasing majorite contents and splits into (800)/(008) doublets from Mj74Py26 to majorite. This splitting of (h00) reflection is additional evidence for the phase transition from the cubic to tetragonal symmetry, which was also observed by Parise et al. (1996) and Heinemann et al. (1997). Therefore, the appearance of new peaks assigned to the tetragonal symmetry and the broadening and splitting of (800)/(400) peaks suggest that the phase transition from cubic to tetragonal structure occurs in the Mj74Py26 garnet, which provides more convincing evidences than Parise et al. (1996). The disagreement with Heinemann et al. (1997), Sinogeikin et al. (1997), and Gwanmesia et al. (2000), who reported that the Mj75 Py25 garnet has a cubic structure, may be caused by the lower resolution of XRD in their studies. We do not observe any XRD peak from the orthorhombic symmetry for majorite-rich garnets (Rauch et al. 1996). Thus, garnets with the majorite content of more than 74 mol % should have a tetragonal symmetry, which is consistent with an earlier study by Parise et al. (1996).

(a) X-ray diffraction patterns of the (400) and (800) for Mj x Py1−x garnets (x majorite mol %). Two peaks of (800) and (008) doublets for the tetragonal garnets are shown with the dashed curves fitted by the pseudo-voight method. b Variations of unit-cell volumes and lattice parameters as a function of the majorite content. The solid circles indicate the data in the present study, and open circles, squares, triangles and diamonds are from earlier studies (Irifune et al. 1996; Angel et al. 1989; Parise et al. 1996; Heinemann et al. 1997). The solid line is a least square fitting of unit-cell volumes in the present study
The unit-cell lattice parameters and volumes of garnets, which are compatible with those of earlier studies (Angel et al. 1989; Irifune et al. 1996; Parise et al. 1996; Heinemann et al. 1997), are plotted in Fig. 4b. The unit-cell volumes of garnets in the present study show a linear dependence on the garnet compositions, indicating that the phase transition from the cubic to tetragonal symmetry in this system is the second-order improper ferroelastic phase transition, which may affect the elastic properties (Sinogeikin et al. 1997; Liu et al. 2015). The linear composition dependence of lattice parameters for cubic garnets can be explained by an increment of cation radii from 0.53 Å of Al to 0.56 Å of (Mg + Si)/2 through the coupled substitution (Mg2+ + Si4+ = 2Al3+) on octahedral sites in the garnet structure (Heinemann et al. 1997). Lattice parameters of tetragonal garnets are calculated on the basis of majorite structure data (Angel et al. 1989), and the values of the a-axis increase with increasing majorite contents, whereas the values of the c-axis decrease (Fig. 4b).
Microstrain
Microstrain, especially in solid solution materials, has been shown to exert considerable influences on the physical and chemical properties of materials (Boffa Ballaran et al. 1999; Boffa Ballaran and Carpenter 2003; Bosenick et al. 2001; Dapiaggi et al. 2005; Freeman et al. 2006; Du et al. 2016). It is the principal reason for the broadening of XRD peaks in garnet solid solutions with a grain size larger than 500 nm (Dapiaggi et al. 2005; Du et al. 2016). Microstrain can be evaluated from peak widths of XRD profiles using the Williamson–Hall plot (Williamson and Hall 1953). The XRD peak widths are simplified as the full width at half maximum (FWHM), which is determined using a pseudo-Voigt fit of XRD profiles of garnet solid solutions (Dapiaggi et al. 2005; Du et al. 2016). Although the phase transition occurs in this garnet system, we can attain the accurate peak widths of tetragonal garnets (Mj74Py26 majorite) by fitting the XRD peaks as the tetragonal symmetry from 10º to 90º for Bragg angle (2θ) based on our high-resolution XRD patterns, and those of cubic garnets are obtained by fitting their XRD peaks as the cubic symmetry in the same Bragg angle region.
The contribution of microstrain and grain size to XRD peak’ broadening can be derived separately from the Williamson–Hall plot (Williamson and Hall 1953):
where B represents the physical peak broadening, L is the average crystallite size, θ is the Bragg angle, K is a Scherrer constant (0.9; Langford and Wilson 1978), λ is the X-ray wavelength, and ɛ is microstrain. Equation (2) is a typical y = a + bx, \(\frac{K \times \lambda }{L}\) of a being the y intercept (peak broadening due to crystalline size) and 4 × ɛ of b being the plot slope (the peak broadening due to microstrain)The XRD peak broadening of garnet (B garnet) can be determined by:
where W obs represents the observed peak broadening and W ins represents the peak broadening caused by instrument contributions (Dapiaggi et al. 2005; Du et al. 2016). The diffraction pattern of pyrope with an almost perfect crystalline texture can be regarded as a reference for the instrumental contribution on the broadening of X-ray diffraction peaks (Du et al. 2016). We selected the average value for peak widths between 0° and 90° for pyrope as the reference for peak widths in this solid system.
The values of peak widths of garnets are fitted by Eq. (2). Theoretically, the ordinate intercepts \(\frac{K \times \lambda }{L}\) of B garnet × cosθ versus sinθ plot for a number of diffraction indices should provide the value of the average crystallite size of L. However, this interception usually presents a rather large error and does not work well for the grain size larger than 500 nm (Dapiaggi et al. 2005; Du et al. 2016). Considering that the actual grain size of garnets is about 2–5 μm in the present study, microstrain is the principle reason for the observed XRD peak broadening in garnet solid solutions, which was also proposed in the pyrope–grossular system (Dapiaggi et al. 2005; Du et al. 2016). The microstrains for some selected garnets (pyrope and Mj74Py26 garnet) derived from the sintered chunky sample are similar to those attained by the powder sample for the same garnet (Supplementary Material Fig. S1), suggesting that microstrain is mainly resulted by the structural heterogeneity due to the cation substitution in these garnet structures.
Microstrain shows a nonlinear and asymmetrical dependence on compositions for garnets (Fig. 5). It can be seen that microstrain decreases with increasing majorite contents from pyrope to Mj50Py50, and then increases to Mj74Py26 and gradually decreases toward majorite. Especially, the intermediate garnets (Mj39Py61 and Mj59Py41) have almost the same value of the microstrain with that of pyrope. The microstrains of Mj74Py26, Mj80Py20, and Mj20Py80 garnets are slightly larger than those of end-members. The variations of microstrain for garnet solid solutions are probably related to the non-uniform lattice distortions resulted by systematic shifts of atoms from their ideal positions (site-disorder) (Bosenick et al. 2001; Dapiaggi et al. 2005; Giesting and Hofmeister 2002).

Microstrain changes with garnet compositions in the majorite–pyrope system. The solid line is a spline fitting of the present values. Dashed lines represent the combined results of pyrope Mj50Py50 and Mj50Py50 majorite, respectively. The vertical dashed lines with arrows indicate the excess microstrain (Δɛ) between the solid and dashed lines
Raman spectroscopy: symmetry and order–disorder of cations
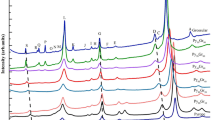
Figure 6a shows the Raman spectra of garnet solid solutions at room conditions. Raman modes of pyrope are well assigned to the vibrational modes allowed by the crystal symmetry (McMillan et al. 1989; Kolesov and Geiger 1998; Giesting and Hofmeister 2002; Giesting et al. 2004; Hofmeister et al. 2004). The Raman mode at 210 cm−1 can be assigned to Mg-translational T(Mg), and 362 and 382 cm−1 modes are assigned to SiO4-rotational R(SiO4). The Raman modes at 562–648 cm−1 and those above 800 cm−1 (e.g., 928 cm−1) can be attributed to the internal stretching and bending of Si–O in the SiO4 tetrahedra (R(SiO4)str.), respectively (McMillan et al. 1989; Kolesov and Geiger 1998). As shown in Fig. 6b, all the Raman modes are nearly independent of the majorite content, suggesting that Mg2+ + Si4+ = 2Al3+ substitution has little effects on the vibrational entropy of garnets in the majorite–pyrope system as suggested by McMillan et al. (1989). Remarkably, with increasing majorite contents from pyrope to majorite, new Raman modes at 595, 795, 890, and 972 cm−1 are first observed in Mj74Py26. Among these modes, 595, 795, and 890 cm−1 show a slight blue-shifting with increasing majorite contents toward the tetragonal majorite, while 972 cm−1 shows a small red-shifting. Thus, Raman modes at around 595 cm−1 for Mj74Py26 majorite should be reasonably assigned to Sioctahedral–O–Sitetrahedral linkage vibrations in the tetragonal structure for the majorite-rich garnets, and those at around 795, 890, and 972 cm−1 can be attributed to R(SiO4)str. for the tetragonal majoritic garnets (McMillan et al. 1989; Kolesov and Geiger. 1998). The newly appearing Raman modes in the majorite-rich garnets may be related to the local structure distortions of cubic garnets due to the cation substitutions. Hence, all the new Raman modes, present at Mj74Py26–majorite but absent in the cubic garnets, further suggest that a phase transition from cubic to tetragonal structure occurs in the majorite–pyrope solid solutions.

a Raman spectra of garnets along the majorite–pyrope binary. The marked stars represent the new appearing Raman modes. b Wavenumber shifts of Raman modes in a function of the majorite content
Another interesting feature of Raman spectra is the width of Raman modes. The peak width of the vibrational Raman spectrum is closely related to the degree of order, and broadening of Raman modes occurs when order defects appear in the materials due to cation substitutions, vacancies, etc. (Michel et al. 1976). For the solid solutions, point defects resulted by cation substitutions are the cause of peak broadening in Raman spectra. As shown in Fig. 6a, Raman modes of synthetic end-members of pyrope and majorite are very sharp, indicating that the cations in pyrope and majorite are well ordered, which is consistent with previous Raman and nuclear magnetic resonance (NMR) studies (McMillan et al. 1989; Phillips et al. 1992). Figure 7 shows peak widths (FWHM) of the strongest R(SiO4)str. mode in the intensity as a function of majorite content. These peak widths show a nonlinear dependence of garnet compositions, and reach a maximum near the intermediate garnets. The widths of Raman modes of intermediate garnets, such as Mj39Py61, Mj59Py41, and Mj74Py26, are significantly higher than those of the other garnets, suggesting the disorder of cations occurs in these intermediate garnets.

Raman mode widths (FWHM) for garnets along the majorite–pyrope binary. Solid circles represent FWHM values in the present study, which are got through the Lorentz fitting the strongest R(SiO4)str. mode. The dashed line represents previously calculated entropy (S) at room condition for garnets in the same system (Giesting et al. 2004)
Since the crystal chemical substitution in the majorite–pyrope system is formally through Mg2+ + Si4+ = 2A13+ on octahedral sites in the garnet structure, Raman mode broadening could be associated with Mg, Si, and A1 disorder over octahedral sites in intermediate garnets (McMillan et al. 1989; Phillips et al. 1992). The ionic radius of aluminum (0.54 Å) is smaller than that of magnesium (0.69 Å) but larger than silicon (0.43 Å). It is conceivable that cations through the charge balance substitution of Mg and Si would try to satisfy its bonding requirements to fit into the site that was occupied by Al by becoming statically or dynamically disordered. Theoretically, the degree of disorder of cations in octahedral sites will increase with increasing majorite contents or cation substitutions. Thus, Mj50Py50 garnet should have a maximum of the degree for cation disorders in octahedral sites or total cation disorder in theory, and other garnets such as Mj90Py10 and Mj59Py41 close to the end-members and intermediate Mj50Py50 garnet may have a partial disorder of cations in octahedral sites. This phenomenon is well interpreted by the variations of Raman mode widths for garnets as shown in Figs. 6 and 7. End-members of majorite and pyrope have well-ordered cations due to their sharp Raman modes, while intermediate Mj39Py61 and Mj59Py41 garnets have a maximum of cation disorder due to the most broadened Raman modes. Garnets such as Mj90Py10 and Mj74Py26, close to both end-members and intermediate garnets, are partially disordered for cations in octahedral sites because their Raman mode widths are sharper than those of the intermediate garnets and wider than those of the end-member garnets. In Fig. 7, the calculated entropy of garnets by Giesting et al. (2004) shows a nonlinear dependence of garnet compositions due to the variation of peak widths of Raman modes of these garnets.
End-members (pyrope and majorite) with well-ordered cations and Mj50Py50 garnet with totally disordered cations have a minimum value of the lattice microstrains compared with those of other garnet solid solutions. This result is consistent with the variation of the microstrain for pyrope–grossular garnets (Du et al. 2016). The reason for intermediate garnets with a minimum microstrain is unclear, which probably resulted from the cation total disorder in these garnet structure. This requires further study to clarify this question. With increasing majorite contents from pyrope to Mj50Py50 garnet, intermediate Mj20Py80 garnet becomes partial cation disorder in the octahedral sites from well-ordered cations for pyrope, and result in a slightly larger microstrain than that of pyrope, thus possessing a large excess microstrain. The Mj50Py50 garnet finally becomes totally disordered regarding its cations in octahedral sites, and has a similar microstrain with that of pyrope. On the other hand, with increasing pyrope contents from another end-member majorite to the Mj50Py50 garnet, Mj90Py10, Mj80Py20, and Mj74Py26 garnets would become partially disordered and result in excess microstrains. Therefore, we cannot clearly see the correlation between microstrain and cation disorder in the majorite–pyrope garnets.
Conclusions
In summary, we used a high-pressure technique to fabricate high-quality garnets in the majorite–pyrope system and studied the phase transition, microstrain, and order–disorder of cations. We clearly find that the phase transition from cubic to tetragonal structure occurs in this solid system. Newly appearing peaks for garnet solid solutions between Mj74Py26 and majorite in both XRD and Raman spectra patterns indicate that garnets with the majorite content of more than 74 mol % are tetragonal.
The lattice microstrain is found to be the principal reason for the broadening of XRD peaks in these garnet solid solutions, which is consistent with previous studies (Dapiaggi et al. 2005; Du et al. 2016). It is worth noting that the microstrain of majorite–pyrope garnets shows a nonlinear and asymmetrical dependence of the chemical composition, which may be caused by the structural heterogeneity due to the cation substitution in the garnet structure. The intermediate garnets have a similar microstrain with that of the end-members, while those between end-members and intermediate compositions possess a larger microstrain. This result is consistent with that in pyrope–grossular garnets (Du et al. 2016).
The width variation of Raman peaks of garnets in this system also shows a nonlinear dependence of garnet chemical compositions, and is related to the cation disorder due to the cation substitution. The widths of Raman modes of intermediate garnets are considerably broader than those of majorite and pyrope, suggesting cation disorder occurs near the middle garnets of this system, which agrees well with previous Raman scattering and NMR studies (McMillan et al. 1989; Phillips et al. 1992).
References
Akaogi M, Akimoto S (1977) Pyroxene-garnet solid solution equilibria in the systems Mg4Si4O12–Mg3Al2Si3O12 and at high pressures and temperatures. Phys Earth Planet Inter 15:90–106
Angel RJ, Finger LW, Hazen RM, Kanzaki M, Weidner DJ, Liebermann RC, Veblen DR (1989) Structure and twinning of single-crystal MgSiO3 garnet synthesized at 17 GPa and 1800 °C. Am Mineral 74:509–512
Boffa Ballaran T, Carpenter MA (2003) Line broadening and enthalpy: some empirical calibrations of solid solution behaviour from IR spectra. Phase Transit 76:137–154
Boffa Ballaran T, Carpenter MA, Geiger CA, Koziol AM (1999) Local structural heterogeneity in garnet solid solutions. Phys Chem Miner 26:554–569
Bosenick A, Dove MT, Heine V, Geiger CA (2001) Scaling of thermodynamic mixing properties of garnet solid solutions. Phys Chem Miner 27:445–452
Dapiaggi M, Geiger CA, Artioli G (2005) Microscopic strain in synthetic pyrope–grossular solid solutions determined by synchrotron X-ray powder diffraction at 5 K: the relationship to enthalpy of mixing behavior. Am Mineral 90:506–509
Du W, Clark SM, Walker D (2016) Excess mixing volume, microstrain, and stability of pyrope–grossular garnets. Am Mineral 101:193–204
Freeman CL, Allan NL, van Westrenen W (2006) Local cation environments in the pyrope–grossular Mg3Al2Si3O12–Ca3Al2Si3O12 garnet solid solution. Phys Rev B 74:13420301–13420309
Giesting PA, Hofmeister AM (2002) Thermal conductivity of disordered garnets from infrared spectroscopy. Phys Rev B 65:14430501–14430516
Giesting PA, Hofmeister AM, Wopenka B, Gwanmesia GD, Jolli BL (2004) Thermal conductivity and thermodynamics of majoritic garnets: implications for the transition zone. Earth Planet Sci Lett 218:45–46
Gwanmesia GD, Liu J, Chen G, Kesson S, Rigden SM, Liebermann RC (2000) Elasticity of pyrope (Mg3Al2Si3O12)–majorite (Mg4Si4O12) garnet solid solution. Phys Chem Miner 27:445–452
Heinemann S, Sharp TG, Seifert F, Rubie DC (1997) The cubic-tetragonal phase transition in the system majorite (Mg4Si4O12)–pyrope (Mg3Al2Si3O12) and garnet symmetry in the earth’s transition zone. Phys Chem Miner 24:206–221
Hofmeister AM, Giesting PA, Wopenka B, Gwanmesia GD, Jolliff BL (2004) Vibrational spectroscopy of pyrope–majorite garnets: structure implications. Am Mineral 89:132–146
Hunt SA, Dobson DP, Li L, Weidner DJ, Brodholt JP (2010) Relative strength of the pyrope–majorite solid solution and the flow-law of majorite containing garnets. Phys Earth Planet Inter 179:87–95
Irifune T, Ringwood AE (1987) Phase transformations in primitive MORB and pyrolite compositions to 25 GPa and some geophysical implications. In: Manghnani MH, Syono Y (eds) High pressure research in mineral phys-ics. Terra Scientific, Tokyo
Irifune T, Koizumi T, Ando J (1996) An experimental study of the garnet–perovskite transformation in the system MgSiO3–Mg3Al2Si3O12. Phys Earth Planet Inter 96:147–157
Kolesov BA, Geiger CA (1998) Raman spectra of silicate garnets. Phys Chem Miner 25:142–151
Langford JI, Wilson AJC (1978) Scherrer after sixty years: a survey and some new results in the determination of crystallite size. J Appl Crystallogr 11:102–113
Li L, Weidner DJ, Brodholt J, Price GD (2007) The effect of cation-ordering on elastic properties of majorite: an ab initio study. Earth Planet Sci Lett 256:28–35
Liu ZD, Irifune T, Greax S, Arimoto T, Shinmei T, Higo Y (2015) Elastic wave velocity of polycrystalline Mj80Py20 garnet to 21 GPa and 2000 K. Phys Chem Miner 42:213–222
Liu ZD, Irifune T, Nishi M, Tange Y, Arimoto T, Shinmei T (2016) Phase relations in the system MgSiO3–Al2O3 up to 52 GPa and 2000 K. Phys Earth Planet Inter 257:18–27
McMillan P, Akaogi M, Ohtani E, Williams Q, Nieman R, Sato R (1989) Cation disorder in garnets along the Mg4Si4O12–Mg3Al2Si3O12 join: an infrared, Raman, and NMR study. Phys Chem Miner 16:428–435
Michel D, Collongues R, Jorba Y, Collongues R (1976) Study by Raman spectroscopy of order–disorder phenomena occurring in some binary oxides with fluorite-related structures. J Raman Spectrosc 5:163–180
Nakatsuka A, Yoshiasa A, Yamanaka T, Ohtaka O, Katsura T, Ito E (1999) Symmetry change of majorite solid-solution in the system Mg3Al2Si3O12–MgSiO3. Am Mineral 94:1135–1143
Novak GA, Gibbs GV (1971) The crystal chemistry of the silicate garnets. Am Mineral 56:791–825
Parise JB, Wang Y, Gwanmesia GD, Zhang J, Sinelnikov Y, Chmielowski J, Weidner DJ, Liebermann RC (1996) The symmetry of garnets on the pyrope (Mg3Al2Si3O12)–majorite (MgSiO3) join. Geophys Res Lett 23:3799–3802
Phillips BL, Howell DA, Kirkpatrick RJ, Gasparik T (1992) Investigation of cation order in MgSiO3-rich garnet using 29Si and 27A1 MAS NMR spectroscopy. Am Mineral 77:704–712
Rauch M, Keppler H, Häfner W, Poe B, Wokaun AA (1996) pressure-induced transition in MgSiO3-rich garnet revealed by Raman spectroscopy. Am Mineral 81:1289–1292
Ringwood AE, Major A (1971) Synthesis of majorite and other high pressure garnets and perovskites. Earth Planet Sci Lett 12:411–418
Shannon RD (1976) Revised effective ionic radii. Acta Crystallogr A32:751–767
Sinogeikin SV, Bass JD, B OʼNeill, Gasparik T (1997) Elasticity of tetragonal end-member majorite and solid solution in the system Mg4Si4O12–Mg3Al2Si3O12. Phys Chem Miner 24:115–121
Wang Y, Gasparik T, Liebermann RC (1993) Modulated microstructure in synthetic majorite. Am Mineral 78:1165–1173
Williamson GK, Hall WH (1953) X-ray broadening from filled aluminium and wolfram. Acta Metall 1:22–31
Acknowledgments
The authors thank J. B. Parise and Y. M. Zhou for their valuable suggestions for X-ray diffraction analysis. The authors thank T. Boffa Ballaran for her help in X-ray diffraction measurements. We thank both editor and two reviewers for their constructive comments. The present study is supported by the Grant-in-Aid for Scientific Research (S) by JSPS to T. Irifune (Grant No. 25220712).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, Z., Du, W., Shinmei, T. et al. Garnets in the majorite–pyrope system: symmetry, lattice microstrain, and order–disorder of cations. Phys Chem Minerals 44, 237–245 (2017). https://doi.org/10.1007/s00269-016-0852-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-016-0852-3