
Abstract
Chordoma is a rare and aggressive bone tumor. An accurate investigation of tumor heterogeneity is necessary for the development of effective therapeutic strategies. This study aims to assess the poorly understood tumor heterogeneity of chordomas and identify potential therapeutic targets. Single-cell RNA sequencing was performed to delineate the transcriptomic landscape of chordomas. Six tumor samples of chordomas were obtained, and 33,737 cells passed the quality control test and were analyzed. The main cellular populations identified with specific markers were as follows: chordoma cells (16,052 [47.6%]), fibroblasts (6945 [20.6%]), mononuclear phagocytes (4734 [14.0%]), and T/natural killer (NK) cells (3944 [11.7%]). Downstream analysis of each cell type was performed. Six subclusters of chordomas exhibited properties of an epithelial-like extracellular matrix, stem cells, and immunosuppressive activity. Although few immune checkpoints were detected on cytotoxic immune cells such as T and NK cells, a strong immunosuppressive effect was exerted on the Tregs and M2 macrophages. In addition, the cellular interactions were indicative of enhancement of the TGF-β signaling pathway being the main mechanism for tumor progression, invasion, and immunosuppression. These findings, especially from the analysis of molecular targeted therapy and tumor immune microenvironment, may help in the identification of therapeutic targets in chordomas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chordoma is a rare, aggressive, and locally invasive bone tumor. It originates from notochordal remnants [1]. Chordomas are treated by bloc excision, which is the standard treatment option. In patients with recurrence or patients who undergo subtotal or piecemeal excision, radiotherapy is recommended [2]. Many patients with chordoma undergo morbid surgeries and eventually die from tumor recurrence; the 5- and 10-year survival rates are 67.6% and 39.9%, respectively [3, 4]. Thus, the treatment strategies are limited [5]. Novel and effective treatment methods are needed.
Tumor heterogeneity is depicted by different cellular populations in the complex tumor microenvironment (TME) of chordomas. These include cancer cells, vascular cells, fibroblasts, and immune cells. Intertumoral and intratumoral heterogeneity refers to heterogeneity between patients with the same histological type, and within a single patient, respectively. Tumor heterogeneity is a key challenge in tumor treatment resistance [6]. Clinical studies on molecular targeted therapy (MTT) in chordomas have identified potential therapeutic targets: PDGFR, EGFR, HER2, VEGFR, etc. [7]. However, the therapeutic effect of these MTTs is limited, and the response rate of apatinib and sorafenib is only approximately 3.7%. Moreover, sorafenib has significant toxicity; 77.8% and 4.8% of patients develop Grades 3 and 4 toxicities, respectively [8]. Immune checkpoint inhibitors are an effective treatment option for cancers such as sarcomas, but their efficacy is limited to mismatch repair-deficient tumors [9]. However, programmed cell death-1 or programmed cell death-ligand 1 (PD-L1) inhibitors have clinical benefits in some patients. Tumor heterogeneity is the main reason for the diverse responses to these drugs [10]. The complex TME explains the high heterogeneity. TMEs comprise cellular populations, such as cancer cells, vascular cells, fibroblasts, and immune cells [11]. The TME of chordomas consists of immune cell populations, including immune checkpoints [12] and vascular endothelial cells [13]. However, it is difficult for bulk gene analyses to provide an accurate reflection of the TME [14]. Therefore, the heterogeneity of chordomas is not well studied, and novel assessment methods are needed.
Single-cell RNA sequencing (scRNA-seq) may elucidate the mechanisms underlying carcinogenesis and the molecular features of cancers. It may provide a clearer description of the TME at the cellular level, allowing more effective therapeutic strategies for tumors [15]. scRNA-seq is used to delineate the complex tumor heterogeneity in several malignancies such as glioblastomas [16], liver cancers [17], lung cancers [18], renal cell carcinoma [19], and head and neck cancers [20]. However, the knowledge on tumor heterogeneity and cellular interactions in chordomas is limited. We used an scRNA-seq sequencing platform (10 × Genomics) to describe the tumor landscape of chordomas. This study aimed to assess tumor heterogeneity and identify potential therapeutic targets in chordomas.
Materials and methods
Chordoma samples
Six patients who underwent surgery at Xuanwu Hospital, Capital Medical University, Beijing, China, from July 2019 to April 2020 were included in this study. None of the patients received preoperative radiation therapy, chemotherapy, or other targeted therapy. Using the 2013 World Health Organization classification of bone tumors, all the patients were diagnosed with classic chordomas [21] (Fig. S1A). On immunohistochemistry [22], all chordoma cells had strongly positive TBXT (brachyury) expression (Fig. S1B). The samples were obtained from fresh tumors intraoperatively. Written informed consent was obtained from all the patients. The study was approved by the Ethics Committee of the Xuanwu hospital of Capital Medical University (No.2016033).
sc-RNA sequencing
Single-cell transcriptomic sequencing was performed (Capitalbio Technology Corporation, http://www.capitalbiotech.com). Following the manufacturer’s instructions for Single Cell 30 Library and Gel Bead Kit V2 (10 × Genomics), cell suspensions were loaded on a Chromium Single Cell Controller (10 × Genomics, San Francisco, CA) to generate single-cell gel beads in emulsion. After Drop-seq droplet collection, cDNA amplification and sequencing library preparations were carried out as described previously [17], and the libraries were sequenced on Illumina HiSeq X Ten. The libraries from one batch of droplets were sequenced separately for Drop-seq data from chordoma cells.
Cell filtration, clustering, and downstream analysis
Downstream analysis was done using the Seurat package (version 3.2.0) [23] in R software (version 4.0.2), which helped in preprocessing to clustering, dimension reduction, visualization, and differential gene expression. In processing, raw gene expression matrices were imported and filtered using the following standards: (1) cells expressing at least 200 genes and genes expressed by at least 3 cells and (2) cells with a mitochondrial percentage below 25% (Fig. S2A, B). A total of 33,737 high-quality cells were included in the downstream analysis. Furthermore, the Seurat package was used to normalize and integrate expression data and remove the batch effect. The new integrated matrix was used to scale, run the principal component analysis, and visualize the landscape with UMAP.
Cell type annotation
We adopted 19 clusters and identified all cluster markers using the Seurat package. Cell types were annotated to known biological cell types using canonical marker genes. The SingleR package [24] (version 1.3.6), CellMarker website [25], and inferCNV package (version 1.5.0) were used. The TBXT gene, a specific feature of chordomas, was also used to annotate the tumor cells.
Functional enrichment analysis
After the annotation of each cell type, a functional enrichment analysis was performed to identify differentially expressing genes between different clusters for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). This analysis illustrated the biological processes and potential functions of different cells using the clusterProfiler package (version 3.17.0) [26] and the org.Hs.eg.db package (version 3.11.4). The p-value cutoffs of GO and KEGG were both 0.05. The top 10 terms of the results were visualized in a bar or dot plot.
Pseudotime
Trajectory analysis was performed using the monocle package (version 2.17.0) [27]. Tumor cells, mononuclear phagocytes, and fibroblasts were included. The following parameters of each group were analyzed: lowerDetectionLimit = 0.5, min_expr = 0.1, and num_cells_expressed ≥ 10. For visualization, the plot_cell_trajectory function was used to plot the potential trajectory according to pseudotime, Seurat clusters, and the data.
Cellular communications
The CellChat package (version 0.0.1) was used to analyze cell-to-cell interactions [28]. Most ligand–receptor interactions were mainly identified using the KEGG signaling pathway database and recent peer-reviewed experimental studies. The main steps of inference of intercellular communications included: (1) identification of differentially expressed signaling genes, (2) calculation of an ensemble average, and (3) calculation of intercellular communication probability.
Results
Overall landscape of the chordoma tumor samples
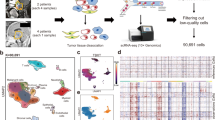
The raw sequencing data could be obtained at the Genome Sequence Archive for Human (GSA-Human), and the access ID is HRA000513 (https://ngdc.cncb.ac.cn/gsa-human/s/GokpMdli). Six tumor samples (from four male and two female patients) were included in the scRNA-seq profiles (Fig. 1A). The patients’ age range was 17–49 years. The tumor sites included the following: the sacrum (n = 1), mobile spine (n = 3), and skull base (n = 2). Details of the characteristics of six chordomas are shown in Supplemental Table 1. After strict filtering, 33,737 cells were included in the final analysis. After normalization of gene expression and principal component analysis, these cells were divided into 18 clusters using the UMAP method (Fig. 1B). The 18 clusters were equally distributed among the six samples, indicating a smaller batch-corrected effect (Fig. S3A). The clusters were further assigned to seven known cell lineages, which were chordoma cells (6392 [47.6%]), mononuclear phagocytes (4734 [14.0%]), T/natural killer (NK) cells (3944 [11.7%]), fibroblasts (6945 [20.6%]), endothelial cells (404 [1.2%]), B cells (780 [2.3%]), and neutrophils (878 [2.6%]) (Fig. 1B–D). The marker genes for the seven cellular populations are shown in Fig. 1B, and the characteristic genes are shown in Supplemental Table 2. Downstream analyses of the B cells, neutrophils, and endothelial cells were not performed due to their small numbers. The variation in the cellular populations was investigated between patients (Fig. 1E) and within patients (Fig. S3) and indicated a potential clinical significance, especially in tumor location. Primary tumor location was a strong prognostic factor and was related to the extent of surgical resection [29].

Overall landscape of chordoma tumor sample, A Schematic diagram of the scRNA-seq analysis workflow, B UMAP clustering for 33,737 cells and cell type identification, C Marker genes for seven distinct cell types, D Heatmap of the top five DEGs of each cell type, E Proportion of identified cell type in samples, scRNA, single-cell RNA sequencing; DEGs, differentially expressed genes
Transcriptomic tumor heterogeneity of tumor cells in chordoma
A total of 16,052 tumor cells were identified according to the chordoma-specific marker, TBXT. These chordomas cells were further assigned to six clusters with differently expressed genes (DEGs) (Fig. 2A and B). There was a high degree of within-patient (Fig. 2A) and between-patient (Fig. S4A) tumor heterogeneity; this means that we found six different clusters with different phenotypes in the overall study population. Meanwhile, each patient had a tumor with specific cellular characteristics. These six tumor subclusters varied with tumor location (Fig. S4B). The GO analysis investigated the biological function of DEGs, which mainly influence immune responses, such as leukocyte migration, regulation, chemotaxis, regulation of B-cell activation, and humoral immune response (Fig. 2C). These results suggest that the tumor heterogeneity explained the varied immunologic tumor microenvironment. Next, the expression of major histocompatibility complex class I genes and PD-L1/PD-L2 expression on chordoma cells (Fig. S4C) were analyzed. However, most tumor cells did not express major histocompatibility complex genes or PD-L1—these findings discourage the use of programmed cell death-1 inhibitors for chordomas.

Transcriptomic tumor heterogeneity of tumor cells in chordoma, A Subclusters of tumor cells, B Heatmap of the top five DEGs, C GO terms for DEGs, D Pseudotime analysis of tumor cells, E MTT target expression in each cluster, DEGs, differentially expressed genes; GO, Gene Ontology; MTT, molecular targeted therapy
There were 6950 chordoma cells in subcluster 0; these cells had the largest tumor population (43.3%). They were mainly involved in the extracellular matrix (ECM) organization by GO analysis (Fig. S4D), which was characterized by CTGF, COL2A1, COL6A3, and COL5A2 (Table S3). They were considered matrix tumor cells (mTCs, mTCs–C0–CTGF). Cluster 1 (4408 cells [27.5%]) displayed a high expression of genes related to metabolism of metal ions: MT1X, MT2A, and MT1G. The enrichment analysis revealed metal ion-related pathways (Fig. S4D). Cluster 1 cells were considered epithelial tumor cells (eTCs-C1-IGFBP3). Cluster 4 was involved in nuclear division (Fig. S4D), i.e., active tumor cell proliferation. The biomarkers for cancer stem cells (CSCs) were expressed in this cluster. Biomarkers such as STMN1[30, 31], UBE2C[32], and PTTG1 [33] [34] are associated with tumor cell survival, clonality, and tumorigenicity in several malignancies. This subcluster was considered to consist of stem tumor cells (sTCs, mTCs–C4–STMN1). The pseudotime analysis showed that subcluster 4 was in the early tumor stage. Clusters 2, 3, and 5 did not express CDKN2A, which is absent in several chordomas [35] (Fig. S4E). Next, we conducted a pseudotime analysis of six clusters to investigate their developmental trajectories: clusters 2, 3, and 5 were in a more advanced stage (Fig. 2D, Fig. S4F). A close relationship between CDKN2A loss and tumor immune microenvironment changes was seen in a prior study [36]. Subclusters 2 and 5, which had CDKN2A loss, were involved in immune functions. As mentioned above, chordomas showed great tumor heterogeneity, which indicates that the pathogenesis and phenotype might be varied. Regarding the clinical treatment, different patients with a risk of recurrence responded differently to the same targeted drug for chordomas [37]. Therefore, MTT plays a crucial role in the personalized treatment of chordoma. The MTT target expression in chordomas was analyzed. This included EGFR, HER2, PDGFR, VEGF, VEGFR2, and stem cell factor receptor (KIT) (Fig. 2E). VEGF and VEGFR2 were the most upregulated, indicating their potential as therapeutic targets.
T/NK cell clustering and subtype analysis
Unsupervised clustering was performed for 3944 T/NK cells; they were divided into five subclusters with their unique signature genes (Fig. 3A, B). Cluster 2 was composed of NK cells (374 cells, 9.5%) marked with KRF1, and others were T cells (3570 cells [90.5%]) with CD3E expression (Fig. 3C). High tumor heterogeneity was associated with T/NK cells among patients (Figure S5a) and tumor location (Fig. S5b). C0, C1, C3, and C4 were CD4 + T (2,298 [64.4%]), CD8 + T (953 [26.7%]), CD4- and CD8- double-negative (160 [4.5%]), and Treg (159 [4.4%]) cells, respectively. The specific markers for the immune cells identified above were CD4, CD8, and foxp3, respectively (Fig. 3C). A cluster of double-negative T cells was found (Fig.S5C), which was involved in systemic inflammation and tumor damage [38]. Only 160 double-negative T cells were detected, and these were not included in the downstream analysis. The C1-CD8 + T and C2-NK + showed a high level of cytotoxic activity, which was characterized by GZMA, GZMK, GNLY, PRF1, and IFNγ (Fig. 3B, Table S4). Moreover, few immune checkpoints were expressed on these cytotoxic immune cells (Fig. 3D). There was overexpression of CTLA4, TIGIT, and TIM3 on Tregs (Fig. 3D), which are the main immunosuppressive mechanisms and therapeutic targets for chordomas.

Clustering and subtype analysis of T and NK cells clustering and subtype analysis, A Clustering for T cells and NK cells, B Heatmap of the top 10 DEGs of subclusters of T/NK cells, C Expression of known marker genes of T and NK cells. D Expression of the immune checkpoints, NK, natural killer; DEGs, differently expressed genes
Mononuclear phagocyte clustering and subtype analysis
To investigate the heterogeneity among macrophages, 3698 tumor-associated macrophages (TAMs) were clustered into six subgroups (Fig. 4A): C0 (1414 [29.9%] cells), C1 (1061 [22.4%] cells), C2 (807 [17.0%] cells), C3 (743 [15.7%] cells), C4 (367 [7.8%] cells), and C5 (342 [7.2%] cells). C0 and C1 were the most populated, and they were associated with M2 subtype polarization because they exhibited high levels of CD68 + CD163 + CD204 + expression (Fig. 4B). In addition, VEGFA was heavily expressed in these cells in cluster 1 (Table S3). M2-subtype TAMs secrete VEGFA, which promotes tumor growth [39]. In the GO analysis, this cluster was involved in the regulation of angiogenesis (Fig. 4C). Subcluster 1 was composed of inflammatory chemokines such as CCL3, CXCL2, and CXCL3 (Table S5), consistent with the GO analysis findings (Fig. 4C). The release of these proteins by TAMs may promote inflammatory response and tumor development. Clusters 0 and 1 represented an M2-like TAM cluster. Moreover, the genes S100A8, S100A9, and S100A12, which encode calcium-binding proteins, were expressed by the cells in cluster 2 (Table S3) [40]. Activated mononuclear cells can release these proteins, which promote inflammatory responses in vivo. Thus, these data indicate that C3 may have a pro-inflammatory and anti-tumor role in chordomas. C4 and C5 indicate nuclear division and neutrophil activation, respectively (Fig. 4C). CD47 and TIM3 were expressed on TAMs (Fig. 4D); however, other immune checkpoints were not (data not shown).

Clustering and subtype analysis of mononuclear phagocytes, A Subclustering of mononuclear phagocytes, B Biomarkers for expression of M2-TAMs expression, C GO analysis for subclusters, D Violin plots showing the expression of CD47 and HAVCR2, TAMs, tumor-associated macrophages; GO, Gene Ontology
Distinct fibroblast subpopulations in human chordomas
There were 6945 fibroblasts identified with the specific markers, and these cells were further divided into six subclusters (Fig. 5A). All six subclusters expressed high levels of canonical fibroblast markers such as ACTA2 (a-SMA), COL1a2, and PDGFRb, confirming their identity as fibroblasts (Fig. 5C). These subclusters exhibited distinct transcriptomic signatures (Fig. 5B, Table S6). There were 2600 cells in subcluster 0, which had the largest population of fibroblasts (37.4%). The subclusters were characterized by ECM signatures, including collagen molecules (COL1A1, COL3A1, COL4A1), POSTN, and LUM. Furthermore, the GO analysis for this subtype was related to ECM and collagen fibril organization (Fig. 5D). They were considered as matrix cellular-associated fibroblasts (mCAFs, mCAFs–C0–POSTN; Fig. 5B, C). Subcluster 1 consisted of 1185 fibroblasts and was the most populated (17.1%). The significant genes that were upregulated in this subtype were IL1RL1, CCL3, CCL4l2, CCL8, and others (Table S6). The GO analysis confirmed that this subtype was involved in cell chemotaxis and T-cell activation; they were considered inflammatory CAFs (iCAFs, iCAFs–C1–IL1RL1; Fig. 5B, C). The C2 subcluster consisted of 910 fibroblasts and was marked with CRABP2, which was associated with osteogenic differentiation. In the GO analysis, C2 was involved in ossification and cartilage development; therefore, they were considered bone development regulation CAFs (bCAFs, bCAFs–c2–IL1RL1; Fig. 5B, C). Subcluster 3 was made of 796 fibroblasts. The marked genes were CST1 and ANGPTL4. There was a relationship between the GO analysis of this C3 and regulation of vasculature development. The C3 was then regarded as vascular CAFs (vCAFs, vCAFs-c3-MCAM; Fig. 5B, C). However, C5 and C3 had similar functions because the marker genes of C5 were characterized by microvasculature signature genes (MCAM, MYH11, GJA4, and RGS5). Subcluster 5 fibroblasts expressed mainly epithelium-specific marker genes such as KRT19 and KRT8; they were considered epithelial-to-mesenchymal transition (EMT)-like CAFs (eCAFs, eCAFs–c5–KRT19; Fig. 5B, C).

Distinct fibroblast subpopulations in human chordomas, A UMAP plots of total fibroblasts color-coded for six clusters, B Heatmap expression showing the top five DEGs of each cluster, C UMAP plots color-coded for the expression of known fibroblast markers and specific marker genes for distinct fibroblast subclusters: C0, C1, and C2, D GO analysis of DEGs in distinct fibroblast clusters, DEGs, differentially expressed genes; GO, Gene Ontology
The TGFβ signaling pathway was enriched by the interplay among the CD4 + T cells, fibroblasts, and macrophages
We first analyzed the overall cellular interactions based on ligand–receptor pairs. The most active pathway in the TME of chordomas was the TGFβ signaling pathway (Fig. 6A, Fig. S6). TGFB1-TGFBR1/TGFBR2 and TGFB1-ACVR1/TGFBR1 contributed most to this communication network (Fig. 6D), which was consistent with the results of previous studies in which TGFB1 was a potent TGFβ ligand in cancers [41]. Moreover, TGFβ signaling played a key role in tumor progression by its different effects on multiple cell types, including malignant and non-cancerous cells within the TME [42]. This showed that TGFβ was mainly produced and released by fibroblasts and macrophages in chordomas (Fig. 6B). However, this TGFβ signaling pathway network was very active among macrophages, fibroblasts, tumor cells, and CD4 + T cells (Fig. 6C). Although few Tregs were detected, they expressed some immune checkpoints that exerted strong immunosuppressive activity (Fig. 3D). The TGFβ signaling pathway was one main reason for transforming CD4 + T cells into Tregs in the TME of chordomas. Cytotoxic immune cells, such as CD8 + T cells and NK cells, did not express immune checkpoints, resulting in no effect on TGFβ (Fig. 3D) [43, 44]. The chordoma cells exhibited properties of the ECM, epithelial cells, and stem cells (Fig. S4D). The enhanced TGFβ signaling was associated with malignant biological processes of ECM, tumor stem cell properties, and EMT [45,46,47]. Therefore, the malignant cells were mainly influenced by the TGFβ pathway in chordomas.

TGFβ signaling pathway was enriched in the interplay between CD4 + T cells, fibroblasts, and macrophages. A Dot plots showing the outgoing and incoming communication patterns of all the cell types, B Heatmap of the relative importance of each cell group based on the four computed network centrality measures of TGFβ signaling, C Circle plot showing the inferred TGFβ signaling networks among tumor cells, fibroblasts, and immune cells. D Relative contribution of each ligand–receptor pair to the overall communication network of the TGFβ signaling pathway
Discussion
In this study, we first delineated the transcriptomic landscape of chordomas by scRNA-seq. The cellular populations of the TME in chordomas included tumor cells, fibroblasts, and immune cells; this varied within and between tumors, indicating high tumor heterogeneity. Moreover, tumor heterogeneity varied with tumor location, suggesting novel mechanisms for different clinical outcomes based on the chordoma site. Molecular therapy targets and immunotherapy biomarkers were also analyzed, and potentially valuable therapeutic targets were shown. The cellular interactions enhanced the TGFβ signaling pathway: this was the main mechanism for tumor progression and invasion and immunosuppression in chordomas. This study is the first to investigate tumor heterogeneity at the single-cell level, and it provides new insights into mechanisms of tumor development and possible therapeutic strategies for chordomas.
Chordoma cells accounted for approximately half of the total cellular populations and were made up of six subclusters. These subclusters conferred malignant cell properties such as ECM, epithelial-like, stem cell, and immunosuppression. There were 4408 (27.5% of total tumor cells) chordoma cells in subcluster 1, which were related to epithelial cells. Previous studies revealed a dual epithelial–mesenchymal differentiation of chordomas [48], and these subclusters were likely to resist chemotherapy and radiotherapy. Biomarkers (CD133+, CD15+, WNT5, ABTG2, and MYCBP) of CSCs are found in chordomas [49], but these markers were absent in the tumors in the present study. Instead, the biomarkers of CSCs, such as STMN1, UB2C, and PTTG1 in hepatocellular carcinoma, breast cancer, and prostate tumors were detected [30,31,32,33,34]. The pathological mechanisms of these markers should be investigated in future studies. In addition, VEGF and VEGFR-2 were the most upregulated in chordomas. These results are consistent with the clinical evidence that chordomas respond objectively to sorafenib and apatinib [7].
Our results showed a complex immune TME of chordomas, consisting of macrophages, T cells, and NK cells. The CD8+ T cells and NK cells showed a high cytotoxic activity, characterized by GZMA, GZMK, GNLY, PRF1, and IFNγ. Two cytotoxic immune cells were devoid of immune checkpoints. However, a strong immunosuppressive activity was observed, which was mainly exerted by Tregs and macrophages. CTLA4, TIGIT, and TIM3 were highly expressed by Tregs in our study. Tregs have been detected in human chordoma samples and associated with poor clinical outcomes due to their strong immunosuppressive activity [50, 51]. TAMs mainly exhibited the M2-subtype, which is involved in angiogenesis and immunosuppression. As CD47 and TIM3 are highly expressed by TAMs, immunotherapy may suppress the pro-tumor development role of TAMs in chordomas.
Cellular interactions showed that the TGFβ signaling pathway played a key role in tumor development, tumor invasion, immunosuppression, and CSCs. According to previous reports, fibroblasts and macrophages are the main sources of TGFβ in chordomas [52,53,54]. Chordoma cells are strongly influenced by TGFβ in several malignant biological factors such as ECM, CSC properties, and epithelial-like characteristics [55, 56]. Furthermore, in the present study, the TGFβ signaling pathway also negatively affected the immune cells, which were CD4+ T cells. The CD4+ T cells were the largest immune cell population in chordomas. CD4+ T cells may be transformed into Tregs or Th0 cells under the influence of certain cytokines or chemokines [57]. CD4+ T cells were affected by the TGFβ signaling pathway and developed into Tregs in this study. Together, these results indicate that the TGFβ signaling pathway is a fundamental mechanism for tumor development and immunosuppression in chordomas.
This study had some limitations. First, the results and conclusions were based on sequencing data and were not validated by experiments and clinical trials. However, the primary study objective was to provide a landscape of chordomas, and our results showed this tumor’s heterogeneity. Second, the TGFβ signaling pathway was found to play a critical role in chordoma progression by promoting tumor invasion, immunosuppression, and CSCs. There are no existing reports on TGFβ signaling pathway in chordomas. Therefore, the results of the TGFβ signaling pathway in chordomas may not be conclusive and need further research.
Our findings provide a large transcriptomic landscape and details of the single-cell resolution of chordomas. This study is an established resource for elucidating chordoma diversity. However, details of the mechanisms and efficacy of therapeutic options still need to be further explored in experimental and clinical studies.
Code availability
All scripts used are available at https://github.com/restore1997/chordoma.
References
McMaster ML et al (2001) Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control 12(1):1–11
Walcott BP et al (2012) Chordoma: current concepts, management, and future directions. Lancet Oncol 13(2):e69-76
Tarpey PS et al (2017) The driver landscape of sporadic chordoma. Nat Commun 8(1):890
Thanindratarn P et al (2019) Advances in immune checkpoint inhibitors for bone sarcoma therapy. J Bone Oncol 15:100221
Stacchiotti S, Sommer J, Chordoma G (2015) Global consensus, Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol 16(2):71–83
Dagogo-Jack I, Shaw AT (2018) Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 15(2):81–94
Meng T et al (2019) Molecular targeted therapy in the treatment of chordoma: a systematic review. Front Oncol 9:30
Bompas E et al (2015) Sorafenib in patients with locally advanced and metastatic chordomas: a phase II trial of the French Sarcoma Group (GSF/GETO). Ann Oncol 26(10):2168–2173
Tawbi HA et al (2017) Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18(11):1493–1501
Guo M et al (2019) Epigenetic heterogeneity in cancer. Biomark Res 7:23
Potter SS (2018) Single-cell RNA sequencing for the study of development, physiology and disease. Nat Rev Nephrol 14(8):479–492
Zou MX et al (2019) Clinical impact of the immune microenvironment in spinal chordoma: immunoscore as an independent favorable prognostic factor. Neurosurgery 84(6):E318-e333
Morimoto Y et al (2019) Prognostic significance of VEGF receptors expression on the tumor cells in skull base chordoma. J Neuro-oncol 144(1):65–77
Wang Y et al (2020) Changing technologies of RNA sequencing and their applications in clinical oncology. Front Oncol 10:447
Zhang Y, Zhang Z (2020) The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol 17(8):807–821
Neftel C et al (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178(4):835-849.e21
Zhang M et al (2020) Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol 73(5):1118–1130
Maynard A et al (2020) Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 182(5):1232-1251.e22
Pan XW et al (2020) Identification of a novel cancer stem cell subpopulation that promotes progression of human fatal renal cell carcinoma by single-cell RNA-seq analysis. Int J Biol Sci 16(16):3149–3162
Puram SV et al (2017) Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171(7):1611-1624.e24
Jo VY, Fletcher CD (2014) WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology 46(2):95–104
Wang LM et al (2020) A novel isocitrate dehydrogenase 1 G131D mutation in glioblastoma. Chin Med J (Engl) 134(4):486–488
Stuart T et al (2019) Comprehensive integration of single-cell data. Cell 177(7):1888-1902.e21
Aran D et al (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20(2):163–172
Zhang X et al (2019) Cell Marker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 47(D1):D721–D728
Yu G et al (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5):284–287
Trapnell C et al (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32(4):381–386
Jin S, Guerrero-Juarez CF (2021) Inference and analysis of cell-cell communication using Cell Chat. Nat Commun 12(1):1088
Zheng W et al (2019) Application of nomograms to predict overall and cancer-specific survival in patients with chordoma. J Bone Oncol 18:100247
Zhang R et al (2020) STMN1 upregulation mediates hepatocellular carcinoma and hepatic stellate cell crosstalk to aggravate cancer by triggering the MET pathway. Cancer Sci 111(2):406–417
Obayashi S et al (2017) Stathmin1 expression is associated with aggressive phenotypes and cancer stem cell marker expression in breast cancer patients. Int J Oncol 51(3):781–790
Shuliang S et al (2013) Involvement of ubiquitin-conjugating enzyme E2C in proliferation and invasion of prostate carcinoma cells. Oncol Res 21(3):121–127
Parte S et al (2019) PTTG1: a unique regulator of stem/cancer stem cells in the ovary and ovarian cancer. Stem Cell Rev Rep 15(6):866–879
Huang S et al (2018) Interleukin-6/signal transducer and activator of transcription 3 promotes prostate cancer resistance to androgen deprivation therapy via regulating pituitary tumor transforming gene 1 expression. Cancer Sci 109(3):678–687
Cottone L et al (2020) Frequent alterations in p16/CDKN2A identified by immunohistochemistry and FISH in chordoma. J Pathol Clin Res 6(2):113–123
Arora A et al (2020) Pan-cancer identification of clinically relevant genomic subtypes using outcome-weighted integrative clustering. Genome Med 12(1):110
Hung YP et al (2020) Dedifferentiated chordoma: clinicopathologic and molecular characteristics with integrative analysis. Am J Surg Pathol 44(9):1213–1223
Brandt D, Hedrich CM (2018) TCRαβ(+)CD3(+)CD4(-)CD8(-) (double negative) T cells in autoimmunity. Autoimmun Rev 17(4):422–430
Hwang I et al (2020) Tumor-associated macrophage, angiogenesis and lymphangiogenesis markers predict prognosis of non-small cell lung cancer patients. J Transl Med 18(1):443
Heilmann RM et al (2019) Mucosal expression of S100A12 (calgranulin C) and S100A8/A9 (calprotectin) and correlation with serum and fecal concentrations in dogs with chronic inflammatory enteropathy. Vet Immunol Immunopathol 211:64–74
Nüchel J et al (2018) TGFB1 is secreted through an unconventional pathway dependent on the autophagic machinery and cytoskeletal regulators. Autophagy 14(3):465–486
Derynck R, Turley SJ, Akhurst RJ (2021) TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol 18(1):9–34
Takeuchi M et al (1997) On the mechanisms by which transforming growth factor-beta 2 alters antigen-presenting abilities of macrophages on T cell activation. Eur J Immunol 27(7):1648–1656
Kobie JJ et al (2003) Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res 63(8):1860–1864
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196
Miettinen PJ et al (1994) TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 127(6 Pt 2):2021–2036
Kornberg TB (2017) Distributing signaling proteins in space and time: the province of cytonemes. Curr Opin Genet Dev 45:22–27
Gulluoglu S et al (2016) The molecular aspects of chordoma. Neurosurg Rev 39(2):185–96
Tuysuz EC et al (2019) Distinctive role of dysregulated miRNAs in chordoma cancer stem-like cell maintenance. Exp Cell Res 380(1):9–19
Zou MX et al (2018) Clinicopathologic implications of CD8(+)/Foxp3(+) ratio and miR-574-3p/PD-L1 axis in spinal chordoma patients. Cancer Immunol Immunother 67(2):209–224
Zou MX et al (2019) The relationship between tumor-stroma ratio, the immune microenvironment, and survival in patients with spinal chordoma. Neurosurgery 85(6):E1095-e1110
Hildenbrand R et al (1998) Transforming growth factor-beta stimulates urokinase expression in tumor-associated macrophages of the breast. Lab Invest 78(1):59–71
Wei Y et al (2017) Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J Clin Invest 127(10):3675–3688
Correction: (2019) Tumor-secreted LOXL2 activates fibroblasts through FAK signaling. Mol Cancer Res, 17(10): 2141
Scheel C, Weinberg RA (2012) Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 22(5–6):396–403
Ramachandran A et al. (2018) TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. 7
Stadhouders R, Lubberts E, Hendriks RW (2018) A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun 87:1–15
Acknowledgements
We acknowledge the contributions of specific colleagues, institutions, or agencies that aided the authors' efforts.
Funding
CAMS/PUMC Research Project #201920200501, Human Brain Tissue Bank Platform for Neurological Diseases.
Author information
Authors and Affiliations
Contributions
ZC involved in conception and design; WD and BZ took part in collection and assembly of data; WD and XL involved in data analysis and interpretation; all authors involved in manuscript writing and approval of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
262_2022_3152_MOESM1_ESM.pdf
Fig. S1 Immumohistochemical staining, A Hematoxylin-eosin staining results of chordoma tissues, B Immunohistochemistry of Brachyury staining of chordoma tissues (PDF 891 kb)
262_2022_3152_MOESM2_ESM.pdf
Fig. S2 Quality control process A Violin plots showing cell infiltration using nFeature_RNA, nCount_RNA, and percent_mt, B Dot plots showing the feature-feature relationships (PDF 507 kb)
262_2022_3152_MOESM3_ESM.pdf
Fig. S3 Visualization of all the cells based on sample and site, A UMAP plots of all the cells colored by samples, B UMAP plots of all the cells colored by site, C Bar plots showing the proportion of the identified cell types in various locations (PDF 1186 kb)
262_2022_3152_MOESM4_ESM.pdf
Fig. S4 Supplementary transcriptomic heterogeneity of tumor cells, A UMAP plot of tumor cells colored by samples, B UMAP plot of tumor cells colored by site, C Violin plots showing the expression of the immune checkpoints, D Bar plots showing the functional enrichment analysis of GO terms for each cluster of tumor cells, E Violin plots showing the expression of CDKN2A, F The ordering of tumor cells along pseudotime in a two-dimensional state space (colored by pseudotime), GO, Gene Ontology (PDF 1660 kb)
262_2022_3152_MOESM5_ESM.pdf
Fig. S5 Clustering and subtype analysis of supplementary T and NK cells, A UMAP plots of T and NK cells colored by samples, B UMAP plots of T and NK cells colored by site (PDF 5298 kb)
262_2022_3152_MOESM6_ESM.pdf
Fig. S6 The significant signaling pathways in the interplay between distinct cells, A The alluvial plot showing the outgoing (left) and incoming (right) signaling patterns of distinct cells, B Circle plot showing the significant inferred signaling networks (PDF 3809 kb)
Rights and permissions
About this article

Cite this article
Duan, W., Zhang, B., Li, X. et al. Single-cell transcriptome profiling reveals intra-tumoral heterogeneity in human chordomas. Cancer Immunol Immunother 71, 2185–2195 (2022). https://doi.org/10.1007/s00262-022-03152-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-022-03152-1