
Abstract
The incidence of neuroendocrine tumours (NETs) is increasing, but curative therapeutic options are limited because diagnosis is often delayed until the tumour has metastasized. Peptide receptor radionuclide therapy (PRRT) is among the most effective therapeutic options for metastatic NETs because of targeted delivery of radioactivity to the tumour via the somatostatin receptor (SSTR) and relatively low systemic toxicity. However, current PRRT regimes result in palliation rather than cure, and higher doses of PRRT that might achieve remission would also be too toxic to the patients. Therefore, there is a need to improve PRRT of NETs by combining it with other agents to achieve maximum benefits from the internal radiation therapy, while sparing non-target organs from radiation toxicity. Here we review various current and potential combination strategies to improve 177Lu-octreotate-based PRRT of NET, some of which could also apply to other radionuclide therapies. These strategies include co-administered drugs that improve delivery of the radiopharmaceutical via increased tumour perfusion or through increased SSTR density at tumour surface. Other combinations are aimed at enhancing the biological effects of the radiation-induced DNA damage in tumour cells or generating additional DNA damage burden to effectively increase the cytotoxicity of PRRT. We also propose an algorithm for stratifying NET patients to receive or not combination therapies with PRRT. Considering that PRRT and many of these combination agents are already used for treating patients with NET and other cancers, the proposed strategies to improve the efficacy of PRRT could be rapidly translated into the clinic.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuroendocrine tumours (NETs) are a heterogeneous group of rare tumours that arise from the diffuse neuroendocrine cells throughout the body, but occur more frequently in the gastroenteropancreatic and lung tissues [1]. NETs present a wide range of clinical presentations depending on their origin from among 15 or more types of precursor NET cells, and the hormones and bioactive substances they secrete, which determine their functioning or non-functioning status [2]. Unlike most other cancers, there has been a constant increase in the incidence of NETs over the last decades, reaching 5.86 and 6.98 new cases per 100,000 population in Canada and in the USA, respectively [3, 4]. While surgery is a curative option, it is not available for about 60% of NET patients either presenting with or developing metastases during follow-up [3]. Since NETs often overexpress somatostatin receptors (SSTRs), the first-line treatment consists in the administration of long-acting somatostatin analogues (SSAs) which have been shown to prolong progression-free survival with long-lasting control of hormonal symptoms and growth inhibition of NET in phase 3 randomized clinical trials [5, 6]. Conventional chemotherapy is better suited for more aggressive NETs, such as those of grade 3, and neuroendocrine carcinoma [7], but it has a limited efficacy against the majority of NETs, which are well-differentiated tumours [8, 9]. Some biotherapies, such as everolimus and sunitinib, are now established treatments against pancreatic NETs, but yield modest objective response rates [10, 11]. The peptide receptor radionuclide therapy (PRRT), a systemic targeted radionuclide therapy utilizing radiolabelled SSAs, has been developed as a palliative treatment for NETs over the last three decades [12, 13].
Peptide receptor radionuclide therapy
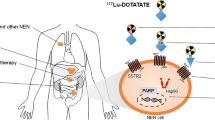
The cellular target of PRRT is SSTR, a member of the G protein–coupled family of receptors, and has five subtypes (SSTR 1–5), of which SSTR2 is overexpressed by the majority of NETs [14]. In normal and NET cells, a transient binding by somatostatin to the extracellular N-terminal domain of the SSTR results in G protein phosphorylation events at the intracellular C-terminal domain of the receptor (Fig. 1) [15]. This leads to the inhibition of various cAMP- or calcium channel–mediated downstream events that suppress various cellular responses including hormonal secretion and growth of NET cells [16]. Somatostatin-bound receptors are internalized through the β-arrestin pathway, and the free intracellular SSTR is either degraded or recycled back to the plasma membrane [17] (Fig. 1). Unlike somatostatin, the SSAs such as octreotide or lanreotide resist the proteolytic degradation from endosomal proteases [17, 18], remain longer in the systemic circulation, and can undergo repeated cycles of engagement with SSTRs [19] (Fig. 1).

Pathways of action of somatostatin, somatostatin analogues, and radiolabelled somatostatin analogues. Somatostatin or 177Lu-octreotate binds to somatostatin receptors (SSTRs) on the neuroendocrine tumour (NET) cell surface. Upon binding, SSTR is activated and G proteins will dissociate into α and βγ subunits. Gα will inhibit adenylyl cyclase involved in cAMP production and Gβγ will inhibit Ca2+ channel resulting in inhibition of hormone secretion. After binding, G protein kinases will phosphorylate the SSTR, which allows binding of β-arrestin and subsequent endocytosis of the somatostatin-coupled receptors. In the early endosomes, the somatostatin will be rapidly degraded by peptidases, β-arrestin will be released, and receptors will now be free either to be recycled rapidly to the surface or slowly through the trans-Golgi network or to be degraded by lysosomal enzymes. Somatostatin analogues, such as octreotide and its derivatives, will resist degradation by peptidases. The dissociation from β-arrestin will therefore be delayed. Once dissociated, the receptors are again free to be recycled or degraded. On the other hand, the analogue can be found in the lysosome or the nucleus. Radiolabelled analogues will emit radiation wherever they are. In the trajectory of the radiation, reactive oxygen species (ROS) will be created. ROS and the radiation itself will induce DNA damage, such as DNA single-strand or double-strand breaks. These breaks will inhibit the progression in cell cycle. If the DNA damage levels are low, the cell will repair its DNA, cell cycle will resume, and the cell will survive. If the damage is too high, the cell will undergo apoptosis
SSAs, mostly octreotide and octreotide derivatives, have been labelled with various radionuclides for molecular imaging and PRRT of NETs [20]. 111In-DTPA-octreotide has been used for both imaging and therapy, while [90Y-DOTA,D-Phe1,Tyr3]octreotide (a.k.a. 90Y-DOTATOC) and [177Lu-DOTA0,Tyr3]octreotate (a.k.a. 177Lu-octreotate or 177Lu-DOTATATE) have been the most studied PRRT radiopharmaceuticals [21]. Among these, 177Lu-octreotate has become the preferred PRRT agent, because 177Lu emits medium-energy (177–498 keV) β particles having an average penetration of 0.25 mm or about 125 cells deep (and maximum penetration of ~ 2 mm), which is associated with a higher efficacy than Auger electron–emitting 111In-DTPA-octreotide, and lower toxicity than 90Y-octreotide, which releases more penetrating β particles. Further, the low abundance of medium-energy γ photons (208 keV) of 177Lu facilitates post-therapy imaging and dosimetry. The emitted particles cause radiation-induced DNA strand breaks and indirect reactive oxygen–mediated DNA damage, both of which need to be repaired for survival of cancer cells (Fig. 1). If not repaired, DNA damage can lead to apoptosis or senescence of the PRRT-treated cancer cells. The toxic effect of radiation emanating from 177Lu starts from the moment it binds to the SSTR on plasma membrane and continues throughout the process of the degradation of the peptide components of 177Lu-octreoate in lysosomes and endosomes (Fig. 1). As such, the radionuclide liberated from the peptide and receptor could continue to accumulate in the cell and cause toxic radiation damage for several days until it decays or is eliminated from the cell (Fig. 1), similar to what has been shown for 111In in the cells treated with 111In-DTPA-octreotide [22].
In a randomized-controlled phase 3 trial, 177Lu-octreotate PRRT has proven its efficacy in treating SSTR2-expressing midgut NET with longer progression-free survival (PFS) and improved quality of life as compared with long-acting octreotide [23, 24]. PRRT efficacy figures also appear favourable when compared with those obtained with chemotherapy and biotherapies, such as tyrosine kinase and mTOR inhibitors [25, 26]. Despite 177Lu-octreotate PRRT clearly being one of the most successful targeted therapies for NETs so far, complete remissions are anecdotal and a risk of significant toxicity remains an obstacle for administration of higher radioactivity to all patients [24, 27, 28]. Also, many patients with NET are currently ineligible to PRRT because they do not meet the current clinical criterion which is to have an uptake greater than that of the liver on SSTR imaging (111In-octreotide scintigraphy or PET/CT with 68Ga-labelled SSA). Other patients are excluded because they have a high-grade NET (e.g. grade 3, Ki-67 > 20%), which is associated with a poorer prognosis. Therefore, many approaches are being explored to improve therapeutic effects of PRRT, including in those patients with a lower tumour uptake or higher-grade NET. Some of these approaches, such as use of α-emitting radionuclides, combination of radionuclides, radiolabelled somatostatin antagonists, and dosimetry-based personalized regimes, have recently been described or reviewed [29,30,31,32,33]. Here, we review different combination treatments that could enhance efficacy of 177Lu-octreotate PRRT for treatment of NET. These approaches are broadly divided in three categories, namely the improved tumour perfusion, SSTR upregulation, and radiosensitization (Table 1 and Fig. 2).

Strategies to improve therapeutic efficacy of PRRT. (a) Improving tumour perfusion: Immature and disorganized vasculature in NET results in heterogeneous and inefficient delivery of 177Lu-octreotate. After treatment with TMZ or with anti-vascular endothelial growth factor (α-VEGF), perfusion is improved or vasculature matures, respectively, allowing homogenous and optimal distribution of 177Lu-octreotate, along with improved oxygenation in all parts of the tumour. (b) Upregulation of SSTR: Drugs can induce synthesis or stabilization of SSTR mRNA resulting in a total increase in SSTR mRNA. Drugs can induce the translation of SSTR mRNA resulting into more SSTR being synthesized. Drugs can also increase the recycling of SSTR to the plasma membrane. (c) Radiosensitization with other DNA damaging agents: 177Lu-octreotate in combination with a cytotoxic agent creates an excessive amount of DNA damage that overwhelms the DNA repair pathways and promotes cell death. (d) Radiosensitization with targeted therapies: mTOR inhibitors can sensitize NET to 177Lu-octreotate by blocking a proliferation and survival pathway. Nicotinamide phosphoribosyltransferase inhibitors can sensitize NET to 177Lu-octreotate by blocking NAD recycling disrupting either DNA repair or aerobic respiration. Poly(ADP-ribose) polymerase inhibitors (PARPi) can sensitize NET by blocking selected pathways of DNA repair
Combination strategies to improve PRRT
Improving tumour perfusion
NETs are known to be hypervascular, but tend to gradually lose their high microvascular density as they become more aggressive [67]. The profile of blood vessels changes from the highly organized network in grade 1 NET to unbranched and plump vessels in grade 3 NET [68] (Fig. 2(a)). This is because the new blood vessels synthesized in response to vascular endothelial growth factor (VEGF) secreted by the tumour are highly disorganized, deprived of pericytes, and leaky. The leakage of blood from these fragile vessels combined with poor drainage of lymph and stiffening of the extracellular matrix in and around the tumour enhances pressure in the interstitial space (reviewed in [69, 70]). Eventually, the equalization of pressure inside and outside the blood vessels prevents the extravasation of nutrients, oxygen, and drugs from the blood. The resultant tumour hypoxia triggers the secretion of more proangiogenic factors and formation of more fragile blood vessels creating a vicious cycle of additional constraints in the delivery of drug to all the tumour cells. Moreover, the hypoxic tumour cells also enter a quiescent state and develop drug resistance. Improved delivery of 177Lu-octreotate via better tumour perfusion can increase the uptake and thus the absorbed dose and efficacy of the treatment for the same administered radioactivity (Fig. 2(a) and Table 1).
It has been observed that the anti-angiogenic drugs, which prevent neovascularization, could allow tumour vasculature to mature and become more efficient at the delivery of drugs, as well as radiopharmaceuticals. In parallel, the better tumour perfusion would also improve the delivery of oxygen, which per se is a potent radiosensitizer [71]. The anti-angiogenic agent sunitinib, an inhibitor of multiple tyrosine kinase receptors, has been approved for pancreatic NET therapy by the FDA [10] and has been shown to potentiate external beam radiotherapy (EBRT) in preclinical models and in a phase 2 clinical trial of patients with oligometastases from any primary sites, the head and neck being the most common [72]. The mTOR inhibitor everolimus has also been approved by the FDA for treating NETs. While its efficacy is primarily attributed to its anti-proliferative properties, which will be detailed later, everolimus also has an anti-angiogenic effect [73]. Recently, results from a study in 33 patients with pancreatic NET with liver metastases showed that everolimus induced increased tumour blood volume, which was attributed to improved tumour perfusion [35]. Interestingly, de Jong’s group used MRI imaging to show that the co-administration of alkylating agent temozolomide (TMZ) improved tumour perfusion and efficacy of the PRRT in NCI-H69 tumour–bearing animals [34].
Therefore, in future clinical trials, monitoring of parameters indicative of tumour perfusion, such as quantification of oxygenation, blood volume to the tumour, or VEGF and hypoxia-induced factor blood levels could provide additional mechanistic insight supporting combination therapy of these drugs with PRRT. Thus, the use of chemotherapeutic or anti-angiogenic drugs to improve the delivery of 177Lu-octreotate and oxygen to NETs is a promising potentiating strategy for PRRT that could be rapidly translated into the clinic with these already approved drugs.
SSTR upregulation
One of the principal determinants of the therapeutic effect of 177Lu-octreotate is the presence of SSTR2 in tumours [74]. For a given level of SSTR, increasing administered activity of 177Lu-octreotate may not result in proportionally higher tumour uptake due to saturation of SSTR, which would decrease the tumour-to-organ absorbed dose ratio, and thus the therapeutic index [75]. Conversely, increasing the expression of SSTR2 by the NET cells could result in a more effective therapy at equal or lower administered activities of 177Lu-octreotate, with limited additional risk of toxicity. Here, we discuss a variety of drugs, as well as radiation, which have been shown to upregulate expression of SSTR in NETs (Fig. 2(b) and Table 1) and which are candidates for potentiation of 177Lu-octreotate PRRT.
Hormones, growth factors, and somatostatin analogues
Many studies have shown the upregulation of SSTR2 levels in the brain, pancreas, ovaries, and liver cells treated with hormones and growth factors, such as β-estradiol, gastrin, epidermal growth factor, follicle-stimulating hormone, insulin, and growth hormone [76,77,78,79]. Oestrogen has been shown to upregulate transcription of SSTR2 in breast cancer, which led to better characterization of the promoter regions of SSTR2 gene [80,81,82], and this phenomenon could potentially be exploited for improved expression of SSTR2 in NETs with other agents, especially considering the reported presence of oestrogen receptors in some gastroenteropancreatic (GEP) and pulmonary NETs [83, 84] (Fig. 2(b)). Interestingly, SSA therapy itself has been shown to upregulate SSTR in different models. For example, the SSTR2 upregulation was shown after chronic exposure to SSA in pituitary cells [85], and an increased uptake of octreotide was shown in vivo in a rat model of pancreatic AR42J cells [36]. The upregulation of SSTR2 by octreotide alone or in combination with decitabine, trichostatin, or AT-101 was also seen in different human prostate cancer cell lines [86, 87].
In fact, many NET patients are prescribed long-acting SSAs for symptomatic control or cytostatic effect, and current protocols require long-acting SSAs to be stopped for 3–4 weeks prior to administration of PRRT, to avoid possible saturation of SSTR. However, in two clinical studies including 35 and 12 NET patients with stable disease, the continuous treatment with long-acting SSA did not reduce—or sometimes even increased—the uptake of 68Ga-octreotate in tumours, but significantly reduced its uptake in the liver and spleen [37, 38]. This enhanced tumour-to-background ratio was suggested to be due to SSTR2 upregulation in NET and a possible saturation of SSTRs in healthy tissues. Desensitization and downregulation of SSTR could also contribute to decrease the uptake in the latter. With a better understanding of the kinetics and mechanisms of SSTR regulation and trafficking by SSAs, the timing and dosage of the latter could be optimized so that PRRT administration would coincide with the peak of SSTR upregulation in tumours and/or SSTR downregulation/saturation in healthy tissues, i.e. the best tumour-to-organ uptake ratio. In summary, many of the above-described SSTR level–altering agents, more specifically long-acting SSAs, which tend to be well tolerated by the patients, could be useful as combination therapy to enhance efficacy of PRRT.
Epigenetic drugs
Epigenetic drugs that alter the expression of genes have already been shown to alter the expression of SSTR in different cellular and animal models of NET and other cancers. In different NET cells (BON-1, QGP-1, and KRJ-1) and in PC3 prostate cancer cells, DNA methylation inhibitor 5-aza-2′-deoxycytidine (decitabine) and three histone deacetylase inhibitors (HDACi), such as tacedinaline, romidepsin, and valproic acid, were shown to upregulate SSTR2 expression and increase the uptake of radiolabelled octreotide; and in BON-1 cells, the combination of decitabine and tacedinaline synergistically upregulated SSTR2 expression [44,45,46,47]. An increased SSTR2 expression in response to valproic acid was exploited for intracellular delivery of lethal doses of SSA-linked camptothecin and improved cell killing in BON-1 cells and xenograft model [47]. Thus, the combination with epigenetic drugs is a promising approach to improve PRRT with a growing body of preclinical evidence of induced SSTR upregulation. However, the clinical translation of this approach will need to be made carefully as epigenetic drugs have a broad range of effects on transcription, and their side effects are frequent.
Chemotherapy
Many common chemotherapeutic agents have been shown to modulate the expression of SSTR. 5-Fluorouracil (5-FU), camptothecin, cisplatin, mitomycin C, and doxorubicin were shown to reduce the uptake of radiolabelled SSAs in four different pancreatic adenocarcinoma–derived cell lines. In contrast, gemcitabine, a nucleoside analogue which inhibits ribonucleotide reductase and induces cell death, caused an increase in the uptake of 111In-DOTA-lanreotide after a period of recovery [48]. Gemcitabine for 4 days also increased the specific uptake of 177Lu-octreotate in vitro in Capan-2 (human adenocarcinoma), AR42J (rat pancreatic cancer), and NCI-H69 (human pulmonary NET) cells after a recovery period of 4 days and increased the toxicity of 177Lu-octreotate in Capan-2 cells [49]. In a patient with metastatic rectal NET treated with capecitabine (CAP, a 5-FU precursor) and TMZ, an increased tumour expression of SSTR2 has been observed on 68Ga-octreotate PET/CT [88]. However, a preclinical study in NCI-H69 cells did not reveal the upregulation of SSTR2 by TMZ [34], and a comparative double-arm clinical study in 20 patients with GEP-NET suggested that pretreatment with CAP and TMZ did not significantly increase the uptake of 177Lu-octreotate [89]. Thus, the kinetics of SSTR upregulation in NET by different chemotherapeutics needs to be carefully calibrated to exploit this option for potentiation of PRRT. Further, the dosing and length of chemotherapy used for this purpose should be minimized to avoid synergistic toxicity.
MicroRNAs
The microRNA or miRNA are known to control gene expression. In non-NET cancers, the levels of miR-185 were shown to be inversely correlated with the expression of SSTR2 in 20 patients with growth hormone–secreting pituitary adenoma; and miR-185 mimics or inhibitors could directly downregulate or upregulate, respectively, SSTR2 expression in vitro in rat pituitary adenoma GH3 cells [90]. Thus, miRNA-level manipulations have the potential to upregulate SSTR, but there is a need to demonstrate the feasibility of this approach in NET models.
Radiation
Radiation has been shown to upregulate SSTR2 and increase radiolabelled SSA uptake [39, 42, 43, 91,92,93]. Oddstig et al. noted an increase in SSTR2 mRNA levels following external beam radiation therapy (EBRT) of NCI-H69 cells [43], suggesting a molecular response following irradiation that leads to the increased expression. Dalmo et al. recently showed that pretreatment with a low activity of 177Lu-octreotate enhances the efficacy of a subsequent higher therapeutic activity of 177Lu-octreotate in a midgut NET (GOT1) xenograft model [41]. The mechanism of this priming effect was not linked to an increased expression of SSTR2 mRNA, although intratumoural uptake of 177Lu-octreotate was significantly increased, suggesting that this effect could be mediated by other pathways of increased intracellular accumulation of 177Lu-octreotate, such as increased SSTR recycling. Interestingly, the increased uptake of radiolabelled SSA was also looked at spatially as there was a heterogeneous SSTR2 expression. The same xenograft model was treated with 177Lu-octreotate and an increased uptake was seen 7 days post-treatment not only in the central region of the tumour that received most of the injected activity per gram (%IA/g) and thus, most of the absorbed dose, but also in peripheral regions which received half of %IA/g and absorbed dose relative to the central region [40]. The heterogeneous 177Lu-octreotate uptake within the tumour seemed linked to the difference in proliferation rate as demonstrated by Ki-67 staining, which was highly expressed in the peripheral region, where the uptake was lower. The molecular mechanisms of SSTR2 regulation by radiation are still unclear, but the accumulating evidence of the phenomenon in preclinical models makes radiation a prime candidate for clinical evaluation.
Radiosensitization
PRRT being an internal radiotherapy, drugs that are used for potentiating EBRT can be considered first-line candidates to potentiate PRRT. However, not all radiosensitization strategies used in EBRT may be equally effective in PRRT because NET biology is different from that of cancers commonly treated with EBRT—most NETs exhibiting a more indolent behaviour—and because of the differences in the delivery of radiation between PRRT (low rate and continuous) and ERBT (high rate and fractionated). Two strategies have been used in clinical trials to increase the radiosensitivity of NET to PRRT: DNA-damaging chemotherapeutics and targeted biotherapies (Fig. 2(c, d), respectively, and Table 1).
Chemotherapy
Chemotherapeutics have been used successfully for decades as radiosensitizers in EBRT [94]. A few clinical studies have examined their effectiveness as radiosensitizers for PRRT (Fig. 2(c)). The firsts of these to be studied clinically were 5-FU and its oral prodrug CAP. 5-FU, an anti-metabolite of DNA synthesis which inhibits thymidine synthase, is known to be a potent radiosensitizer for EBRT [95]. Three groups have used this rationale to co-administer 5-FU or CAP to NET patients treated with PRRT [50,51,52, 96]. In a retrospective study of 68 NET patients treated with 5-FU (200 mg/m2/24 h) starting 4 days prior to 177Lu-octreotate administration and continuing the infusion for 3 weeks thereafter, a group from Melbourne showed that 68% of patients with previously progressive disease had a stabilization of their disease or a partial response, while the combination was well tolerated [53]. They suggested that the combination was more effective in 18F-fluorodeoxyglucose (FDG)-avid NETs, thought to represent more aggressive or rapidly proliferating NETs [54]. The results from two randomized clinical trials of PRRT with or without CAP currently underway in Italy (NCT02736448, NCT02736500) will provide more information as to the suitability of this approach to enhance PRRT.
More recently, another Australian group evaluated the combination of four cycles of 7.9GBq 177Lu-octreotate with CAP and TMZ (a.k.a. CAPTEM) in 30 patients with grade 1 or 2 pancreatic NETs. These studies showed that combining 177Lu-octreotate with 1500 mg/m2 of CAP for 14 days starting 5 days before each cycle along with 200 mg/m2 of TMZ for the last 5 days of CAP appears as safe as 177Lu-octreotate alone in the short and long terms [55, 56]. After a 4-year follow-up, the combination therapy achieved a complete remission rate of 13% in pancreatic NETs, which is substantially better than previous results of PRRT alone in this NET patient subpopulation [26, 57]. A multicentre phase 2 randomized clinical trial is currently comparing the efficacy of PRRT versus CAP + TMZ or a combination of the three (NCT02358356). In Bison’s animal study, TMZ by itself potentiated 177Lu-octreotate by radiosensitizing the tumours and also by increasing perfusion to the tumour [34], as detailed above. In yet another preclinical study, the combination of carboplatin, a DNA cross-linking agent, and etoposide, an inhibitor of the DNA topoisomerase II, with 177Lu-octreotate exhibited a longer period of tumour control in a mouse model of human small cell lung cancer (NCI-H69) and prolonged survival of the animals, which also translated into a clinical response in one patient with pulmonary NET [58].
Although peptide receptor chemoradionuclide therapy is promising and is being studied in multicentre clinical trials, there is a need to identify the specific subpopulation of NETs that will benefit the most from this combination as compared with PRRT alone.
Targeted therapies
Molecularly targeted drugs that improve the tumour perfusion and the radiopharmaceutical delivery, as described above, are also candidates for radiosensitizing NET cells to 177Lu-octreotate therapy (Fig. 2(d) and Table 1). Everolimus has a direct radiosensitization mechanism through its inhibition of the mammalian (or mechanistic) target of rapamycin (mTOR) that regulates cell growth and proliferation in NETs. Since both PRRT and everolimus have been proven effective as monotherapies against NET [11], their combination was tested by two groups. Two animal studies conducted by the Rotterdam group showed the surprising results that everolimus induced metastases and reduced the efficacy of 177Lu-octreotate when used in combination in their rat model of pancreatic tumour cell line CA20948 [59, 60]. They hypothesized that the suppressive effect of everolimus on the immune system could partly explain the increased metastases. However, this observation has been made in only one animal model derived from a non-human NET cell line, and no clinical data is available to support that everolimus promotes metastatic progression in patients. In an Australian phase 1 study (NETTLE) in 16 patients, the combination of everolimus and PRRT appeared well tolerated. The maximum dose of everolimus was 7.5 mg/day and the median follow-up was 34 months [61].
The inhibitors of poly(ADP-ribose) polymerase (PARPi) are being used in the clinic for the therapy of BRCA-mutant ovarian cancers to target PARP1 and other members of the PARP family involved in DNA repair pathways [97, 98]. Recent preclinical studies show that PARPi sensitize different NET cells and U2OS sarcoma cells expressing SSTR2 to 177Lu-octreotate [63, 64]. Both the studies showed that the inhibition of PARP lead to a greater accumulation of DNA damage after 177Lu-octreotate treatment as seen with markers such as 53BP1 or γH2AX, which in turn increase apoptosis. A nicotinamide phosphoribosyltransferase (NAMPT) inhibitor depletes cells in nicotinamide adenine dinucleotide (NAD) [99], which is essential in energy metabolism, DNA repair, gene expression, and stress response [100], and was shown to block tumour growth and to potentiate the effect of 177Lu-octreotate in a midgut NET cell line (GOT1) [62]. The target of NAMPT, NAD, is the substrate for PARP, making the results from these studies consistent and highlighting NAD metabolism as an important target for radiosensitization of NET in patients. Indeed, this is especially relevant for those who suffer from the carcinoid syndrome, as serotonin and NAD are both derived from tryptophan, which is consumed by serotonin overproduction. This could further lower intracellular and blood NAD levels [101]. While both NAMPT and PARP inhibitors are promising as radiosensitizers, they are both potentially hematotoxic [102, 103]. Therefore, their combination with PRRT should be attempted prudently in early clinical studies.
Another study showed that sonidegib that targets the Hedgehog pathway, which is implicated in gene regulation of proliferation, migration, and invasion, extended the progression-free survival in a rat model of midgut NET (GOT1 xenografts) when combined with 177Lu-octreotate [65]. With the advantage of specificity of targeted drugs to a given pathway, it would be interesting to combine PRRT with more than one targeted drug. For example, since the combination of sonidegib with 177Lu-octreotate activates the mTOR pathway [65], adding an mTOR inhibitor such as everolimus could further enhance the therapeutic response in this model.
Multiple combination
While each of the three broad categories of combination treatments described above could individually enhance PRRT, a combination of agents derived from two different proposed approaches could synergistically enhance PRRT without increasing toxicity. In fact, Jin et al. demonstrated that a combination of the radiosensitizer 5-FU with epigenetics drugs tacedinaline or decitabine can upregulate SSTR2 and radiosensitize the NET cells [66]. Interestingly, this study also revealed that each of these agents alone acted as both a radiosensitizer and an SSTR-upregulating agent. Moreover, the upregulation of SSTR2 by these agents varied with initial SSTR levels expressed by each cell line. For example, non-expresser QGP1 had no induction of SSTR2, while low expresser BON-1 cells had robust upregulation and medium to high expresser (NCI-H727 and GOT1) had mild upregulation of SSTR2. On the other hand, Fueger et al. had observed the downregulation of SSTR2 by 5-FU in different cancer cell models [48]. These opposing effects on SSTR2 upregulation could be explained by different timing of assessment of SSTR2 status, or due to inherent differences in NET versus other cell models. Nonetheless, these studies also highlight the need to select patients with lower initial levels of SSTR, as they will benefit the most from SSTR upregulation strategies.
A clinical algorithm for selecting patients for PRRT-enhancing combinations
Here, we propose a strategy to streamline NET patients who could benefit from combination-augmented PRRT (Fig. 3). Currently, the eligibility of NET patients to receive PRRT is largely based on radiolabelled SSA uptake on SSTR imaging, and those patients with poor tumour uptake (i.e. lower than liver uptake) are being denied PRRT. Our review highlights that some of the patients may become eligible for PRRT if a combination therapy results in a significantly increased tumour uptake. As a first step, an initial 68Ga-SSA PET/CT could be used to determine the tumour uptake, and a strategy to enhance PRRT delivery would be attempted in patients with a low-uptake NET. Currently, no tool can reliably predict which NET would respond better to increased perfusion versus SSTR2 upregulation. Since the fast-growing tumours (e.g. high FDG uptake or Ki-67) generally have a more disrupted vasculature, they would be good candidates to a perfusion-enhancing strategy. On the other hand, slow-growing tumours (e.g. low FDG uptake or Ki-67), which are expected to have normal vasculature, should be subjected to SSTR-upregulating agents first. The success of increased perfusion or SSTR upregulation should be verified with a repeat 68Ga-SSA PET/CT re-assessing eligibility to PRRT.

Clinical decision algorithm for the selection of patients to undergo combination therapy
Thereafter, the patients with slow-growing tumours (e.g. low FDG uptake or Ki-67) could be subjected to PRRT alone, as they appear less likely to derive significant added benefits versus the increased risk of toxicity of a radiosensitizing combination. On the other hand, the patients with faster growing tumours (e.g. high FDG uptake or Ki-67) may have a favourable benefit-risk balance regarding the application of a radiosensitization strategy along with PRRT, since these tumours are more sensitive to chemotherapeutics and potential side effects would be more acceptable in the setting of a more aggressive NET conferring a worse prognosis.
Apart from tumour uptake and grading, novel analyses of circulating RNA transcripts and gene cluster, which have been shown to predict response to PRRT with a high accuracy (using a so-called PRRT predictive quotient derived from NET transcriptome and grade), could potentially help selecting the most appropriate combination treatments and monitoring their efficacy [104]. It is even possible that genomic signatures predictive of response to specific combination strategies exist, but they need to be identified and validated in conjunction with PRRT for future application to select eligible patients.
Among different combination strategies to improve PRRT that we have described, the ones which are most likely to reach the clinic in the near future are those already studied in clinical trials, such as the radiosensitizing agents CAP and TMZ. The combination of PRRT with PARPi is also likely to be prioritized based on promising preclinical results and the fact that PARPi is already approved by the FDA for other malignancies. Among the perfusion-enhancing agents, sunitinib, the anti-VEGF already approved for pancreatic NET, needs to be examined for its capacity to increase the delivery of 177Lu-octreotate in patients. SSTR upregulators, such as radiation (e.g. priming PRRT or EBRT) and epigenetics drugs, which have shown benefits in multiple preclinical studies, should be excellent candidates for clinical translation.
Conclusion
PRRT is currently one of the most effective palliative treatments for inoperable NETs. Combining PRRT with synergistic drugs that have ideally minimally overlapping toxicities could potentiate PRRT through several mechanisms such as increased tumour perfusion, SSTR upregulation, and radiosensitization. Moreover, a combination of multiple PRRT potentiators could be used, in a personalized fashion, to achieve the highest likelihood of therapeutic benefits for patients suffering from NET. These PRRT-enhancing approaches could be considered for other, non-NET cancers that have the potential to overexpress SSTR. Finally, some could be directly relevant to other radionuclide therapies, such as the rapidly emerging prostate-specific membrane antigen radioligand therapy of prostate cancer (i.e. perfusion-increasing, radiosensitization) or inspire analogous strategies (i.e. target upregulation via hormonal manipulations, irradiation, or other drugs).
References
Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of tumours of the digestive system: World Health Organization; 2010.
Kidd M, Modlin IM, Bodei L, Drozdov I. Decoding the molecular and mutational ambiguities of gastroenteropancreatic neuroendocrine neoplasm pathobiology. Cell Mol Gastroenterol Hepatol. 2015;1:131–53. https://doi.org/10.1016/j.jcmgh.2014.12.008.
Hallet J, Law CH, Cukier M, Saskin R, Liu N, Singh S. Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer. 2015;121:589–97. https://doi.org/10.1002/cncr.29099.
Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017. https://doi.org/10.1001/jamaoncol.2017.0589.
Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–63. https://doi.org/10.1200/JCO.2009.22.8510.
Caplin ME, Pavel M, Cwikla JB, Phan AT, Raderer M, Sedlackova E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:224–33. https://doi.org/10.1056/NEJMoa1316158.
Sorbye H, Welin S, Langer SW, Vestermark LW, Holt N, Osterlund P, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol: Off J Eur Soc Med Oncol. 2013;24:152–60. https://doi.org/10.1093/annonc/mds276.
Wong MH, Chan DL, Lee A, Li BT, Lumba S, Clarke SJ, et al. Systematic review and meta-analysis on the role of chemotherapy in advanced and metastatic neuroendocrine tumor (NET). PLoS One. 2016;11:e0158140. https://doi.org/10.1371/journal.pone.0158140.
Lamarca A, Elliott E, Barriuso J, Backen A, McNamara MG, Hubner R, et al. Chemotherapy for advanced non-pancreatic well-differentiated neuroendocrine tumours of the gastrointestinal tract, a systematic review and meta-analysis: a lost cause? Cancer Treat Rev. 2016;44:26–41. https://doi.org/10.1016/j.ctrv.2016.01.005.
Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–13. https://doi.org/10.1056/NEJMoa1003825.
Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–23. https://doi.org/10.1056/NEJMoa1009290.
Krenning EP, Kwekkeboom DJ, Bakker WH, Breeman WA, Kooij PP, Oei HY, et al. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med. 1993;20:716–31.
Werner RA, Weich A, Kircher M, Solnes LB, Javadi MS, Higuchi T, et al. The theranostic promise for neuroendocrine tumors in the late 2010s - where do we stand, where do we go? Theranostics. 2018;8:6088–100. https://doi.org/10.7150/thno.30357.
Zamora V, Cabanne A, Salanova R, Bestani C, Domenichini E, Marmissolle F, et al. Immunohistochemical expression of somatostatin receptors in digestive endocrine tumours. Dig Liver Dis. 2010;42:220–5. https://doi.org/10.1016/j.dld.2009.07.018.
Cambiaghi V, Vitali E, Morone D, Peverelli E, Spada A, Mantovani G, et al. Identification of human somatostatin receptor 2 domains involved in internalization and signaling in QGP-1 pancreatic neuroendocrine tumor cell line. Endocrine. 2017;56:146–57. https://doi.org/10.1007/s12020-016-1026-2.
Pyronnet S, Bousquet C, Najib S, Azar R, Laklai H, Susini C. Antitumor effects of somatostatin. Mol Cell Endocrinol. 2008;286:230–7. https://doi.org/10.1016/j.mce.2008.02.002.
Zhao P, Canals M, Murphy JE, Klingler D, Eriksson EM, Pelayo JC, et al. Agonist-biased trafficking of somatostatin receptor 2A in enteric neurons. J Biol Chem. 2013;288:25689–700. https://doi.org/10.1074/jbc.M113.496414.
Roosterman D, Kempkes C, Cottrell GS, Padilla BE, Bunnett NW, Turck CW, et al. Endothelin-converting enzyme-1 degrades internalized somatostatin-14. Endocrinology. 2008;149:2200–7. https://doi.org/10.1210/en.2007-1628.
Koenig JA, Kaur R, Dodgeon I, Edwardson JM, Humphrey PP. Fates of endocytosed somatostatin sst2 receptors and associated agonists. Biochem J. 1998;336(Pt 2):291–8.
Kolby L, Wangberg B, Ahlman H, Tisell LE, Fjalling M, Forssell-Aronsson E, et al. Somatostatin receptor subtypes, octreotide scintigraphy, and clinical response to octreotide treatment in patients with neuroendocrine tumors. World J Surg. 1998;22:679–83.
Brabander T, Teunissen JJ, Van Eijck CH, Franssen GJ, Feelders RA, de Herder WW, et al. Peptide receptor radionuclide therapy of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2016;30:103–14. https://doi.org/10.1016/j.beem.2015.10.005.
Wang M, Caruano AL, Lewis MR, Meyer LA, VanderWaal RP, Anderson CJ. Subcellular localization of radiolabeled somatostatin analogues: implications for targeted radiotherapy of cancer. Cancer Res. 2003;63:6864–9.
Strosberg J, Wolin E, Chasen B, Kulke M, Bushnell D, Caplin M, et al. Health-related quality of life in patients with progressive midgut neuroendocrine tumors treated with (177)Lu-Dotatate in the phase III NETTER-1 trial. J Clin Oncol. 2018;36:2578–84. https://doi.org/10.1200/JCO.2018.78.5865.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376:125–35. https://doi.org/10.1056/NEJMoa1607427.
Kim S-J, Pak K, Koo P, Kwak J, Chang S. The efficacy of 177Lu-labelled peptide receptor radionuclide therapy in patients with neuroendocrine tumours: a meta-analysis. Eur J Nucl Med Mol Imaging. 2015:1–7. https://doi.org/10.1007/s00259-015-3155-x.
Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124–30. https://doi.org/10.1200/JCO.2007.15.2553.
Bodei L, Kidd M, Paganelli G, Grana CM, Drozdov I, Cremonesi M, et al. Long-term tolerability of PRRT in 807 patients with neuroendocrine tumours: the value and limitations of clinical factors. Eur J Nucl Med Mol Imaging. 2015;42:5–19. https://doi.org/10.1007/s00259-014-2893-5.
Bergsma H, Lom KV, Konijnenberg M, Kam B, Teunissen J, Herder W, et al. Therapy-related hematological malignancies after peptide receptor radionuclide therapy with 177Lu-DOTA-Octreotate: incidence, course & predicting factors in patients with GEP-NETs. J Nucl Med: Off Publ, Soc Nucl Med. 2017. https://doi.org/10.2967/jnumed.117.189712.
Navalkissoor S, Grossman A. Targeted alpha particle therapy for neuroendocrine tumours: the next generation of peptide receptor radionuclide therapy. Neuroendocrinology. 2019;108:256–64. https://doi.org/10.1159/000494760.
Radojewski P, Dumont R, Marincek N, Brunner P, Macke HR, Muller-Brand J, et al. Towards tailored radiopeptide therapy. Eur J Nucl Med Mol Imaging. 2015;42:1231–7. https://doi.org/10.1007/s00259-015-3030-9.
Fani M, Nicolas GP, Wild D. Somatostatin receptor antagonists for imaging and therapy. J Nucl Med: Off Publ, Soc Nucl Med. 2017;58:61S–6S. https://doi.org/10.2967/jnumed.116.186783.
Del Prete M, Buteau FA, Arsenault F, Saighi N, Bouchard LO, Beaulieu A, et al. Personalized (177)Lu-octreotate peptide receptor radionuclide therapy of neuroendocrine tumours: initial results from the P-PRRT trial. Eur J Nucl Med Mol Imaging. 2019;46:728–42. https://doi.org/10.1007/s00259-018-4209-7.
Sundlov A, Sjogreen-Gleisner K, Svensson J, Ljungberg M, Olsson T, Bernhardt P, et al. Individualised 177Lu-DOTATATE treatment of neuroendocrine tumours based on kidney dosimetry. Eur J Nucl Med Mol Imaging. 2017. https://doi.org/10.1007/s00259-017-3678-4.
Bison SM, Haeck JC, Bol K, Koelewijn SJ, Groen HC, Melis M, et al. Optimization of combined temozolomide and peptide receptor radionuclide therapy (PRRT) in mice after multimodality molecular imaging studies. EJNMMI Res. 2015;5:62. https://doi.org/10.1186/s13550-015-0142-y.
D’Onofrio M, Cingarlini S, Ortolani S, Crosara S, DER R, Vallerio P, et al. Perfusion CT changes in liver metastases from pancreatic neuroendocrine tumors during everolimus treatment. Anticancer Res. 2017;37:1305–11. https://doi.org/10.21873/anticanres.11448.
Froidevaux S, Hintermann E, Torok M, Macke HR, Beglinger C, Eberle AN. Differential regulation of somatostatin receptor type 2 (sst 2) expression in AR4-2J tumor cells implanted into mice during octreotide treatment. Cancer Res. 1999;59:3652–7.
Haug AR, Rominger A, Mustafa M, Auernhammer C, Goke B, Schmidt GP, et al. Treatment with octreotide does not reduce tumor uptake of (68)Ga-DOTATATE as measured by PET/CT in patients with neuroendocrine tumors. J Nucl Med: Off Publ, Soc Nucl Med. 2011;52:1679–83. https://doi.org/10.2967/jnumed.111.089276.
Cherk MH, Kong G, Hicks RJ, Hofman MS. Changes in biodistribution on (68)Ga-DOTA-Octreotate PET/CT after long acting somatostatin analogue therapy in neuroendocrine tumour patients may result in pseudoprogression. Cancer Imaging. 2018;18:3. https://doi.org/10.1186/s40644-018-0136-x.
Bernhardt P, Oddstig J, Kolby L, Nilsson O, Ahlman H, Forssell-Aronsson E. Effects of treatment with (177)Lu-DOTA-Tyr(3)-octreotate on uptake of subsequent injection in carcinoid-bearing nude mice. Cancer Biother Radiopharm. 2007;22:644–53. https://doi.org/10.1089/cbr.2007.333.
Oddstig J, Bernhardt P, Lizana H, Nilsson O, Ahlman H, Kolby L, et al. Inhomogeneous activity distribution of 177Lu-DOTA0-Tyr3-octreotate and effects on somatostatin receptor expression in human carcinoid GOT1 tumors in nude mice. Tumour Biol. 2012;33:229–39. https://doi.org/10.1007/s13277-011-0268-0.
Dalmo J, Spetz J, Montelius M, Langen B, Arvidsson Y, Johansson H, et al. Priming increases the anti-tumor effect and therapeutic window of 177Lu-octreotate in nude mice bearing human small intestine neuroendocrine tumor GOT1. EJNMMI Res. 2017;7:6. https://doi.org/10.1186/s13550-016-0247-y.
Oddstig J, Bernhardt P, Nilsson O, Ahlman H, Forssell-Aronsson E. Radiation-induced up-regulation of somatostatin receptor expression in small cell lung cancer in vitro. Nucl Med Biol. 2006;33:841–6. https://doi.org/10.1016/j.nucmedbio.2006.07.010.
Oddstig J, Bernhardt P, Nilsson O, Ahlman H, Forssell-Aronsson E. Radiation induces up-regulation of somatostatin receptors 1, 2, and 5 in small cell lung cancer in vitro also at low absorbed doses. Cancer Biother Radiopharm. 2011;26:759–65. https://doi.org/10.1089/cbr.2010.0921.
Taelman VF, Radojewski P, Marincek N, Ben-Shlomo A, Grotzky A, Olariu CI, et al. Upregulation of key molecules for targeted imaging and therapy. J Nucl Med: Off Publ, Soc Nucl Med. 2016;57:1805–10. https://doi.org/10.2967/jnumed.115.165092.
Veenstra MJ, van Koetsveld PM, Dogan F, Farrell WE, Feelders RA, Lamberts SW, et al. Epidrug-induced upregulation of functional somatostatin type 2 receptors in human pancreatic neuroendocrine tumor cells. Oncotarget. 2016. https://doi.org/10.18632/oncotarget.9462.
Arvidsson Y, Johanson V, Pfragner R, Wangberg B, Nilsson O. Cytotoxic effects of valproic acid on neuroendocrine tumour cells. Neuroendocrinology. 2016;103:578–91. https://doi.org/10.1159/000441849.
Sun L, Qian Q, Sun G, Mackey LV, Fuselier JA, Coy DH, et al. Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide-drug conjugate via activating somatostatin receptor type II. J Drug Target. 2016;24:169–77. https://doi.org/10.3109/1061186X.2015.1066794.
Fueger BJ, Hamilton G, Raderer M, Pangerl T, Traub T, Angelberger P, et al. Effects of chemotherapeutic agents on expression of somatostatin receptors in pancreatic tumor cells. J Nucl Med: Off Publ, Soc Nucl Med. 2001;42:1856–62.
Nayak TK, Atcher RW, Prossnitz ER, Norenberg JP. Enhancement of somatostatin-receptor-targeted (177)Lu-[DOTA(0)-Tyr(3)]-octreotide therapy by gemcitabine pretreatment-mediated receptor uptake, up-regulation and cell cycle modulation. Nucl Med Biol. 2008;35:673–8. https://doi.org/10.1016/j.nucmedbio.2008.05.003.
van Essen M, Krenning EP, Kam BL, de Herder WW, van Aken MO, Kwekkeboom DJ. Report on short-term side effects of treatments with 177Lu-octreotate in combination with capecitabine in seven patients with gastroenteropancreatic neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2008;35:743–8. https://doi.org/10.1007/s00259-007-0688-7.
Hubble D, Kong G, Michael M, Johnson V, Ramdave S, Hicks RJ. 177Lu-octreotate, alone or with radiosensitising chemotherapy, is safe in neuroendocrine tumour patients previously treated with high-activity 111In-octreotide. Eur J Nucl Med Mol Imaging. 2010;37:1869–75. https://doi.org/10.1007/s00259-010-1483-4.
Claringbold PG, Brayshaw PA, Price RA, Turner JH. Phase II study of radiopeptide 177Lu-octreotate and capecitabine therapy of progressive disseminated neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2011;38:302–11. https://doi.org/10.1007/s00259-010-1631-x.
Kong G, Thompson M, Collins M, Herschtal A, Hofman MS, Johnston V, et al. Assessment of predictors of response and long-term survival of patients with neuroendocrine tumour treated with peptide receptor chemoradionuclide therapy (PRCRT). Eur J Nucl Med Mol Imaging. 2014;41:1831–44. https://doi.org/10.1007/s00259-014-2788-5.
Kashyap R, Hofman MS, Michael M, Kong G, Akhurst T, Eu P, et al. Favourable outcomes of (177)Lu-octreotate peptide receptor chemoradionuclide therapy in patients with FDG-avid neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2015;42:176–85. https://doi.org/10.1007/s00259-014-2906-4.
Claringbold PG, Price RA, Turner JH. Phase I-II study of radiopeptide 177Lu-octreotate in combination with capecitabine and temozolomide in advanced low-grade neuroendocrine tumors. Cancer Biother Radiopharm. 2012;27:561–9. https://doi.org/10.1089/cbr.2012.1276.
Kesavan M, Claringbold PG, Turner JH. Hematological toxicity of combined 177Lu-octreotate radiopeptide chemotherapy of gastroenteropancreatic neuroendocrine tumors in long-term follow-up. Neuroendocrinology. 2014;99:108–17. https://doi.org/10.1159/000362558.
Claringbold PG, Turner JH. Pancreatic neuroendocrine tumor control: durable objective response to combination 177Lu-octreotate-capecitabine-temozolomide radiopeptide chemotherapy. Neuroendocrinology. 2016;103:432–9. https://doi.org/10.1159/000434723.
Lewin J, Cullinane C, Akhurst T, Waldeck K, Watkins DN, Rao A, et al. Peptide receptor chemoradionuclide therapy in small cell carcinoma: from bench to bedside. Eur J Nucl Med Mol Imaging. 2015;42:25–32. https://doi.org/10.1007/s00259-014-2888-2.
Pool SE, Bison S, Koelewijn SJ, van der Graaf LM, Melis M, Krenning EP, et al. mTOR inhibitor RAD001 promotes metastasis in a rat model of pancreatic neuroendocrine cancer. Cancer Res. 2013;73:12–8. https://doi.org/10.1158/0008-5472.CAN-11-2089.
Bison SM, Pool SE, Koelewijn SJ, van der Graaf LM, Groen HC, Melis M, et al. Peptide receptor radionuclide therapy (PRRT) with [(177)Lu-DOTA(0),Tyr(3)]octreotate in combination with RAD001 treatment: further investigations on tumor metastasis and response in the rat pancreatic CA20948 tumor model. EJNMMI Res. 2014;4:21. https://doi.org/10.1186/s13550-014-0021-y.
Claringbold PG, Turner JH. NeuroEndocrine Tumor Therapy with Lutetium-177-octreotate and Everolimus (NETTLE): a phase I study. Cancer Biother Radiopharm. 2015;30:261–9. https://doi.org/10.1089/cbr.2015.1876.
Elf AK, Bernhardt P, Hofving T, Arvidsson Y, Forssell-Aronsson E, Wangberg B, et al. NAMPT inhibitor GMX1778 enhances the efficacy of 177Lu-DOTATATE treatment of neuroendocrine tumors. J Nucl Med: Off Publ, Soc Nucl Med. 2016. https://doi.org/10.2967/jnumed.116.177584.
Nonnekens J, van Kranenburg M, Beerens CE, Suker M, Doukas M, van Eijck CH, et al. Potentiation of peptide receptor radionuclide therapy by the PARP inhibitor olaparib. Theranostics. 2016;6:1821–32. https://doi.org/10.7150/thno.15311.
Purohit NK, Shah RG, Adant S, Hoepfner M, Shah GM, Beauregard JM. Potentiation of (177)Lu-octreotate peptide receptor radionuclide therapy of human neuroendocrine tumor cells by PARP inhibitor. Oncotarget. 2018;9:24693–706. https://doi.org/10.18632/oncotarget.25266.
Spetz J, Langen B, Rudqvist N, Parris TZ, Helou K, Nilsson O, et al. Hedgehog inhibitor sonidegib potentiates 177Lu-octreotate therapy of GOT1 human small intestine neuroendocrine tumors in nude mice. BMC Cancer. 2017;17:528. https://doi.org/10.1186/s12885-017-3524-x.
Jin XF, Auernhammer CJ, Ilhan H, Lindner S, Nolting S, Maurer J, et al. Combination of 5-fluorouracil with epigenetic modifiers induces radiosensitization, somatostatin receptor 2 expression and radioligand binding in neuroendocrine tumor cells in vitro. J Nucl Med: Off Publ, Soc Nucl Med. 2019. https://doi.org/10.2967/jnumed.118.224048.
Cavalcanti E, Ignazzi A, De Michele F, Caruso ML. PDGFRalpha expression as a novel therapeutic marker in well-differentiated neuroendocrine tumors. Cancer Biol Ther. 2019;20:423–30. https://doi.org/10.1080/15384047.2018.1529114.
Yazdani S, Kasajima A, Tamaki K, Nakamura Y, Fujishima F, Ohtsuka H, et al. Angiogenesis and vascular maturation in neuroendocrine tumors. Hum Pathol. 2014;45:866–74. https://doi.org/10.1016/j.humpath.2013.09.024.
Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–18. https://doi.org/10.1200/JCO.2012.46.3653.
Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. 2017. https://doi.org/10.1007/s10456-017-9562-9.
Stepien K, Ostrowski RP, Matyja E. Hyperbaric oxygen as an adjunctive therapy in treatment of malignancies, including brain tumours. Med Oncol. 2016;33:101. https://doi.org/10.1007/s12032-016-0814-0.
Kleibeuker EA, Ten Hooven MA, Verheul HM, Slotman BJ, Thijssen VL. Combining radiotherapy with sunitinib: lessons (to be) learned. Angiogenesis. 2015;18:385–95. https://doi.org/10.1007/s10456-015-9476-3.
Villaume K, Blanc M, Gouysse G, Walter T, Couderc C, Nejjari M, et al. VEGF secretion by neuroendocrine tumor cells is inhibited by octreotide and by inhibitors of the PI3K/AKT/mTOR pathway. Neuroendocrinology. 2010;91:268–78. https://doi.org/10.1159/000289569.
Kratochwil C, Stefanova M, Mavriopoulou E, Holland-Letz T, Dimitrakopoulou-Strauss A, Afshar-Oromieh A, et al. SUV of [68Ga]DOTATOC-PET/CT predicts response probability of PRRT in neuroendocrine tumors. Mol Imaging Biol: MIB: Off Publ Acad Mol Imaging. 2015;17:313–8. https://doi.org/10.1007/s11307-014-0795-3.
Ilan E, Sandstrom M, Wassberg C, Sundin A, Garske-Roman U, Eriksson B, et al. Dose response of pancreatic neuroendocrine tumors treated with peptide receptor radionuclide therapy using 177Lu-DOTATATE. J Nucl Med: Off Publ, Soc Nucl Med. 2015;56:177–82. https://doi.org/10.2967/jnumed.114.148437.
Slama A, Videau C, Kordon C, Epelbaum J. Estradiol regulation of somatostatin receptors in the arcuate nucleus of the female rat. Neuroendocrinology. 1992;56:240–5.
Vidal C, Rauly I, Zeggari M, Delesque N, Esteve JP, Saint-Laurent N, et al. Up-regulation of somatostatin receptors by epidermal growth factor and gastrin in pancreatic cancer cells. Mol Pharmacol. 1994;46:97–104.
Riaz H, Dong P, Shahzad M, Yang L. Constitutive and follicle-stimulating hormone-induced action of somatostatin receptor-2 on regulation of apoptosis and steroidogenesis in bovine granulosa cells. J Steroid Biochem Mol Biol. 2014;141:150–9. https://doi.org/10.1016/j.jsbmb.2014.02.001.
Nelson LE, Sheridan MA. Insulin and growth hormone stimulate somatostatin receptor (SSTR) expression by inducing transcription of SSTR mRNAs and by upregulating cell surface SSTRs. Am J Physiol Regul Integr Comp Physiol. 2006;291:R163–9. https://doi.org/10.1152/ajpregu.00754.2005.
Kimura N, Takamatsu N, Yaoita Y, Osamura RY, Kimura N. Identification of transcriptional regulatory elements in the human somatostatin receptor sst2 promoter and regions including estrogen response element half-site for estrogen activation. J Mol Endocrinol. 2008;40:75–91. https://doi.org/10.1677/JME-07-0108.
Pscherer A, Dorflinger U, Kirfel J, Gawlas K, Ruschoff J, Buettner R, et al. The helix-loop-helix transcription factor SEF-2 regulates the activity of a novel initiator element in the promoter of the human somatostatin receptor II gene. EMBO J. 1996;15:6680–90.
Xu Y, Berelowitz M, Bruno JF. Characterization of the promoter region of the human somatostatin receptor subtype 2 gene and localization of sequences required for estrogen-responsiveness. Mol Cell Endocrinol. 1998;139:71–7.
Zimmermann N, Lazar-Karsten P, Keck T, Billmann F, Schmid S, Brabant G, et al. Expression pattern of CDX2, estrogen and progesterone receptors in primary gastroenteropancreatic neuroendocrine tumors and metastases. Anticancer Res. 2016;36:921–4.
Sica G, Wagner PL, Altorki N, Port J, Lee PC, Vazquez MF, et al. Immunohistochemical expression of estrogen and progesterone receptors in primary pulmonary neuroendocrine tumors. Arch Pathol Lab Med. 2008;132:1889–95. https://doi.org/10.1043/1543-2165-132.12.1889.
Presky DH, Schonbrunn A. Somatostatin pretreatment increases the number of somatostatin receptors in GH4C1 pituitary cells and does not reduce cellular responsiveness to somatostatin. J Biol Chem. 1988;263:714–21.
Liu Z, Marquez M, Nilsson S, Holmberg AR. Incubation with somatostatin, 5-aza decitabine and trichostatin up-regulates somatostatin receptor expression in prostate cancer cells. Oncol Rep. 2008;20:151–4.
Degirmenci M, Erdogan AP, Bulut G, Atmaca H, Uzunoglu S, Karaca B, et al. Octreotide in combination with AT-101 induces cytotoxicity and apoptosis through up-regulation of somatostatin receptors 2 and 5 in DU-145 prostate cancer cells. Tumour Biol. 2016;37:4939–44. https://doi.org/10.1007/s13277-015-4331-0.
Basu S, Ostwal V. Observation on enhanced avidity on somatostatin receptor targeted 68Ga-DOTATATE PET-CT following therapy with everolimus and capecitabine-temozolamide: is redifferentiation akin phenomenon a reality in neuroendocrine tumors? Nucl Med Commun. 2016;37:669–71. https://doi.org/10.1097/MNM.0000000000000507.
Thakral P, Sen I, Pant V, Gupta SK, Dureja S, Kumari J, et al. Dosimetric analysis of patients with gastro entero pancreatic neuroendocrine tumors (NETs) treated with PRCRT (peptide receptor chemo radionuclide therapy) using Lu-177 DOTATATE and capecitabine/temozolomide (CAP/TEM). Br J Radiol. 2018;91:20170172. https://doi.org/10.1259/bjr.20170172.
Fan X, Mao Z, He D, Liao C, Jiang X, Lei N, et al. Expression of somatostatin receptor subtype 2 in growth hormone-secreting pituitary adenoma and the regulation of miR-185. J Endocrinol Investig. 2015;38:1117–28. https://doi.org/10.1007/s40618-015-0306-7.
Melis M, Forrer F, Capello A, Bijster M, Bernard BF, Reubi JC, et al. Up-regulation of somatostatin receptor density on rat CA20948 tumors escaped from low dose [177Lu-DOTA0,Tyr3]octreotate therapy. Q J Nucl Med Mol Imaging. 2007;51:324–33.
Behe MKS, Pqsken M, Gross M, Alfke H, Keil B, et al. Irradiation-induced upregulation of somatostatin and gastrin receptors in vitro and in vivo. Eur J Nucl Med Mol Imaging. 2004;31:S237–8.
Capello A, Krenning E, Bernard B, Reubi JC, Breeman W, de Jong M. 111In-labelled somatostatin analogues in a rat tumour model: somatostatin receptor status and effects of peptide receptor radionuclide therapy. Eur J Nucl Med Mol Imaging. 2005;32:1288–95. https://doi.org/10.1007/s00259-005-1877-x.
Driessen CM, de Boer JP, Gelderblom H, Rasch CR, de Jong MA, Verbist BM, et al. Induction chemotherapy with docetaxel/cisplatin/5-fluorouracil followed by randomization to two cisplatin-based concomitant chemoradiotherapy schedules in patients with locally advanced head and neck cancer (CONDOR study) (Dutch Head and Neck Society 08-01): a randomized phase II study. Eur J Cancer. 2016;52:77–84. https://doi.org/10.1016/j.ejca.2015.09.024.
Rich TA, Shepard RC, Mosley ST. Four decades of continuing innovation with fluorouracil: current and future approaches to fluorouracil chemoradiation therapy. J Clin Oncol. 2004;22:2214–32. https://doi.org/10.1200/JCO.2004.08.009.
Kong G, Johnston V, Ramdave S, Lau E, Rischin D, Hicks RJ. High-administered activity In-111 octreotide therapy with concomitant radiosensitizing 5FU chemotherapy for treatment of neuroendocrine tumors: preliminary experience. Cancer Biother Radiopharm. 2009;24:527–33. https://doi.org/10.1089/cbr.2009.0644.
Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18:610–21. https://doi.org/10.1038/nrm.2017.53.
Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017;31:101–26. https://doi.org/10.1101/gad.291518.116.
Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. 2009;29:5872–88. https://doi.org/10.1128/MCB.00112-09.
Yaku K, Okabe K, Hikosaka K, Nakagawa T. NAD metabolism in cancer therapeutics. Front Oncol. 2018;8:622. https://doi.org/10.3389/fonc.2018.00622.
Shah GM, Shah RG, Veillette H, Kirkland JB, Pasieka JL, Warner RR. Biochemical assessment of niacin deficiency among carcinoid cancer patients. Am J Gastroenterol. 2005;100:2307–14. https://doi.org/10.1111/j.1572-0241.2005.00268.x.
LaFargue CJ, Dal Molin GZ, Sood AK, Coleman RL. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019;20:e15–28. https://doi.org/10.1016/S1470-2045(18)30786-1.
Hovstadius P, Larsson R, Jonsson E, Skov T, Kissmeyer AM, Krasilnikoff K, et al. A phase I study of CHS 828 in patients with solid tumor malignancy. Clin Cancer Res. 2002;8:2843–50.
Bodei L, Kidd MS, Singh A, van der Zwan WA, Severi S, Drozdov IA, et al. PRRT genomic signature in blood for prediction of (177)Lu-octreotate efficacy. Eur J Nucl Med Mol Imaging. 2018;45:1155–69. https://doi.org/10.1007/s00259-018-3967-6.
Acknowledgments
We would like to thank Marine A. Merlin for helpful discussion to improve the manuscript.
Funding
This work was supported by research funding to J.M.B. and G.M.S. from the Canadian Cancer Society Research Institute (grant no. 705327) and the Carcinoid-NeuroEndocrine Tumor Society of Canada,; and to J.M.B. from the Education and Research Foundation for Nuclear Medicine and Molecular Imaging, Quebec Bio-Imaging Network, Fondation du CHU de Québec – Université Laval and Fonds de Recherche du Québec – Santé. S.A. received a scholarship from the Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest related to this work. J.M.B. has received honoraria for invited conferences from Ipsen, Novartis, and Siemens Healthineers.
Ethical approval
Not applicable. This article does not contain any studies with human participants or animals performed by any of the authors other than those previously published and cited in this review article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Oncology – General
Rights and permissions
About this article

Cite this article
Adant, S., Shah, G.M. & Beauregard, JM. Combination treatments to enhance peptide receptor radionuclide therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging 47, 907–921 (2020). https://doi.org/10.1007/s00259-019-04499-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-019-04499-x