
Abstract
Background
Peptide receptor radionuclide therapy (PRRT) utilizes somatostatin receptor (SSR) overexpression on neuroendocrine tumors (NET) to deliver targeted radiotherapy. Intensity of uptake at imaging is considered related to efficacy but has low sensitivity. A pretreatment strategy to determine individual PRRT response remains a key unmet need. NET transcript expression in blood integrated with tumor grade provides a PRRT predictive quotient (PPQ) which stratifies PRRT “responders” from “non-responders”. This study clinically validates the utility of the PPQ in NETs.
Methods
The development and validation of the PPQ was undertaken in three independent 177Lu-PRRT treated cohorts. Specificity was tested in two separate somatostatin analog-treated cohorts. Prognostic value of the marker was defined in a cohort of untreated patients. The developmental cohort included lung and gastroenteropancreatic [GEP] NETs (n = 72) from IRST Meldola, Italy. The majority were GEP (71%) and low grade (86% G1-G2). Prospective validation cohorts were from Zentralklinik Bad Berka, Germany (n = 44), and Erasmus Medical Center, Rotterdam, Netherlands (n = 42). Each cohort included predominantly well differentiated, low grade (86–95%) lung and GEP-NETs. The non-PRRT comparator cohorts included SSA cohort I, n = 28 (100% low grade, 100% GEP-NET); SSA cohort II, n = 51 (98% low grade; 76% GEP-NET); and an untreated cohort, n = 44 (64% low grade; 91% GEP-NET). Baseline evaluations included clinical information (disease status, grade, SSR) and biomarker (CgA). NET blood gene transcripts (n = 8: growth factor signaling and metabolism) were measured pre-therapy and integrated with tumor Ki67 using a logistic regression model. This provided a binary output: “predicted responder” (PPQ+); “predicted non-responder” (PPQ-). Treatment response was evaluated using RECIST criteria [Responder (stable, partial and complete response) vs Non-Responder)]. Sample measurement and analyses were blinded to study outcome. Statistical evaluation included Kaplan-Meier survival and standard test evaluation analyses.
Results
In the developmental cohort, 56% responded to PRRT. The PPQ predicted 100% of responders and 84% of non-responders (accuracy: 93%). In the two validation cohorts (response: 64–79%), the PPQ was 95% accurate (Bad Berka: PPQ + =97%, PPQ- = 93%; Rotterdam: PPQ + =94%, PPQ- = 100%). Overall, the median PFS was not reached in PPQ+ vs PPQ- (10–14 months; HR: 18–77, p < 0.0001). In the comparator cohorts, the predictor (PPQ) was 47–50% accurate for SSA-treatment and 50% as a prognostic. No differences in PFS were respectively noted (PPQ+: 10–12 months vs. PPQ-: 9–15 months).
Conclusion
The PPQ derived from circulating NET specific genes and tumor grade prior to the initiation of therapy is a highly specific predictor of the efficacy of PRRT with an accuracy of 95%.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Management of neuroendocrine tumors (NETs) has proved problematic since the disease is often diagnosed when metastatic and therapeutic options are limited [1]. More recently, however, a number of novel targeted therapies have been introduced [2]. These include somatostatin analogues (SSAs), everolimus, sunitinib and targeted radionuclide therapies [3]. All therapeutic strategies demonstrate variable outcomes and adverse events because no scientific tools exist to predict individual efficacy [4].
Peptide receptor radionuclide therapy (PRRT), which utilizes the octreotide derivative 177Lu-DOTA-Tyr3,Thr8-octreotide or 177Lu-octreotate [5], is widely used in Europe and has recently been introduced into the USA [6]. Diverse non-controlled studies in pancreatic, small bowel and bronchopulmonary (BP) NETs have demonstrated it is effective (disease control rates: 50–80%) with objective responses and a positive impact on survival parameters [7,8,9,10]. A recent phase III, randomized, controlled trial of midgut NETs, progressive on standard-dose octreotide LAR (NETTER-1) demonstrated 177Lu-octreotate to be more effective than high-dose octreotide LAR for PFS [11].
A key unmet need is the requirement to identify who will benefit or fail therapy. This is important since PRRT is likely to become generally available, and clinically heterogeneous patient cohorts will be treated. Local study protocols may include subjects with stable or progressive disease, low or high grade tumors, both FDG-negative and positive tumors as well as disease at different stages and with different intensities of somatostatin receptor (SSR) expression. Typically, inclusion is based on detectable (≥Krenning 2) SSR expression [13] but intensity of uptake does not accurately predict an individual’s response. Other clinical parameters such as tumor grading and biomarkers (e.g. CgA) have been investigated [4] but are disease prognostic and none objectively or accurately predicts the outcome of therapy [14, 15].
A liquid biopsy that provides real-time assessment of NET-specific transcripts in circulating blood has generated predictive indices that can distinguish stable vs progressive disease or treatment susceptibility of an individual tumor [16]. Recently, we described a strategy to predict the efficacy of 177Lu-octreotate PRRT [17] by developing an algorithm that integrates blood-derived NET-specific gene transcripts (growth factor signaling and metabolic regulation) with tissue Ki67 values. This generates a PRRT Predictive Quotient (PPQ) with two prediction outputs: “PRRT-responder” or negative: “PRRT-non-responder”.
Historically, biomarker discovery and usage has generated confusion because of the lack of criteria/standardization [18]. Development and validation studies undertaken on intended use samples from multiple independent sites per standardized approaches, e.g. Standards for Reporting Diagnostic Accuracy (STARD) [19] and a high accuracy for treatment stratification (negative predictive value), are fundamental prerequisites for predictive marker studies. In this study, we assessed and confirmed the accuracy of the PPQ, and demonstrated that it was specific to PRRT treatment and did not function as a prognostic.
Materials and methods
Study design
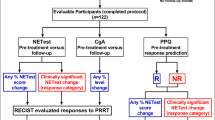
We developed and validated the PPQ as a predictor of PRRT response in three separate cohorts. To determine the PPQ specificity (i.e., whether it only predicted PRRT treatment), we evaluated biomarker predictions in two somatostatin analog-treated cohorts. To examine whether it functioned as a prognostic marker (measured disease progression), we determined whether it predicted mPFS in untreated patients included in a separate watch-and-wait cohort (Fig. 1).

STARD diagram outlining the study. PPQ =PRRT predictive quotient; R =responder, These are biomarker “positive” patients. NR= non-responder, These are biomarker “negative” patients
The initial (Meldola) cohort [17] provided the “Development” cohort. To validate the PPQ, samples were prospectively collected from two independent sites (Validation cohorts) prior to PRRT. Differences in median progression free survival (mPFS) were compared between PPQ “responder” and “non-responder” patients to assess utility of prediction. To test PPQ specificity and if it was prognostic, pre-treatment samples from three different “comparator cohorts” including non-radiolabeled SSA treatment and a watch-and-wait program were evaluated.
Patients
PRRT
Subjects included individuals with GEP and BP-NETs (n = 178) enrolled for 177Lu-octreotate-based PRRT on the basis of SSR expression at imaging. Patients were enrolled between July 2012 and June 2016 at three sites: Nuclear Medicine and Radiometabolic Units, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori IRST IRCCS, Meldola, Italy; Theranostics Center for Molecular Radiotherapy and Imaging, Zentralklinik Bad Berka, Bad Berka, Germany; and the Radiology and Nuclear Medicine Department, Erasmus Medical Center, Rotterdam, The Netherlands. The Meldola group provided the initial 54 patients used for PPQ development [17]. The current study is the summated analysis of the evaluable patients in the Meldola group (n = 72), Bad Berka (n = 44) and Rotterdam (n = 42).
Non-PRRT
These included three different cohorts. Cohort I (previously published [20]) included GEP-NET patients (n = 28) prospectively collected at the University of Warmia and Masuria, Olztyn, Poland between January 2014 and July 2015 (Supplemental Table 1). These were treated with SSAs and pre-treatment blood was available to assess the PPQ. Cohort II (Supplemental Table 2) comprised 51 SSA-treated GEP- and BP-NETs prospectively collected in the United States for a Registry (www.clinicaltrials.gov #NCT02270567). Cohort III (Supplemental Table 2) included a separate cohort of untreated 44 GEP- and BP-NETs in a watch-and-wait program from the Registry.
Study design and procedures
All participants provided informed consent for PRRT and molecular genomic translational analysis, authorized by their respective ethics committees (PRRT: IRST 100.06, EudraCT: 2011–002891-18, 04/08/2011; transcripts: IRST B007 [70/12], 10/10//2012; WIRB: 20,150,174; MEC-2014-309, 24/07/2014, NL48623.078.14). Inclusion criteria were: histological confirmation of a NET, SRS positive lesions, all with Krenning≥2; all with preserved renal and hematological function. Patients with all grades (GEPNET, G1-G3; lung: TC/AC/SCLC/NEC) were included in the study. Treatment was undertaken pursuant to pertinent clinical criteria including previous treatments and risk factors for delayed toxicity [7, 17, 21] (Table 1).
Assessment of therapeutic response
PRRT
Response was assessed per RECIST 1.1 criteria. CT (or MRI) was performed at baseline (within 3 months of PRRT) and ~2–3 and ~6–9 months after PRRT per protocol. 68Ga-SSA-PET or OctreoScan® was performed at baseline as an inclusion criterion. Response was defined as either disease control (partial or complete response or stabilization-censored “0”) or progression (treatment failure-censored as “1”). The latter included all patients who completed at least one PRRT cycle. Patients who progressed or died during PRRT treatment or follow-up were included. Follow-up and PFS were assessed from cycle 1.
No PRRT
For the SSA cohorts, RECIST (1.0) criteria were used to determine response (stable disease or treatment failure defined by progression). For the watch-and-wait cohort, RECIST (1.0) criteria were used to determine whether subjects were stable or had progressed during the follow-up period.
Blood collection
A sample of 10 ml whole blood were collected in 2 × 5 ml EDTA-K2 tubes and snap-frozen at baseline. Plasma CgA samples were collected at the same time point in PPT plasma preparations tubes.
Proliferation index
Ki67 was assessed by counting Mib-1 antibody positivity in hot-spot zones in either a primary or metastasis. The highest Ki67 value was considered to define the grade [22]. Tumors with Ki67 ≤ 20% (Grade 1 or 2) were categorized as “Low”. Tumors with Ki67 > 20% (Grade 3 or poorly differentiated) were categorized as “High”. For lung NETs, typical, atypical and NOS carcinoids, per histological evaluation, were grouped as “Low” [23], SCLC and NECs were categorized as “High”.
Transcript analysis and PRRT predictive quotient
Samples were deidentified prior to send-out from participating institutions. All samples were blinded to treatment and outcome prior to transcript analysis. The PPQ, a blood-based classifier [24, 25], is described in detail in the Supplemental Methods [17]. In brief, circulating expression of genes involved in growth factor expression and metabolism are amplified by PCR (Fig. 2). Expression levels are summated and scored as “low” or “high”. Using a logistic regression model [24, 26], the gene expression data is integrated with tumor grade to generate a prediction classifier. Samples are scored as either biomarker “positive” or “negative”. PPQ-positive identifies individuals predicted to respond (disease stabilization or partial/complete response). PPQ-negative are predicted not to respond to PRRT.

Overview of PPQ. PCR measurement of gene expression from 1 ml of blood. Two gene clusters are evaluated: NET growth factor signaling (n = 4) and NET metabolism regulation (n = 4). Summated gene expression (n = 8) is normalized to the housekeeping gene (ALG9). Individual genes exhibit expression values ranging from 0 to 104 [17]. Summated gene PCR values (n = 8) ≥5.9 are scored “1”, otherwise “0”. Each blood sample therefore has only one possible binary score. Tissue is evaluated by histology and graded (Ki-67). Tumors are categorized as either “Low” or “High”. Low include all G1 and G2 tumors (Ki67 ≤ 20%) or typical/atypical lung NETs. Low are scored “0”, high are scored “1”. Each tumor sample can therefore only have one possible score. High comprised all grade 3 (Ki67 > 20%) tumors, including NET G3, and PDNECs (e.g. SCLC). The scored (“1 or 0”) blood and tissue scores are incorporated into a logistic regression model which has two outputs— “R” (responder) or “NR” (non-responder). These represent the predictive quotient (PPQ) predicted responses to PRRT. An “R” value is expected to respond to therapy and an NR value is anticipated not to benefit from PRRT. AC =atypical carcinoid (bronchopulmonary NET), NR= non-responder, PDNEC =poorly differentiated neuroendocrine carcinoma, R =responder, SCLC =small cell lung cancer, TC= typical carcinoid (bronchopulmonary NET)
CgA assay
CgA was measured using NEOLISA™ Chromogranin A kit (Euro Diagnostica AB, Malmö, Sweden). The upper limit of normal was 108 ng/ml [20].
Clinical parameters
Other factors analyzed included: age, gender, tumor site (lung, pancreas, gastrointestinal tract, carcinoid of unknown primary), time since diagnosis, grading (low grade [G1/G2, well-differentiated, or bronchial typical or atypical carcinoid] versus or high grade [G3, poorly differentiated]), the baseline status (progressive or stable disease at start of PRRT), ECOG status, syndromic presence or absence, the extent of disease based on imaging (localized and moderate versus extensive), SRS intensity of uptake (Krenning grade 2 versus 3-4), 18FDG-positivity, and use of SSA therapy.
Statistical analyses
Prism 6.0 for Windows (GraphPad Software, La Jolla CA USA, www.graphpad.com) and MedCalc Statistical Software version 12.7.7 (MedCalc Software bvba, Ostend, Belgium; http://www.medcalc.org; 2013) were utilized [17]. The efficacy of PRRT was defined by RECIST 1.1 as either disease control (PR + CR + SD) or progressive. The accuracy of the PPQ was directly assessed at each of two time-points, initial follow-up (~2–3 months) and final follow-up (~6–9 months) after culmination of PRRT. Kaplan-Meier survival curves (PFS) were generated and analyzed. Log-rank (Mantel–Cox) and hazard ratios (Mantel–Haenszel) were calculated. The utility of PPQ as a predictor of PRRT treatment were evaluated by comparing mPFS between PPQ-positive and negative groups in each of the cohorts. Sensitivity, specificity, PPV and NPV were calculated. Logistic and multiple regression analyses were undertaken to identify clinical parameters that could be used as predictors of PRRT. The predictive accuracy of PPQ and the biomarker CgA (elevated versus normal) as well as each of the clinical parameters were compared. Decision curve analysis [27] was used to compare clinical benefits of PPQ versus CgA and grade. A metric accuracy of 80% is the acceptable cut-off [28] for the clinical utility of a biomarker.
Results
PRRT-patient demographics
One hundred seventy-eight patients were enrolled of which 158 (88.8%) were evaluable for response (Fig. 1). Among the 20 excluded, eight withdrew consent; in five a pre-treatment blood was not collected; five were excluded for medical reasons and two were lost to follow-up. Patient demographics for each cohort are included in Table 2.
Developmental cohort: Meldola (n = 72)
The PPQ was initially developed in 54 patients enrolled at IRST-Meldola [17]. The overall accuracy was 94% (97% responders and 91% non-responders) for PRRT response prediction. In the present study, we evaluated the PPQ in the entire cohort of 76 Meldola patients (four drop-outs). The median time from PRRT cycle I to post-treatment assessment was 14 months (range: 1–33). The mPFS for the cohort (responders/non-responders) was 18 months (95%CI: 16–18) (Fig. 3). PRRT response occurred in 42 (56%) at the second follow-up time point. The mPFS for this cohort was not-reached. In those with progressive disease, the mPFS was 8 months (95%CI: 7–10). Regression analysis identified no clinical parameters were associated with treatment response (odds ratios: 0.67 [SSA use, p = 0.53] – 3.13 [grading, p = 0.13]) except for ECOG status (OR = 3.7, p = 0.009) (Table 3). Sub-analysis identified a worse ECOG status was associated with a significantly earlier PFS (10 months vs. undefined, chi2 = 8.9, p = 0.0029).

Evaluation of progression free survival in each of the three cohorts. Median progression free survival (mPFS) for Bad Berka and Meldola cohorts was 18 months. It was not reached in the Rotterdam cohort. FUP1 = initial follow-up (FUP) evaluation ~2–3 months after the last PRRT cycle. FUP2 = 2nd follow-up evaluation ~6–9 months after the last PRRT cycle
The PPQ accurately predicted responders at initial (100%) and final (100%) follow-up (Fig. 4). Non-responders were predicted in 65% (initial) and 84% (final) (Fishers, p = NS between initial and final). At the final follow-up, PRRT response was correctly predicted in 67/72 (93%); PRRT-responders in 100% and non-responders in 84% (Fig. 4). Assessment of mPFS identified that this was not reached in PPQ+ subjects. In the PPQ-negative group, the mPFS was 8 months. This difference was highly significant (HR 36.4, p < 0.0001) (Fig. 5A). The PPQ sensitivity was 100%, the NPV was 100%.

Evaluation of PPQ at two time-points: Biomarker-positive “responder” PPQ was overall 97% (range: 94–100%) accurate for predicting responders at the initial follow-up and 97% (range: 94–100%) accurate at final follow-up. Biomarker-negative PPQ was 61% (range: 47–70%) accurate for predicting non-responders at initial follow-up and 89% accurate (range: 84–100%) at final follow-up. Overall, the PPQ was 94% accurate (149/158) for predicting responders and non-responders. Initial evaluation was undertaken ~2–3 months after the last PRRT cycle. Second evaluation was undertaken ~6–9 months after the last PRRT cycle. M-Meldola Cohort, B-Bad Berka Cohort, Rt-Rotterdam Cohort

PRRT Prediction Quotient for PFS prediction in PRRT-treated and non-treated cohorts. PRRT cohorts: A Development Cohort: Meldola: Positive PPQ (biomarker positive) prediction pre-therapy was associated with mPFS which was not reached. A negative PPQ prediction (biomarker negative) was associated with a mPFS of 8 months. This difference was significant (HR 36.4, p < 0.0001). B Validation Cohort I: Bad Berka: Positive PPQ prediction was associated with mPFS which was not reached. A negative PPQ prediction was associated with a mPFS of 14 months (HR 17.7, p < 0.0001). C Validation Cohort II: Rotterdam: Positive PPQ prediction was associated with mPFS which was not reached. A negative PPQ prediction was associated with an mPFS 9.7 months. This was significantly different (HR 92, p < 0.0001). No-PRRT cohorts: D Comparator Cohort I: SSA treatment: In prediction-responders, the mPFS was 10 months. For those predicted not to respond, the mPFS was 11 months. This was not significantly different (HR 0.75, p = NS). E Comparator Cohort II: SSA treatment II: In prediction-responders, the mPFS was 10 months. For those predicted not to respond, the mPFS was 15 months. This was not significantly different (HR 2.2, p = NS). F Comparator Cohort III: Watch-and-wait: In prediction-responders, the mPFS was 12 months. For those predicted not to respond, the mPFS was 9 months. This was not significantly different (HR 1.36, p = NS). PPQ-positive = biomarker-positive (responder), PPQ-negative = biomarker-negative (non-responder)
Validation cohort I: Bad Berka (n = 44)
The median time from PRRT cycle I to final treatment assessment was 14 months (range: 2–23). The mPFS for the entire cohort was 18 months (95%CI: 17–23) (Fig. 3). PRRT response occurred in 28 (64%). The mPFS for this cohort was not reached. In those with progressive disease, the median PFS was 14 months (95%CI: 8–17). No baseline clinical parameters were associated with treatment response (odds ratios 0.57 [SSA, p = 0.42] – 3.68 [disease extent, p = 0.07]) except for gender (OR = 0.25, p = 0.04) (Table 3). No gender-related differences in mPFS were, however, identified.
The PPQ accurately predicted responders at initial (97%) and final (97%) follow-up. Non-responders were predicted in 47% (initial) and 93% (final) (Fishers, p = 0.01) (Fig. 4). At final follow-up, 42/44 (95%) were correctly predicted: PRRT-responders in 97% and non-responders in 93% (Fig. 4). Survival analysis identified the mPFS was not reached in PPQ+. For “non-responders”, the mPFS was 14 months (HR 17.7, p < 0.0001) (Fig. 5B). The sensitivity of PPQ was 97%, the NPV was 93%.
Validation cohort II: Rotterdam (n = 42)
The median time from PRRT cycle I to the final treatment assessment was 13.7 months (range: 5–20). The mPFS for the entire cohort was not reached (Fig. 3). PRRT response was 33 (79%) at the final follow-up (mPFS not-reached). For progressive disease, the median PFS was 9.7 months (95%CI: 9.1–10.9). No baseline clinical parameters predicted treatment response (odds ratios 0.65 [SSA, p = 0.60] – 2.1 [gender, p = 0.40]) except for age (OR = 0.9, p = 0.035) (Table 3). Individuals >62 years had significantly longer mPFS Chi2 = 9.0, p = 0.0029).
The PPQ accurately predicted responders at initial (94%) and final (94%) follow-up (Fig. 4). Non-responders were predicted in 70% (initial) and 100% (final) (Fishers, p = NS). At final follow-up, 40/42 (95%) were correctly predicted: PRRT-responders in 95% and non-responders in 100% (Fig. 4). An evaluation of survival in PPQ+ identified the mPFS was not reached. For “non-responders”, the mPFS was 9.7 months (HR 92, p < 0.0001) (Fig. 5C). The sensitivity of the test was 94%, the NPV was 95%.
Specificity of the PPQ as a predictive marker
In SSA cohort I (n = 28), PPQ+ was associated with disease stabilization in 8 (53%) and progressive disease in 6 (47%, p = NS). No differences in mPFS (10 [95%CI: 9–10] vs. 11 months [95%CI: 10–11], p = NS) were noted (Fig. 5D). The sensitivity and NPV were 53% and 46%, respectively. In the second SSA cohort (n = 51), PPQ+ was associated with disease stabilization in 15 (50%) and progressive disease in 9 (43%, p = NS). No differences in mPFS (10 [95%CI: 6–12] vs. 15 months [95%CI: 8–15], p = NS) were noted (Fig. 5E). The sensitivity and NPV were 50% and 47%. In the watch-and-wait cohort (n = 44), PPQ+ was associated with stable disease in 15 (44%) and progressive disease in 7 (64%, p = NS). No differences in mPFS (12 [95%CI: 6–13] vs. 9 [95%CI: 6–9] months, p = NS) were noted (Fig. 5F). The sensitivity and NPV were 44% and 27%, respectively.
Predictive utility of elevated CgA
Meldola cohort
Elevated CgA (>108 ng/ml) was evident in 55% that responded and in 69% that failed PRRT (p = NS) (Table 4). The concordance correlation co-efficient for CgA was 0.13 (95%CI: –0.09 to 0.34].
Bad Berka cohort
Elevated levels were present in 61% of responders and 69% that failed PRRT (p = NS). The CgA concordance correlation was 0.067 (95%CI: –0.19 to 0.32].
Rotterdam cohort
Elevated CgA were present in 90% that failed PRRT and 81% of responders (p = NS). The concordance correlation was 0.10 (95%CI: –0.04 to 0.23].
Comparison between PRRT prediction quotient and grade/SRS/FDG
PPQ correctly predicted 149 (94.3%) of 158 patients at final follow-up (Table 4, Fig. 6A). This was significantly better than CgA (49%), grade assessment (73%), status at time of PRRT (63%), FDG-positivity (55%), intensity of uptake (63%) or disease extent (34%) (all parameters Fisher’s: p < 0.0001). The concordance correlations were: PPQ: 0.86 (95%CI: 0.82–0.9); CgA: 0.07 (95%CI: -0.06-0.19); grade: 0.15 (95%CI: 0.04–0.26); status: 0.14 (95%CI: 0.03–0.24); FDG: 0.10 (95%CI: –0.08 to 0.27); intensity of uptake: 0.04 (95%CI: –0.03 to 0.11); and extent: 0.13 (95%CI: 0.01–0.25).

PRRT predictive quotient (PPQ) in the three PRRT cohorts for PFS prediction. A Accuracy of prediction for the PPQ and each clinical criterion. The PPQ was significantly more accurate (p < 0.0001) than any other variable measured. B The metrics of the PPQ for response were sensitivity: 97.1%, specificity: 88.9%, PPV: 94.4% and NPV: 94.1%. Dotted line represents the 80% cut-off for biomarker accuracy. PPV =positive predictive value, NPV =negative predictive value
The PPQ diagnostic metrics for response were sensitivity: 97.1%, specificity: 88.9%, PPV: 94.4% and NPV: 94.1% (Fig. 6B). Multivariate regression analysis retained only PPQ as a predictive factor (p < 0.0001). The PPQ explained 86 ± 4% of the variance (R2 = 0.86; F-ratio 165.6, p < 0.0001).
Decision curve analysis of the PPQ as a companion diagnostic
Decision curve analysis quantified the clinical benefit of PPQ (Fig. 7), identifying it exhibited >90% standardized predictive benefit up to a risk threshold of 80%. The benefit of an CgA or grade was equivalent of not using a biomarker (<10% across comparable risk thresholds).

Decision curve analysis. The standardized NET benefit line (y-axis) reflects the predictive usefulness of a biomarker; 1.0 reflects 100% useful, 0.5 is 50% useful; negative values reflect “harm” (negative benefit) related to an intervention based on the results of a biomarker. The x-axis demonstrates the probability (risk) of disease. When a biomarker is not used for intervention, the standardized net benefit = 1.0 and the risk threshold is 0 (none). The gray line (labeled as All), reflects the overall benefit of introducing an intervention, e.g., PRRT to all individuals irrespective of biomarker value. In the treated cohort of 158 patients, the clinical benefit for the PPQ (red line) is >90% up to a disease risk threshold of 0.80. This indicates that the PPQ has significant predictive benefit for PRRT in NETs. In contrast, elevated CgA expression levels (blue line) or grade alone (green line) do not introduce any clinical benefit. Quantitatively, CgA and grade are the same as no biomarker. *p < 0.00001 vs. PPQ (Fisher’s 2-tailed exact test)
Discussion
A key unmet need in using PRRT appropriately is to identify those who will benefit. Current strategies used to prognosticate efficacy include SSR expression at baseline, tumor load and characteristics, e.g., grade, baseline glycolytic tumor metabolism (FDG-PET/CT), CgA especially if abnormal, and the progressive reduction of uptake at interim 177Lu-scan (or 68Ga-SSA-PET/CT) after each therapy cycle. SSR expression measurement by an OctreoScan or 68Ga-DOTATATE/DOTATOC PET/CT, is a cardinal pre-therapy requirement of PRRT, since it provides assessment of disease extent and the presence of the target. Response prediction for tumors with111In-pentetreotide uptake greater than kidney/spleen (Grade 4 of the Krenning scale) is, however, only 60% [12]. Similarly, 68Ga-SSA uptake is non-predictive in an individual patient [29,30,31]. This reflects the implausibility that SSR expression is the only determinant of response to radionuclide therapy [32]; it functions as an inclusion criterion for PRRT.
Grading using morphology or Ki67 has some clinical utility but is limited by tumor heterogeneity, subjective observer variations and a low kappa value. Furthermore, tissue biopsies are rarely obtained from more than one location, cannot be repetitively undertaken and metastases often differ significantly from the primary lesion biopsied for diagnosis [33].
Other “predictive” parameters include: extensive tumor load and reduced performance status (KPS ≤70), both associated with significantly shorter mPFS [15]. Tumor origin may also be relevant. Small intestine tumors are associated with a lower response rate compared to pancreatic NETs [7]. FDG avidity is also associated with a significantly shorter PFS after 177Lu-octreotate [34]. A retrospective analysis of 177Lu-octreotate series reported that baseline CgA values ≥600 ng/ml constituted a risk factor for early progression [35, 36]. All are prognostic factors and none predict response in an individual patient (Fig. 8). The progressive reduction of uptake at interim 177Lu-scans or a reduced tumor-to-spleen SUV ratio at 68Ga-DOTATATE performed after the first PRRT cycle also correlate with an objective response [12, 37]. This, however, does not predict responsiveness before therapy induction.

Clinical predictors of PRRT response. A panoply of prognostic markers has been evaluated as predictors of PRRT response. Somatostatin receptor (SSR) determination through imaging or by immunohistochemistry (IHC) function as inclusion criteria for PRRT. Their utility as a predictor of response is low. The other factors examined are all prognostic in nature and are non-predictive of PRRT response. PFS= progression free survival, ORR =objective response rate, OS= overall survival, KPS= Karnofsky Performance Status
We previously noted that PRRT-responders exhibited different patterns of gene expression to non-responders in pre-treatment blood [17]. Further investigation allowed us to identify a subset of eight genes involved in the regulation of growth factor signaling and metabolism. Integration of data from these two regulatory pathways had an AUC of 0.74 for predicting response. ROC analyses determined a cut-off of 5.9 in gene expression had an >85% specificity for PRRT response prediction [17]. In a separate analysis, we noted that low grade tumors (G1/G2, typical/atypical carcinoids) responded more frequently (77%) than high grade tumors (50%) [17]. The combination of the two variables, namely, gene expression and grading (PPQ) produced an AUC of 0.90 for extrapolation of response. The predictive accuracy in this developmental cohort of 54 subjects was 94% [17].
In the present study, this cohort was expanded to 72 patients. Analysis of the augmented dataset produced a similar result. The PPQ was 93% accurate in predicting responders versus non-responders. A significant treatment effect was therefore evident for biomarker “positive” patients. Predicted non-responders (biomarker negative) exhibited an mPFS of 8 months versus undefined for predicted responders (HR 36.4, p < 0.0001). The sensitivity of the PPQ was 100% (PPV) i.e., identification of 100% therapy responders with an NPV of 100%, and identification of those who would not respond. We conclude that the PPQ was highly effective and exhibited clinical benefit for the determination of PRRT efficacy.
We then prospectively evaluated the PPQ in intended use patients enrolled at two independent sites. The data from each of these separate locations demonstrated the PPQ was effective in 95%. Responders were correctly predicted in 94–97% and non-responders in 93–100%. Particularly interesting was that the PPQ predicted the outcome observed in the follow-up more accurately at the later rather than the earlier follow-up point; this emphasizes its predictive role. Overall, 23 of 124 (19%) identified as exhibiting disease control at initial follow-up were subsequently determined to demonstrate progressive disease at final evaluation. This likely reflects a “lag-time” between “best response” as assessed by CT/MRI and actual tumor molecular dynamics. The PPQ, which captures the underlying cellular biology of a tumor, correctly predicted that 18/23 (78%) would not respond to therapy.
At the second time point, the mPFS for responders was not reached; for non-responders mPFS ranged between 9.7 and 14 months. The Hazard ratio (HR) for the PPQ was 18–92 (p < 0.0001). The sensitivity and NPV were 94–97% and 83–93%, respectively. This identifies the PPQ was highly accurate for determining PRRT efficacy in intended use patients prospectively recruited from two independent sites (Bad Berka, Rotterdam). The high NPV accurately identified individuals in whom PRRT would not be effective; the high sensitivity accurately identified patients in whom PRRT would be effective (Supplemental Table 3).
Separately, we investigated the specificity of the signature in two additional cohorts (n = 128 patients): one from Poland, the second from the United States. The PPQ was measured before therapy in 28 patients from Olsztyn, Poland undergoing SSA treatment. The accuracy of the test was not different in responders (53%) versus non-responders (47%); the HR was 0.6. This identifies the PPQ was not predictive for targeting SSR alone, i.e. treatment with a “cold” somatostatin analog. In the second specificity study, we examined whether the PPQ was prognostic in US patients (n = 100) included in a Registry study (NCT02270567) which contained no PRRT-treated patients. The PPQ was not associated with PFS identifying it and did not function as a prognostic marker over the 18 months evaluated.
The advantages of this study are we examined the PPQ in a large development cohort (n = 72) and independently validated its predictive utility at two different sites in intended use patients (n = 86). Furthermore, we examined whether the PPQ was predictive in patients treated with a different targeted agent (SSA) and if it functioned as a prognostic biomarker. The PPQ neither predicted SSA response nor was prognostic. This confirmed the PPQ was specifically predictive for therapies targeting SSR with a radioligand.
As a predictive marker for PRRT, PPQ was effective in all PRRT protocols which were variable in each of the sites evaluated. For example, some patients in validation cohort I (Bad Berka) were treated with only two intended cycles of PRRT; in the development cohort, some patients were treated with different intended cumulative activities, according to risk factors for toxicity. Individual patients had different stages of disease, including very advanced stages and a proportion (development cohort) were undergoing re-treatment with PRRT after being previously treated with Y- or Lu-PRRT.
No single clinical assessment, including cumulative activity, was consistently associated with the prediction of PRRT responsiveness. The utility of SRS, FDG, grading alone or an elevated CgA ranged between 34 and 73% as predictive markers. Tumor histology (Ki67) predicted response in 73% of participants. While 95–100% of responders had well-differentiated tumors, 14–35% of poorly differentiated tumors also responded to PRRT. Grade alone therefore cannot predict non-response to therapy. CgA was predictive in 49% but the efficacy in individual treatment cohorts ranged between 18 and 100%.
A limitation of the study is that tumor grade evaluation differed between the cohorts. In Meldola, Ki67/grade were mainly assessed at outside institutions prior to referral. The same process occurred in Bad Berka. In contrast, grade evaluation was centralized in Rotterdam (at the Erasmus Medical Center) and, in the majority of cases, was reread prior to PRRT. However, the similar efficacies of the PPQ between the cohorts suggests a centralized analysis may not be required. Nevertheless, a uniform evaluation of grade should be a fundamental requirement for an accurate PPQ and reflects one of the longstanding issues that has previously been extensively discussed in respect of Ki67 [38].
Another limitation is that we evaluated retrospective samples from the comparator cohorts. Nevertheless, the discrepancy in prediction with PRRT was substantial and consistent with the postulate that PPQ is specific for radionuclide therapy. An additional limitation is that one of the studies (Bad Berka) included mixed 177Lu-based protocols. This could be viewed as a confounding factor. However, the observation that the PPQ demonstrated high predictive accuracy even in mixed protocol patients supports the proposal that this is specific for PRRT and that radiation response-associated genes captured in the signature are important factors. Both the growth factor and the metabolomic genes captured by the signature are specifically related to oxidative stress, metabolism and hypoxic signaling [39,40,41]. We consider that elevated expression of these genes in blood identifies tumors that are more radiosensitive given the role of hypoxia, oxidative stress and loss of DNA repair associated with radiation responsiveness [42].
The clinical value of a specific PRRT predictive biomarker consists in its ability to facilitate management decisions on whether to start the treatment based on the likelihood of efficacy [43]. When there is a molecular prediction of failure, an alternative clinical decision could be made to either modulate the treatment (combine with biologics or chemotherapy), or consider sequencing of different treatments. Alternative therapy, particularly if the patient is at risk of toxicity, could also be considered.
We feel that the combination of genetic data (PRRT prediction signature) with sensitive imaging information (68Ga-DOTATATE/DOTATOC PET/CT) for localization and disease extent will result in a multifaceted clinical tool that provides an efficient method to effectively stratify and appropriately treat patients with PRRT.
References
Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61–72.
Pavel M, Valle JW, Eriksson B, Rinke A, Caplin M, Chen J, et al. ENETS consensus guidelines for the standards of care in neuroendocrine neoplasms: systemic therapy - biotherapy and novel targeted agents. Neuroendocrinology. 2017;105:266–80.
Faggiano A, Lo Calzo F, Pizza G, Modica R, Colao A. The safety of available treatments options for neuroendocrine tumors. Expert Opin Drug Saf. 2017;16:1149–61.
Oberg K, Krenning E, Sundin A, Bodei L, Kidd M, Tesselaar M, et al. A Delphic consensus assessment: imaging and biomarkers in gastroenteropancreatic neuroendocrine tumor disease management. Endocrine connections. 2016;5:174–87.
Bodei L, Kwekkeboom DJ, Kidd M, Modlin IM, Krenning EP. Radiolabeled Somatostatin analogue therapy of gastroenteropancreatic cancer. Semin Nucl Med. 2016;46:225–38.
Cives M, Strosberg J. Radionuclide therapy for Neuroendocrine tumors. Curr Oncol Rep. 2017;19:9.
Brabander T, van der Zwan WA, Teunissen JJM, Kam BLR, Feelders RA, de Herder WW, et al. Long-term efficacy, survival, and safety of [177Lu-DOTA0,Tyr3]octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. J Clin Oncol : Off J Am Assoc Cancer Res. 2017;23:4617–24.
Sansovini M, Severi S, Ambrosetti A, Monti M, Nanni O, Sarnelli A, et al. Treatment with the radiolabelled somatostatin analog Lu-DOTATATE for advanced pancreatic neuroendocrine tumors. Neuroendocrinology. 2013;97:347–54.
Ezziddin S, Khalaf F, Vanezi M, Haslerud T, Mayer K, Al Zreiqat A, et al. Outcome of peptide receptor radionuclide therapy with 177Lu-octreotate in advanced grade 1/2 pancreatic neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2014;41:925–33.
Mariniello A, Bodei L, Tinelli C, Baio SM, Gilardi L, Colandrea M, et al. Long-term results of PRRT in advanced bronchopulmonary carcinoid. Eur J Nucl Med Mol Imaging. 2016;43:441–52.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376:125–35.
Kwekkeboom DJ, Kam BL, van Essen M, Teunissen JJ, van Eijck CH, Valkema R, et al. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17:R53–73.
Imhof A, Brunner P, Marincek N, Briel M, Schindler C, Rasch H, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29:2416–23.
Sabet A, Dautzenberg K, Haslerud T, Aouf A, Sabet A, Simon B, et al. Specific efficacy of peptide receptor radionuclide therapy with (177)Lu-octreotate in advanced neuroendocrine tumours of the small intestine. Eur J Nucl Med Mol Imaging. 2015;42:1238–46.
Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. Clin Canc Res: Off J Am Assoc Cancer Res. 2008;26:2124–30.
Kidd M, Drozdov I, Modlin I. Blood and tissue neuroendocrine tumor gene cluster analysis correlate, define hallmarks and predict disease status. Endocr Relat Cancer. 2015;22:561–75.
Bodei L, Kidd M, Modlin IM, Severi S, Drozdov I, Nicolini S, et al. Measurement of circulating transcripts and gene cluster analysis predicts and defines therapeutic efficacy of peptide receptor radionuclide therapy (PRRT) in neuroendocrine tumors. Eur J Nucl Med Mol Imaging. 2016;43:839–51.
Ballman KV. Biomarker: predictive or prognostic? J Clin Oncol. 2015;33:3968–71.
Beaver JA, Tzou A, Blumenthal GM, McKee AE, Kim G, Pazdur R, et al. An FDA perspective on the regulatory implications of complex signatures to predict response to targeted therapies. Clin Cancer Res. 2017;23:1368–72.
Cwikla JB, Bodei L, Kolasinska-Cwikla A, Sankowski A, Modlin IM, Kidd M. Circulating transcript analysis (NETest) in GEP-NETs treated with Somatostatin analogs defines therapy. J Clin Endocrinol Metab. 2015;100:E1437–45.
Bodei L, Cremonesi M, Grana CM, Fazio N, Iodice S, Baio SM, et al. Peptide receptor radionuclide therapy with (177)Lu-DOTATATE: the IEO phase I-II study. Eur J Nucl Med Mol Imaging. 2011;38:2125–35.
Bosman FT. WHO classification of tumours of the digestive system. 4th ed. Lyon: IARC Press, 2010
Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC (ed). Pathology and genetics of tumours of the lung, pleura, thymus and heart. Geneva: WHO Press, 2004
Li XJ, Hayward C, Fong PY, Dominguez M, Hunsucker SW, Lee LW, et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Sci Transl Med. 2013;5:207ra142.
Vachani A, Pass HI, Rom WN, Midthun DE, Edell ES, Laviolette M, et al. Validation of a multiprotein plasma classifier to identify benign lung nodules. J Thorac Oncol. 2015;10:629–37.
Kaijser J, Sayasneh A, Van Hoorde K, Ghaem-Maghami S, Bourne T, Timmerman D, et al. Presurgical diagnosis of adnexal tumours using mathematical models and scoring systems: a systematic review and meta-analysis. Hum Reprod Update. 2014;20:449–62.
Vickers AJ, Van Calster B, Steyerberg EW. Net benefit approaches to the evaluation of prediction models, molecular markers, and diagnostic tests. BMJ. 2016;352:i6. https://doi.org/10.1136/bmj.i6.
Frank R, Hargreaves R. Clinical biomarkers in drug discovery and development. Nat Rev Drug Discov. 2003;2:566–80.
Oksuz MO, Winter L, Pfannenberg C, Reischl G, Mussig K, Bares R, et al. Peptide receptor radionuclide therapy of neuroendocrine tumors with (90)Y-DOTATOC: is treatment response predictable by pre-therapeutic uptake of (68)Ga-DOTATOC? Diagnos Interven Imaging 2014;95:289-300.
Gabriel M, Oberauer A, Dobrozemsky G, Decristoforo C, Putzer D, Kendler D, et al. 68Ga-DOTA-Tyr3-octreotide PET for assessing response to somatostatin-receptor-mediated radionuclide therapy. J Nucl Med: Off Publ, Soc Nucl Med. 2009;50:1427–34.
Blaickner M, Baum RP. Relevance of PET for pretherapeutic prediction of doses in peptide receptor radionuclide therapy. PET Clin. 2014;9:99–112.
Wu Y, Pfeifer AK, Myschetzky R, Garbyal RS, Rasmussen P, Knigge U, et al. Induction of Anti-Tumor Immune Responses by Peptide Receptor Radionuclide Therapy with (177)Lu-DOTATATE in a Murine Model of a Human Neuroendocrine Tumor. Diagnostics (Basel, Switzerland). 2013;3:344–55.
Yang Z, Tang LH, Klimstra DS. Effect of tumor heterogeneity on the assessment of Ki67 labeling index in well-differentiated neuroendocrine tumors metastatic to the liver: implications for prognostic stratification. Am J Surg Pathol. 2011;35:853–60.
Sansovini M, Severi S, Ianniello A, Nicolini S, Fantini L, Mezzenga E, et al. Long-term follow-up and role of FDG PET in advanced pancreatic neuroendocrine patients treated with 177Lu-D OTATATE. Eur J Nucl Med Mol Imaging. 2017;44:490–9.
Ezziddin S, Attassi M, Yong-Hing CJ, Ahmadzadehfar H, Willinek W, Grunwald F, et al. Predictors of long-term outcome in patients with well-differentiated gastroenteropancreatic neuroendocrine tumors after peptide receptor radionuclide therapy with 177Lu-octreotate. J Nucl Med. 2014;55:183–90.
Ezziddin S, Sabet A, Heinemann F, Yong-Hing CJ, Ahmadzadehfar H, Guhlke S, et al. Response and long-term control of bone metastases after peptide receptor radionuclide therapy with (177)Lu-octreotate. J Nucl Med. 2011;52:1197–203.
Haug AR, Auernhammer CJ, Wangler B, Schmidt GP, Uebleis C, Goke B, et al. 68Ga-DOTATATE PET/CT for the early prediction of response to somatostatin receptor-mediated radionuclide therapy in patients with well-differentiated neuroendocrine tumors. J Nucl Med: Off Publ, Soc Nucl Med. 2010;51:1349–56.
Yang Z, Tang LH, Klimstra DS. Gastroenteropancreatic neuroendocrine neoplasms: historical context and current issues. Semin Diagn Pathol. 2013;30:186–96.
Valli A, Rodriguez M, Moutsianas L, Fischer R, Fedele V, Huang HL, et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1alpha-dependent and -independent pathways. Oncotarget. 2015;6:1920–41.
Olsson AH, Yang BT, Hall E, Taneera J, Salehi A, Nitert MD, et al. Decreased expression of genes involved in oxidative phosphorylation in human pancreatic islets from patients with type 2 diabetes. Eur J Endocrinol. 2011;165:589–95.
Day TF, Mewani RR, Starr J, Li X, Chakravarty D, Ressom H, et al. Transcriptome and proteome analyses of TNFAIP8 knockdown cancer cells reveal new insights into molecular determinants of cell survival and tumor progression. Methods Mol Biol. 2017;1513:83–100.
Hill RP. The changing paradigm of tumour response to irradiation. Br J Radiol. 2017;90:20160474.
Kidd M, Modlin IM. Therapy: the role of liquid biopsies to manage and predict PRRT for NETs. Nat Rev Gastroenterol Hepatol. 2017;14:331–2.
Funding
Work was performed with an unrestricted grant from the LuGenIum consortium of independent research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all participants in the study.
Conflict of interest
LB and GP have received consultancy fees from Ipsen and Advanced Accelerator Applications (AAA). EPK and DJK received support from AAA outside the submitted work. MK is employed by Wren Laboratories that undertook the molecular testing. IMM is a consultant for Wren Laboratories. All others have nothing to disclose.
Electronic supplementary material
ESM 1
(DOCX 367 kb)
Rights and permissions
About this article

Cite this article
Bodei, L., Kidd, M.S., Singh, A. et al. PRRT genomic signature in blood for prediction of 177Lu-octreotate efficacy. Eur J Nucl Med Mol Imaging 45, 1155–1169 (2018). https://doi.org/10.1007/s00259-018-3967-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-018-3967-6