
Abstract
Purpose
The aim of this study was to synthesize and preclinically evaluate an 18F-PSMA positron emission tomography (PET) tracer. Prostate-specific membrane antigen (PSMA) specificity, biodistribution, and dosimetry in healthy and tumor-bearing mice were determined.
Methods
Several conditions for the labeling of 18F-PSMA-11 via 18F-AlF-complexation were screened to study the influence of reaction temperature, peptide amount, ethanol volume, and reaction time. After synthesis optimization, biodistribution and dosimetry studies were performed in C57BL6 mice. For proof of PSMA-specificity, mice were implanted with PSMA-negative (PC3) and PSMA-positive (LNCaP) tumors in contralateral flanks. Static and dynamic microPET/computed tomography (CT) imaging was performed.
Results
Quantitative labeling yields could be achieved with >97 % radiochemical purity. The 18F-PSMA-11 uptake was more than 24-fold higher in PSMA-high LNCaP than in PSMA-low PC3 tumors (18.4 ± 3.3 %ID/g and 0.795 ± 0.260 %ID/g, respectively; p < 4.2e-5). Results were confirmed by ex vivo gamma counter analysis of tissues after the last imaging time point. The highest absorbed dose was reported for the kidneys. The maximum effective dose for an administered activity of 200 MBq was 1.72 mSv.
Conclusion
18F-PSMA-11 using direct labeling of chelate-attached peptide with aluminum-fluoride detected PSMA-expressing tumors with high tumor-to-liver ratios. The kidneys were the dose-limiting organs. Even by applying the most stringent dosimetric calculations, injected activities of up to 0.56 GBq are feasible.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostate-specific membrane antigen (PSMA) expression is associated with prostate cancer progression and prognosis [1]. PSMA-targeting positron emission tomography (PET) probes have become available for prostate cancer imaging [2–4]. Promising tracers include radiolabeled antibodies, aptamers, and small molecule inhibitors. Of these, 68Ga-labeled PSMA-ligands have been extensively studied [5–15]. However, 68Ga-labeled compounds are produced with generators that provide limited activity per synthesis. Thus, depending on the age of the generator, only 1–4 patient doses per elution can be produced. In contrast, much larger doses of 18F-labeled tracers can be obtained by one cyclotron production. Thus, 18F-compounds are suitable for off-site production and distribution over longer distances. As such, the introduction of 18F-choline analogs for imaging prostate cancer [16] led to their widespread clinical use in Europe when compared to 11C-choline [17].
Recent data suggest that radiolabeled PSMA ligands detect PSMA-expressing tumors with a higher sensitivity than choline derivatives. Moreover, a significant impact on patient management has been demonstrated [7]. Therefore, an urgent need exists to establish a straightforward method to label PSMA ligands with 18F.
Previous methods to synthesize 18F-labeled PSMA ligands have been successful [18–20]. However, these compounds are patented and not commonly accessible [18, 20], involve complex, multistep syntheses [18, 20], or haven’t been investigated in preclinical models [19]. Development of direct labeling of peptides via aluminum-fluoride (Al18F) chelation allows for a simple, not patented, and therefore freely accessible synthesis of 18F-PSMA PET probes [19, 21–23].
The aims of this study were therefore (1) to establish the synthesis of an 18F-PSMA tracer using direct labeling via Al18F-chelation, and (2) to determine its in-vivo PSMA-specificity, biodistribution, and dosimetry in healthy and tumor-bearing mice. We chose the up-to-date most successful PSMA-targeted peptide tracer, PSMA-11. Complexed to natGa, the Ki value of PSMA-11 in a cell-based assay towards PSMA was determined to be 12.0 ± 2.8 nM and is readily taken up in PSMA-positive LNCaP cells.
Materials and methods
Synthesis development
Chemicals: Glu-urea-Lys (Ahx)-HBED-CC (PSMA-11) was purchased from ABX (Radeberg, Germany). AlCl3.6H2O, sodium acetate trihydrate, and acetic acid were all metal-free. HPLC eluents, water, acetonitrile, and trifluoroacetic acid (TFA) were of high-grade purity. Sep-Pak Accell Plus QMA and Sep-Pak C18-Light cartridges were from Waters. No-carrier-added 18F-fluoride was produced with the PETtrace cyclotron (GE Medical Systems, Uppsala) via an 18O (p,n) 18F reaction.
Preparation of 18F-fluoride: QMA cartridge was conditioned with 5 mL of 0.5 M sodium acetate, followed by 10 mL of water (MilliQ, 18.5 MΩ). 18F-fluoride solution was loaded onto the cartridge and after washing with 5 mL water, Na18F was eluted by 200 μL of acetate buffer, pH 4.5 in one fraction (70–80 MBq), and by 300 μL into a second fraction (600–900 MBq).
Preparation of stock solutions: Sodium acetate/acetic acid buffer 0.5 M, pH 4.5 and 0.2 M, pH 6.3 were also prepared starting from 0.5 M solution of each component and mixing to obtain the desired final pH. PSMA-11 was dissolved in water [1 mg/500 μL] and aliquots were stored frozen (−18 °C). AlCl3.6H2O 0.01 M was prepared in 0.05 M acetate buffer (pH 4) and stored at 4 °C.
Synthesis of [Al18F] PSMA-11: For testing and optimizing the selected radiosynthesis parameters, 100 μL of Na18F (200–300 MBq) solution from the second fraction was added to 3 μL of 0.01 M AlCl3.6H2O (30nM) and 10–45 μL (20–90 μg) of PSMA-11. Acetate buffer and ethanol (30–200 μL) were finally added to a final reaction volume of 300 μL and the mixture was incubated at 50 °C for 5–20 min. Aliquots of the mixture at different reaction time points were injected into high-performance liquid chromatography (HPLC) equipped with a reversed-phase column (Eclipse XDB-C8, 5 mm, 4.6 × 150 mm from Agilent (CA, USA) with isocratic elution (solvent A: 85 % water with 0.1 % TFA, solvent B: 15 % CH3CN with 0.1%TFA) at a flow rate of 1 mL/min. HPLC was connected with a radiometric and UV detector set at 220 nm. The reaction mixture after incubation was trapped onto a Sep Pak C18 light cartridge, washed with saline, and eluted with 500 μL of ethanol. The final product was diluted with 3 mL of saline or acetate buffer 0.2 M pH 6.3 to verify the most suitable condition for stability. Quality control including absence of free fluoride for the final product was performed via radioHPLC. For animal experiments, an aliquot of the product solution containing 10 % ethanol was diluted with sterile saline. To check for stability, the reaction mixture after incubation was trapped onto a Sep Pak C18 light, the cartridge was washed with saline, and finally it was eluted with 500 μL of ethanol and 3 mL of saline or acetate buffer 0.2 M at various pH values to verify the most suitable condition for stability.
Murine tumor models
Animal studies were approved by the UCLA Animal Research Committee and were carried out according to the guidelines of the Division of Laboratory Animal Medicine at UCLA. Six- to seven-week-old male C57BL6 mice were used to determine probe dosimetry. For tumor models, prostate cancer cell lines PC3 (PSMA-negative) and LNCaP (PSMA-positive), gifts from the laboratory of Robert Reiter at UCLA, were resuspended in 50 % phosphate-buffered saline and 50 % MatrigelTM (354234, BD Biosciences) and 1 × 106 cells each were injected subcutaneously on contralateral flanks of four male six- to seven-week-old NOD SCID gamma-null (NSG) mice.
In-vitro stability assay
The in-vitro stability of 18F-PSMA-11 (20 μL) was determined in mouse and human blood plasma (180 μL) at 37 °C at time points 30, 60, 90, and 120 min. At each time point, an aliquot of 50 μL of the incubation mixture was collected, treated with 50 μL ice-cooled MeCN, centrifuged, and the supernatant was analyzed by thin-layer radiochromatography (radio-TLC). Results have been confirmed by radio-HPLC.
Animal PET/CT image acquisition and analysis
C57BL6 and tumor-bearing NSG mice, engrafted with tumor cells approximately 2 weeks prior, underwent 18F-PSMA and 18F-FDG microPET/computed tomography (CT) imaging (GENISYS 8 PET/CT, Sofie Biosciences). The G8 PET/CT is an integrated scanner with a PET subsection optimized for mouse imaging with an energy window of 150–650 keV and peak sensitivity of approximately 14 % at the center of FOV. The intrinsic detector spatial resolution is 1.5 mm full-width at half-maximum (FWHM) in the transverse and axial directions.
The CT section consists of a gantry and flywheel that uses a 50 kVp, 200 μA x-ray source and flat-panel detector. The CT acquires images in a continuous-rotation mode with 720 projections at 55 msec per projection, and reconstructed using a Feldkamp algorithm.
Mice were anesthetized under 2 % isoflurane, placed in a heated G8 imaging chamber and catheterized by the tail vein. The chamber was placed in the PET section of the G8. PET data were acquired at 0–1 h (dynamic), 2 h, 4 h and 6 h after bolus 18F-PSMA-11 injection (approximately 1.4 MBq, 0.21 nmol). Animals were conscious and kept warm in between imaging time points. For 18F-FDG, mice were fasted overnight, followed by intravenous injection of 18F-FDG (approximately 1.4 MBq) and PET/CT imaging after 1 h unconscious uptake. The list-mode data-framing sequence for the 0–1 h dynamic scan was 1 × 10 s, 4 × 15 s, 8 × 30 s, 5 × 60 s, 4 × 300 s, 3 × 600 s.
Maximum-likelihood expectation maximization was used to create the final image volumes as recommended by the vendor. All images were corrected for photon attenuation. Images were analyzed using AMIDE version 1.0.5 [24] and OsiriX version 3.8 imaging software. Images were quantified using 2-mm diameter regions-of-interest (ROI) except for kidneys, bladder, and whole body, which were determined using 3D isocontours, and reported in Bq/mL.
Biodistribution analysis
18F-PSMA-11 biodistribution in wild-type C57BL6 mice was determined by microPET/CT imaging as described and confirmed by ex-vivo analysis. For the latter, each mouse was sacrificed following its last PET imaging. Tissues were collected, weighed, and radioactivity counted on a Packard Cobra II Auto Gamma Counter with decay correction to time of 18F-PSMA-11 injection. Data were normalized to mass of tissue.
Dosimetry analysis
From quantitative 18F-PSMA-11 microPET scans of male C57BL6 mice, the amount of activity in selected organs was quantified and absorbed doses were calculated based on the respective time-integrated activity coefficients. The dose extrapolation to humans involved the scaling of the biodistributions and the subsequent calculation of the absorbed doses from them. The biodistribution scaling was performed by two alternative methods. Method 1 was based on the assumption that the residence time for the same organ is the same in mice and humans [25, 26]. Method 2 considered a relative mass scaling where the specific activity in a certain human organ is equal to the specific activity in the same mouse organ multiplied by the ratio of the body mass of human and mouse [25, 27, 28]. Time-integrated activity coefficients (TIACs or “Residence Times”, RT) were calculated with using the software solution NUKFIT as described by Kletting et al., choosing the optimal fit functions as proposed by the code [29]. The dose calculation was performed for a selected group of organs using OLINDA/EXM V1.1 [30]. Details on the methodology used for extrapolating the mouse data to humans are provided in the supplementary information.
Statistical analysis
Data are presented as mean ± standard deviation. All p values were determined with unpaired, two-tailed T tests, and values less than 0.05 were considered to be statistically significant. GraphPad Prism 6 and Origin 2015 software were used to calculate statistics and generate graphs.
Results
Radioligand synthesis
During the synthesis setup, samples were taken at 5, 10, 15, and 20 min. In most conditions, no changes after 10 min of reaction time were observed. The presence of ethanol dramatically increased the percentage of Al18F binding to the peptide (Fig. 1). In the absence of ethanol, approximately 30 % of the labeling was obtained at 100 °C with 90 μg of peptide. Increasing the ethanol concentration (from 0 to 60 %) allowed higher binding values also in the presence of less peptide amount and lower temperature (50 °C). After incubating 20 μg of peptide in the presence of 150–180 μL (50–60 %) of ethanol, 70–90 % of binding was obtained, with a slight decrease using 200 μL (67 %) of ethanol. A typical chromatogram of the reaction mixture is shown in Fig. 2. An increase of Al3+ concentration from 0.10 to 0.33 nmol/μL led to a decrease of labeling efficiency to approximately 50 % (data not shown). The stability was confirmed in a 3 h stability test. The results showed that radiochemical purity > 97 % was observed in buffered solution at pH 6.8 in comparison with unbuffered saline formulation, which showed a roughly 20 % decrease in radiochemical purity after 3 h.

Percentage of Al18F binding to PSMA-11 in various reaction conditions. Red line demonstrates the decrease of peptide concentration as a function of the ethanol concentration

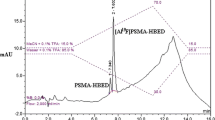
Radio-HPLC chromatogram of the reaction product in the chromatographic condition (20 μg of peptide, 60 % ethanol, 50 °C, shaking). First peak confirmed as Al18F2+ (data not shown), second peak assigned via retention time comparison with DKFZ-PSMA-11
For the final PET imaging studies, aliquoted 25 μg precursor, 3 μL AlCl3 (0.01 M), 260–740 MBq Na18F (100 μL), and 150 μL ethanol were mixed and incubated for 10 min at 50 °C on a shaking plate. After purification of [Al18F] PSMA-11 over a C18 Sep Pak light, the product was eluted with 0.5 mL ethanol and diluted with 4.5 mL acetate buffer (0.2 M, pH 6.8). The product solution was filtered through a sterile filter into the final product vial, yielding a radiochemical purity of >99 % with a specific activity of at least >9.9 GBq/μmol. The final product was stable in mouse and human plasma for a time course of up to 2 h with 91 and 94 % intact compound, respectively.
Biodistribution
The highest 18F-PSMA-11 tissue uptake at 1 h post-injection by microPET imaging was in the kidneys in all mice (119.3 ± 16.5 %ID/g) and bladder (77.4 ± 39.1 %ID/g) (Fig. 3). All other tissues exhibited relatively low uptake. Results were confirmed by ex-vivo tissue analysis.

MicroPET/CT imaging. a Volume rendering and tissue biodistribution of 18F-PSMA-11 in C57BL6. b Volume rendering of 18F-PSMA-11 and c MIP image of 18F-FDG in NSG mice engrafted with PC3 (right) and LNCaP (left) tumor cells. Shown are representative microPET/CT images 1 h post-injection of PET probe. Error bars are standard deviations. Tumors are delineated in dashed circles
Time activity curves derived from PET imaging demonstrated fast uptake and retention in kidneys, minimal retention in bone, wash-out from other tissues (linear decline on logarithmic plot), and rapid renal clearance as indicated by significant bladder activity.
Tumor imaging
18F-PSMA-11 uptake was significantly higher in PSMA-high LNCaP (Fig. 3) than in PSMA-low PC3 tumors (55.7 ± 11.8 %ID/g and 3.1 ± 0.9 %ID/g, respectively; p < 3.6e-4). Time-activity curves derived from PET imaging showed early 18F-PSMA-11 saturation in LNCaP tumors. Probe uptake was 24 times higher in LNCaP than in PC3 tumors. Time-activity curves in PC3 tumors were nearly identical to those for background tissues such as muscle and liver, and were in general only 1–2 times higher. Results were confirmed with ex-vivo gamma counter analysis of tissues after the last imaging time point. In contrast, 18F-FDG uptake only tended to differ between LNCaP and PC3 tumors (3.1 ± 0.6 %ID/g and 4.2 ± 0.7 %ID/g, respectively; p = 0.07).
Dosimetric calculations
Absorbed doses for 18F-PSMA-11 in humans were extrapolated from mouse PET biodistribution data using two extrapolation methods, method 1 and method 2 (see supplemental file). The time-integrated activity coefficients (TIACs) are summarized in Table 1. A full list of the corresponding mean absorbed doses is provided in Table 2.
The highest TIAC was observed for the kidneys and bone based on method 1 (mean RTkidney: 0.141 ± 0.033 h, mean RTbone: 0.045 ± 0.013 h). Based on method 2, the highest TIACs were observed for the bone, muscle, and kidneys (mean RTbone: 0.114 ± 0.028 h, mean RTmuscle: 0.077 ± 0.018 h, and mean RTkidney: 0.042 ± 0.009 h). The total absorbed doses are summarized in Table 2. The highest absorbed dose was 8.87 × 10−2 mGy/MBq and 2.87 × 10−2 mGy/MBq for the kidneys in method 1 and method 2, respectively. All other organs showed significantly lower absorbed doses (Fig. 4). Kidneys were the dose-limiting organ, and on average, the maximum administered human activity limit is calculated to be 564 MBq (Method 1) and 1,742 MBq (Method 2) (FDA Code of Federal Regulations 21CFR361.1). In addition, the effective dose per unit activity was calculated. However, the quantity “effective dose” can only be applied to the description of stochastic radiation effects and organ-absorbed doses of less than 1 Gy. The mean extrapolated effective doses are 8.59 × 10−3 ± 7.46 × 10−4 mSv/MBq (Method 1) and 6.23 × 10−3 ± 5.74 × 10−4 mSv/MBq (Method 1). This corresponds to effective doses of 1.72 mSv (Method 1) and 1.25 mSv (Method 2) for an administered activity of 200 MBq.

Non-decay-corrected time activity curves determined by microPET imaging and accounting for total organ weights
Discussion
Here we report the successful synthesis of 18F-PSMA-11 via Al18F-complexation. High radiochemical purity was achieved and the tracer was stable in mouse and human plasma with 91 and 94 % intact compound after 2 h incubation at 37 °C. Specificity for PSMA was confirmed by the significantly higher uptake in PSMA-positive relative to PSMA-negative xenografts. In contrast, 18F-FDG verified tissue viability, but was not able to reliably differentiate between the two tumor types. Biodistribution and dosimetry studies identified the kidneys as the critical organ. The most conservative calculations would permit human administration of up to 564 MBq. This results in much lower effective doses than those associated with other clinically available PSMA-targeting PET probes. However, results acquired in mice can only be extrapolated to humans with caution.
In contrast to other promising 18F-labeled PSMA-directed compounds [31, 32] 18F-PSMA-11 will be freely accessible, as this ligand is developed without commercial interest. Especially in Europe, Asia, and South America where the radiopharmaceutical is very often not adequately reimbursed, this may be of major importance and a significant driver for clinical adoption.
Moreover, the synthesis of 18F-PSMA-11 is a very simple one-pot, one-step synthesis following the recently published findings by Malik et al [19]. The simplicity is beneficial compared to the more complex multistep syntheses of 18F-DCFBX [20] and 18F-DCFPyL [18].
We found ethanol to strongly influence labeling efficiency and achieved the highest yield with 60 % ethanol in the reaction mixture. This is because ethanol and other polar solvents like acetonitrile and isopropanol facilitate the binding by breaking the hydration sphere of the metal in solution.
Biodistribution of 18F-PSMA-11 by microPET imaging of non-tumor-bearing C57BL6 mice demonstrated fast renal clearance as confirmed by kidney and bladder uptake within the first hour of probe administration. At later time points the kidney signal arises from the renal cortex, which is not unexpected since PSMA is expressed in renal tubules [33]. Bone uptake was at least one order of magnitude lower than kidney uptake, as confirmed by ex-vivo analysis of femur bone versus bone marrow. This suggests a minimal degree of de-fluorination of 18F-PSMA-11, as PSMA is only expressed in renal tubules, prostate epithelium, and some intestinal tissues [33] but not in the bone. Whether this will affect human image quality and diagnostic information has yet to be determined.
The high 18F-PSMA-11 uptake in LNCaP and low uptake in PC3 tumors is explained by the high and low PSMA expression of the corresponding tumors [3, 34]. Both xenografts demonstrated 18F-FDG avidity, confirming viable tumor tissue and further supporting the target selectivity of 18F-PSMA-11. The tendency towards higher 18F-FDG uptake values in the PC3 cells most likely reflects the previously published higher aggressiveness and shorter doubling time [35] compared to LNCaP [36].
The favorable biodistribution resulted an estimated maximum effective dose of 1.7 mSv for an injection of 200 MBq 18F-PSMA-11. Thus, the radiation dose is significantly lower than of currently used 18F- and 68Ga-labeled compounds [32, 37].
The substantially higher prostate cancer detection rates of 68Ga-PSMA compared to the established 18F-choline [7, 38] has already resulted in its rapid clinical acceptance in Europe. The favorable biodistribution and dosimetry, the reliable and simple synthesis and the high PSMA selectivity of 18F-PSMA-11 via Al18F-complexation underline the high translational potential of this compound. No safety concerns arise from the use of aluminum chloride in the PET drug preparation, although aluminum is considered to have neurotoxic effects at higher doses. Normal serum aluminum concentration is measured to be 0.1 μmol/L [39]. Our synthesis starts with 0.03 μmol AlCl3, whereas most is washed out during the C18 cartridge purification step.
As the availability of 68Ga-labeled PSMA ligands is limited due to the shorter half-life and the generator size-dependent product output, there is a need for cyclotron-produced PSMA PET tracers such as 18F-PSMA-11. Despite a minimal amount of defluorination, the preclinical results strongly suggest that the clinical translation of 18F-PSMA-11 is feasible and warranted. Future studies comparing 18F-PSMA-11 with 68Ga-PSMA-11, 18F-DCFBX, and 18F-DCPyL are mandatory to elucidate which of the currently available compounds provides superior specific activities, biodistribution, and tumor-to-background ratios.
Conclusion
The clinical need for early detection of prostate cancer in patients with biochemical recurrence and the selectivity of PSMA ligands drive their development and translation. Synthesis of 18F-PSMA-11 by direct-labeling of chelate-attached-peptides with aluminum fluoride provides high radiochemical purity and product yields.
References
Ghosh A, Heston WD. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J Cell Biochem. 2004;91:528–39. doi:10.1002/jcb.10661.
Eder M, Schafer M, Bauder-Wust U, et al. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug Chem. 2012;23:688–97. doi:10.1021/bc200279b.
Weineisen M, Schottelius M, Simecek J, et al. 68Ga- and 177Lu-labeled PSMA I&T: optimization of a PSMA targeted theranostic concept and first proof of concept human studies. J Nucl Med. 2015. doi:10.2967/jnumed.115.158550.
Mease RC, Foss CA, Pomper MG. PET imaging in prostate cancer: focus on prostate-specific membrane antigen. Curr Top Med Chem. 2013;13:951–62.
Schiavina R, Ceci F, Romagnoli D, et al. Ga-PSMA-PET/CT-guided salvage retroperitoneal lymph node dissection for disease relapse after radical prostatectomy for prostate cancer. Clin Genitourin Cancer. 2015. doi:10.1016/j.clgc.2015.06.004.
Budaus L, Leyh-Bannurah SR, Salomon G, et al. Initial experience of Ga-PSMA PET/CT imaging in high-risk prostate cancer patients prior to radical prostatectomy. Eur Urol. 2015. doi:10.1016/j.eururo.2015.06.010.
Morigi JJ, Stricker PD, van Leeuwen PJ, et al. Prospective comparison of 18F-fluoromethylcholine versus 68Ga-PSMA PET/CT in prostate cancer patients who have rising PSA after curative treatment and are being considered for targeted therapy. J Nucl Med. 2015;56:1185–90. doi:10.2967/jnumed.115.160382.
Jadvar H. PSMA PET, in prostate cancer. J Nucl Med. 2015;56:1131–2. doi:10.2967/jnumed.115.157339.
Ceci F, Uprimny C, Nilica B, et al. (68)Ga-PSMA PET/CT for restaging recurrent prostate cancer: which factors are associated with PET/CT detection rate? Eur J Nucl Med Mol Imaging. 2015;42:1284–94. doi:10.1007/s00259-015-3078-6.
Eiber M, Maurer T, Souvatzoglou M, et al. Evaluation of hybrid (6)(8)Ga-PSMA ligand PET/CT in 248 patients with biochemical recurrence after radical prostatectomy. J Nucl Med. 2015;56:668–74. doi:10.2967/jnumed.115.154153.
Kabasakal L, Demirci E, Ocak M, et al. Evaluation of PSMA PET/CT imaging using a 68Ga-HBED-CC ligand in patients with prostate cancer and the value of early pelvic imaging. Nucl Med Commun. 2015;36:582–7. doi:10.1097/MNM.0000000000000290.
Chakraborty PS, Kumar R, Tripathi M, Das CJ, Bal C. Detection of brain metastasis with 68Ga-labeled PSMA ligand PET/CT: a novel radiotracer for imaging of prostate carcinoma. Clin Nucl Med. 2015;40:328–9. doi:10.1097/RLU.0000000000000709.
Avanesov M, Karul M, Derlin T. (68)Ga-PSMA as a new tracer for evaluation of prostate cancer: comparison between PET-CT and PET-MRI in biochemical recurrence. Radiologe. 2015;55:89–91. doi:10.1007/s00117-014-2792-6.
Afshar-Oromieh A, Avtzi E, Giesel FL, et al. The diagnostic value of PET/CT imaging with the (68)Ga-labelled PSMA ligand HBED-CC in the diagnosis of recurrent prostate cancer. Eur J Nucl Med Mol Imaging. 2015;42:197–209. doi:10.1007/s00259-014-2949-6.
Uprimny C, Kroiss A, Nilica B, et al. (68)Ga-PSMA ligand PET versus (18)F-NaF PET: evaluation of response to (223)Ra therapy in a prostate cancer patient. Eur J Nucl Med Mol Imaging. 2015;42:362–3. doi:10.1007/s00259-014-2922-4.
Iwata R, Pascali C, Bogni A, Furumoto S, Terasaki K, Yanai K. [18F]fluoromethyl triflate, a novel and reactive [18F]fluoromethylating agent: preparation and application to the on-column preparation of [18F]fluorocholine. Appl Radiat Isot. 2002;57:347–52.
Evangelista L, Cervino AR, Guttilla A, Zattoni F, Cuccurullo V, Mansi L. (1)(8)F-fluoromethylcholine or (1)(8)F-fluoroethylcholine pet for prostate cancer imaging: which is better? a literature revision. Nucl Med Biol. 2015;42:340–8. doi:10.1016/j.nucmedbio.2014.12.019.
Chen Y, Pullambhatla M, Foss CA, et al. 2-(3-{1-Carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pen tanedioic acid, [18F]DCFPyL, a PSMA-based PET imaging agent for prostate cancer. Clin Cancer Res. 2011;17:7645–53. doi:10.1158/1078-0432.CCR-11-1357.
Malik N, Baur B, Winter G, Reske SN, Beer AJ, Solbach C. Radiofluorination of PSMA-HBED via AlF chelation and biological evaluations in vitro. Mol Imaging Biol. 2015. doi:10.1007/s11307-015-0844-6.
Mease RC, Dusich CL, Foss CA, et al. N-[N-[(S)-1,3-Dicarboxypropyl]carbamoyl]-4-[18F]fluorobenzyl-L-cysteine, [18F]DCFBC: a new imaging probe for prostate cancer. Clin Cancer Res. 2008;14:3036–43. doi:10.1158/1078-0432.CCR-07-1517.
McBride WJ, Sharkey RM, Goldenberg DM. Radiofluorination using aluminum-fluoride (Al18F). EJNMMI Res. 2013;3:36. doi:10.1186/2191-219X-3-36.
McBride WJ, D’Souza CA, Karacay H, Sharkey RM, Goldenberg DM. New lyophilized kit for rapid radiofluorination of peptides. Bioconjug Chem. 2012;23:538–47. doi:10.1021/bc200608e.
D’Souza CA, McBride WJ, Sharkey RM, Todaro LJ, Goldenberg DM. High-yielding aqueous 18F-labeling of peptides via Al18F chelation. Bioconjug Chem. 2011;22:1793–803. doi:10.1021/bc200175c.
Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging. 2003;2:131–7.
Repetto-Llamazares AH, Larsen RH, Mollatt C, Lassmann M, Dahle J. Biodistribution and dosimetry of (177)Lu-tetulomab, a new radioimmunoconjugate for treatment of non-Hodgkin lymphoma. Curr Radiopharm. 2013;6:20–7.
Sparks RB, Aydogan B. Comparison of the effectiveness of some common animal data scaling techniques in estimating human radiation dose. . In: Stelson A, Stabin M, Sparks R, editors. Sixth International Radiopharmaceutical Dosimetry Symposium 1999. p. 705–16.
International Commission on Radiological Protection (ICRP). Publication 89: basic anatomical and physiological data for use in radiological protection—reference values. Ann ICRP. 2002;32:1–278.
Bitar A, Lisbona A, Thedrez P, et al. A voxel-based mouse for internal dose calculations using Monte Carlo simulations (MCNP). Phys Med Biol. 2007;52:1013–25. doi:10.1088/0031-9155/52/4/010.
Kletting P, Schimmel S, Kestler HA, et al. Molecular radiotherapy: the NUKFIT software for calculating the time-integrated activity coefficient. Med Phys. 2013;40:102504. doi:10.1118/1.4820367.
Stabin MG, Sparks RB, Crowe E. OLINDA/EXM: the second-generation personal computer software for internal dose assessment in nuclear medicine. J Nucl Med. 2005;46:1023–7.
Dietlein M, Kobe C, Kuhnert G, et al. Comparison of [F]DCFPyL and [Ga]Ga-PSMA-HBED-CC for PSMA-PET imaging in patients with relapsed prostate cancer. Mol Imaging Biol. 2015. doi:10.1007/s11307-015-0866-0.
Szabo Z, Mena E, Rowe SP, et al. Initial evaluation of [F]DCFPyL for prostate-specific membrane antigen (PSMA)-targeted PET imaging of prostate cancer. Mol Imaging Biol. 2015. doi:10.1007/s11307-015-0850-8.
Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81–5.
Fuchs AV, Tse BW, Pearce AK, et al. Evaluation of polymeric nanomedicines targeted to PSMA: effect of ligand on targeting efficiency. Biomacromolecules. 2015;16:3235–47. doi:10.1021/acs.biomac.5b00913.
Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther. 2011;336:496–505. doi:10.1124/jpet.110.174870.
Horoszewicz JS, Leong SS, Kawinski E, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809–18.
Afshar-Oromieh A, Hetzheim H, Kratochwil C, et al. The novel theranostic PSMA-ligand PSMA-617 in the diagnosis of prostate cancer by PET/CT: biodistribution in humans, radiation dosimetry and first evaluation of tumor lesions. J Nucl Med. 2015. doi:10.2967/jnumed.115.161299.
Afshar-Oromieh A, Zechmann CM, Malcher A, et al. Comparison of PET imaging with a (68)Ga-labelled PSMA ligand and (18)F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur J Nucl Med Mol Imaging. 2014;41:11–20. doi:10.1007/s00259-013-2525-5.
Lavergne V, Ghannoum M, Christie M, et al. Risk factors and consequences of hyperaluminemia in a peritoneal dialysis cohort. Perit Dial Int. 2012;32:645–51. doi:10.3747/pdi.2011.00203.
Acknowledgment
We thank Larry Pang and Andreea Stuparu (members of the Ahmanson Translational Imaging Division) for their support and assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was partly supported by an unrestricted grant from the Movember Foundation—MOVEMBER GAP2 FUNDING AWARD.
Conflict of interest
Johannes Czernin is founder of Sofie Biosciences, which manufactures the Genisys4 scanner used in this manuscript. No other potential conflicts of interest are reported.
Ethical approval
Animal studies were approved by the UCLA Animal Research Committee and were carried out according to the guidelines of the Division of Laboratory Animal Medicine at UCLA. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants.
Author’s contributions
Initials: Stefano Boschi (SB), Jason T. Lee (JTL), Seval Beykan (SeB), Roger Slavik (RS), Liu Wei (LW), Uta Eberlein (UE), Andreas K. Buck (AKB), Filippo Lodi (FL), Gianfranco Coria (GC), Johannes Czernin (JC), Michael Lassmann (ML), Ken Herrmann (KH), Stefano Fanti (SF)
Conception and design: SB, AKB, JC, ML, KH, SF
Development of methodology: SB, JTL, RS, GC, ML, KH, SF
Acquisition of data: SB, JTL, RS, LW, FL, GC
Analysis and interpretation of data: JTL, SeB, RS, ML, KH, SF
Writing, review, and/or revision of the manuscript: all authors
Administrative, technical, or material support: SB, AKB, JC, ML, KH, SF
Supervision: SB, JC, ML, KH, SF
Additional information
Stefano Boschi and Jason T. Lee contributed equally to this work.
Rights and permissions
About this article

Cite this article
Boschi, S., Lee, J.T., Beykan, S. et al. Synthesis and preclinical evaluation of an Al18F radiofluorinated GLU-UREA-LYS(AHX)-HBED-CC PSMA ligand. Eur J Nucl Med Mol Imaging 43, 2122–2130 (2016). https://doi.org/10.1007/s00259-016-3437-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-016-3437-y