
Abstract
Beta-thalassemia is a heterogeneous group of anemic disorders caused by the absence or defective production of beta-globin chains. Their clinical manifestations vary from asymptomatic to severe symptoms leading to a transfusion-dependent anemic state. The genes that cause thalassemia are prevalent in Asian and African populations, particularly concentrated in the Middle East, Mediterranean region, parts of India, and South East Asia. Over time, the disease causes various musculoskeletal abnormalities with complex pathophysiology secondary to chronic anemia. The compensatory mechanisms result in diffuse marrow hyperplasia, yellow to red marrow reconversion, osteopenia, and pathologic fractures. Inability to remove excess iron and inevitable iron overload as a result of multiple blood transfusions in patients with thalassemia major and intermedia is another face of the disease. Musculoskeletal manifestations include osteopenia, coarse trabeculae, bone expansion, synovitis, joint effusion, and metaphyseal dysplasia. These complications have long-lasting effects on the skeletal growth pattern resulting in bone deformity, short stature, premature closure of physes, and predisposition to infection. Additionally, there are radiologic features of iron-chelator therapy, which are unique and unrelated to the disease process itself. Familiarity of radiologists with the imaging features of beta-thalassemia is crucial in both diagnosis and timely management of the disease and its complications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Beta-thalassemia is a group of hereditary anemia secondary to reduction or absence of beta-globin chains. Hemoglobin A (the most common form of adult hemoglobin) is made up of four globin chains—two alphas and two betas [1, 2]. In contrast to the alpha-globin genes with 4 copies on chromosome 16, the beta-globin gene only requires two copies on chromosome 11 for beta-chain production. Beta-thalassemia is an autosomal recessive hereditary disorder that occurs when beta-globin chains are reduced or absent secondary to one allele defect (thalassemia minor) or two allele defects (thalassemia intermedia and thalassemia major). The imbalance in alpha and beta chain production leads to deficient hemoglobin synthesis and accumulation of excess insoluble alpha chains in red blood cells, which causes early destruction. This results in ineffective hematopoiesis, hemolysis, and severe anemia. The genes that cause thalassemia are prevalent in Asian and African populations. Beta-thalassemia is the most common hereditary anemia in the Middle East, Mediterranean region, parts of India, and South East Asia [2, 3].
Thalassemia minor results in mild anemia, which is only detectable on laboratory values with no significant clinical symptoms. On the other end of spectrum, thalassemia intermedia has mild to moderate clinical manifestations and thalassemia major has the most severe symptoms, often leading to chronic transfusion-dependent anemia. Thalassemia major involves different body parts (Table 1) leading to significant morbidity and mortality that reduces life expectancy.
Patients with thalassemia intermedia present with various clinical manifestations ranging from chronic anemia with mild symptoms to a transfusion-dependent state identical to thalassemia major. The severity of clinical symptoms is related to the amount of globin chain imbalance. Compared to thalassemia major, patients with thalassemia intermedia usually have a delayed presentation with mild anemia and infrequent need for blood transfusions. It is assumed that the maintenance of hemoglobin level above 10 g/dl can prevent musculoskeletal manifestations in these patients. Beta thalassemia major leads to a wide range of musculoskeletal system involvement with low back pain and arthralgia as the most common symptoms and growth retardation, fracture, osteoporosis, and kyphosis as the most severe presentations (Table 2). The severity of radiologic manifestations are also affected by the type of thalassemia, time of treatment onset, number of transfusions, and complications related to iron chelating therapy.
Chronic tissue hypoxemia secondary to anemia and compensatory marrow hyperplasia results in bone marrow expansion and extra-medullary hematopoiesis. Iron overload and inability to remove excess iron secondary to multiple transfusions is another cause of musculoskeletal manifestations (Table 1) [12, 13]. While the sequelae of recurrent transfusions has decreased due to optimal treatment regimens and iron chelation therapy, new skeletal findings secondary to iron chelation therapy have become noticeable.
In this review, we will illustrate musculoskeletal imaging manifestations of beta-thalassemia and its treatment-related findings using a multi-modality approach (Table 3). After reading this article, the reader will be able to describe the musculoskeletal manifestations of beta-thalassemia, recognize treatment-related imaging findings, and discuss the role of imaging in diagnosis and management of this disease.
Intra-medullary marrow hyperplasia
As previously described, beta-thalassemia major and intermedia can lead to chronic anemia due to inadequate production of the beta-globin chains. Chronic anemia stimulates intra-medullary bone marrow hyperplasia and expansion. Normally, red marrow is diffusely distributed throughout the fetal skeleton. Over lifetime, the red marrow is gradually replaced by yellow or fatty marrow. The fatty marrow exists within the appendicular skeleton and then advances to involve the axial skeleton. By adulthood, the red marrow is only seen within the axial skeleton, pelvis, sternum, and spine. As a response to chronic anemia, reconversion of yellow to red marrow takes place. In patients with uncontrolled thalassemia, this long-lasting anemia results in marrow expansion with widening of medullary spaces and thinning of the cortical bone. This compensatory mechanism leads to fragile bones and subsequent pathologic fractures involving different parts of the skeleton (Fig. 1).

A 15-year-old girl with β-thalassemia major. a Axial skull CT shows skull thickening, thinning of the cortices, trabecular coarsening, and bone demineralization. b Lateral skull radiograph shows “frontal bossing” (arrow). c AP pelvis radiograph shows marrow expansion, cortical thinning, and coarsening of the trabeculae (arrow)
Skull
In thalassemia patients, the compensatory bone marrow hyperplasia in the skull bone medullary space results in marked widening of the diploe, trabecular coarsening, osteopenia, and enlargement of the middle meningeal vessel impression on the cranial vault [13]. Another feature includes the thinning of the cortices, especially the outer table, which may be completely obliterated. These changes are detectable on skull radiographs and are more severe in patients with thalassemia major, specifically in those under 9 years of age. Nonetheless, these changes do not correlate with the severity of the disease nor do they signify involvement of other parts of the skeleton. In addition, it is not obvious whether these changes are reversible even after proper disease management [4, 5].
The widening of the diploic space mainly occurs in the frontal bone resulting in the characteristic skull shape known as frontal bossing or “tower skull” in advanced disease stages. The extreme skull bone expansion in children with uncontrolled thalassemia major can resemble the shape of a “hot cross bun” or “caput quadratum”. Due to lack of hematopoietic marrow within the occipital bone, this area is spared and the extent of bone marrow hyperplasia and diploic space widening is less pronounced [14].
On skull radiographs and CT in patients with beta-thalassemia major, multiple horizontally oriented radiolucent, and radiopaque lines (particularly within the frontal and parietal bones) are observed, which can be attributed to new bone formation. The combination of diploe sinusoidal revascularization and marrow hyperplasia is the main reason for this radiologic appearance, which was first named the “lamellated skull.” It seems that the “lamellated skull” occurs before the more typical “hair-on-end” appearance. The “hair-on-end” appearance is observed as vertical striations projecting from the outer aspect of the skull. These striations are the thickened trabeculae projecting perpendicular to the cortex of the widened diploic space. This can be identified on lateral skull radiograph, CT, and MRI [14]. Vertical elongated trabeculae within the radiolucent marrow resemble a hair stranded on end. On MRI, as a consequence of red marrow reconversion and reduction in the fat component, the signal intensity of the diploe decreases and becomes hypointense to gray matter on T1-weighted, T2-weighted, and proton density-weighted images. The thickened trabeculae appear as hypointense bands within the expanded marrow space, which causes the “hair-on-end” appearance (Fig. 2). These imaging findings are unusual after 9 years of age, especially in adequately treated patients [15]. While the “hair-on-end” appearance of the skull is often seen with thalassemia, other etiologies of severe anemia including sickle cell disease, iron deficiency anemia, or hemolytic anemia can cause a similar appearance and should be included in the differential diagnosis.

A 19-year-old boy with β-thalassemia major. a Sagittal FLAIR, b axial T2W, and C axial T1W+ Gadolinium show hypointense marrow in comparison to the gray matter in both T2W and T1W sequences consistent with red marrow reconversion and decreased fatty marrow. d Frontal skull radiograph shows marked thickening of the skull bone, thinning of the cortices, trabecular coarsening, and bone demineralization
Facial bones
Intra-medullary bone marrow expansion in facial bones results in under-pneumatization of the frontal and paranasal sinuses, maxillary bone hypertrophy, nasal bridge flattening, and various other orofacial and dental deformities. Radiograph and CT are useful in identifying these changes. The main imaging findings include wall thickening of the frontal bone and paranasal sinuses, obliteration of the paranasal sinuses and nasal cavities, thinning of the cortices, trabecular coarsening, and osteopenia. The ethmoid sinus is usually preserved due to absence of hematopoietic marrow within its walls. The mastoid sinuses are involved in varying degrees depending on the marrow activity in this region (Fig. 3) [16].

A 17-year-old boy with β-thalassemia major. a Axial face CT shows wall thickening of the maxillary bone, thinning of the cortices, trabecular coarsening, and bone demineralization. Note the under-pneumatization of the maxillary sinuses. b Coronal face CT in another patient shows the same findings. Additionally, there is obliteration of the right maxillary sinus, widening of the ostium, and obliteration of the right nasal cavity secondary to antrochoanal polyp
CT is the modality of choice for evaluation of facial bone involvement in patients with beta-thalassemia major who need reconstructive surgery for treatment of facial deformities. CT provides vital information for pre-operative mapping and extent of osseous involvement. Key facial bone deformities include reduction in the height of the mandibular ramus, shortening of tooth roots, and reduction in the total posterior facial height with concomitant increase in anterior facial height. Lateral displacement of the orbits (hypertelorism) associated with maxillary protrusion and ventral displacement of the incisors leads to the pathognomonic “chipmunk face” appearance in patients with beta-thalassemia. This is rarely seen in today’s practice due to early diagnosis and more robust treatment protocols [17,18,19].
Ribs
Like other parts of the body, red marrow hyperplasia is the main reason for changes and deformities in the ribs, which are detectable on chest radiographs. The first and most common finding is the expansion of the ribs, specifically at the costovertebral junctions (Figs. 4 and 5). Other common findings include osteoporosis, which causes a trabeculated appearance of the ribs. Another reported thoracic finding is the “rib within a rib” appearance due to subperiosteal extension of the red marrow through the cortex. This is usually seen in the anterior and middle segments of the ribs. New bone formation superimposed on underlying extensive subperiosteal marrow proliferation can result in a rib osteoma, which is a rare location for this entity [13].

A 17-year male with β-thalassemia. a Contrast-enhanced CT shows cardiomegaly and b extramedullary hematopoiesis. c There is medullary expansion, especially at the costovertebral junction (arrows), leading to cortical thinning, trabecular coarsening, and decreased bone density of the ribs secondary to bone marrow hyperplasia. Incidentally noted bilateral axillary lymphadenopathy was felt to be reactive in this patient with no history of infection or malignancy

A 21-year-old male with poorly controlled β-thalassemia major a PA chest radiograph shows cardiomegaly, osteopenia, expansion of the costovertebral junction, thinning of the cortex, and trabecular thickening. b AP chest radiograph in another patient shows similar findings of rib osteopenia, cortical thinning, and costovertebral junction expansion (*)
In some patients, the red marrow breaks the inner cortex and infiltrates outward to cause extra-medullary hematopoiesis (EMH). EMH appears as prominent lobulated masses covering the anterior or posterior segments of the ribs on chest radiographs (Figs. 6, 7, and 8). The severity of rib changes and deformities depends on the transfusion history of patients. EMH is most frequently seen in the posterior mediastinum in comparison to the anterior mediastinum and pelvic cavity. Intra-canal extension of EMH can cause cord compression, resulting in neurologic findings [20, 21]. These changes are rarely seen in children who start blood transfusion at an early age. Regression of the changes may take place in most cases after proper transfusion [13].

A 42-year-old woman with β-thalassemia major presented with progressive dyspnea. a Axial contrast enhanced CT shows a lobulated right paravertebral mass with septations. There is a smaller hypodense mass near the left costovertebral junction (arrow). b Axial CT demonstrates surgical clips from prior splenectomy. c Coronal image shows the craniocaudal extent of the mass. Biopsy confirmed the diagnosis of EMH

A 29-year-old man with poorly controlled β-thalassemia major. a Axial contrast enhanced CT of the chest shows bilateral, well-defined, lobulated, and mildly enhancing paravertebral soft tissue masses without internal calcification (*), representing EMH. There is a large left pleural effusion. b Radiographic appearance of the bilateral lobulated paraspinal masses on PA chest radiograph (different patient with same pathology)

A 45-year-old man with a history of β-thalassemia major and a mass seen on chest radiograph. a Coronal T2W image of the thorax demonstrates bilateral paravertebral masses, which appear lobulated with heterogenous iso and hyperintense signal (arrows). b Sagittal T1W image demonstrates the right paravertebral mass (*) which is isointense to muscle. c Sagittal STIR image shows the right paravertebral mass (*) with heterogenous iso and hyperintense STIR signal
Spine
Various spinal abnormalities may occur following marrow hyperplasia, iron deposition, and chelation therapy. Common findings on radiographs and CT of the spine include osteopenia, cortical thinning, and increase in the height to width ratio of the vertebral bodies, secondary to medullary expansion (Fig. 9). Severe medullary expansion can cause cord compression, most commonly seen in the thoracic spine. These changes result in several characteristic appearances that are pathognomonic for thalassemia. The first common appearance is the “fish-mouth” vertebral body, which is defined as the biconcavity of the superior and inferior vertebral body end plates as a result of compression microfractures and subchondral bone thinning (similar to findings in sickle cell disease). These changes contribute to spinal kyphosis and scoliosis in these patients [3]. The ground glass appearance of the vertebrae is attributed to coarsening of vertical trabeculae, which appears as high-density vertical stripes positioned perpendicular to horizontal trabeculae [13]. Lastly, the “bone-within-bone” appearance may be observed in lateral spinal radiographs because of end plate depression [22]. These imaging manifestations are usually not seen in well-controlled thalassemia with adequate transfusion.

A 31-year-old woman with β-thalassemia major. a Sagittal CT demonstrates cortical thinning and an increase in the width/height ratio of the vertebral bodies secondary to medullary expansion. b Sagittal T1W and c T2W images reveal diffusely decreased marrow signal intensity as a consequence of red marrow reconversion and reduction in the fat component. The thickened trabeculae appear as hypointense bands within the expanded marrow space. The iron deposition within the bone marrow as a result of iron overload also contributes to signal decay on T2-weighted images
Paraspinal EMH is mainly seen in patients with thalassemia intermedia and its neurological complications usually manifest in the third and fourth decades of life because compensatory marrow hyperplasia needs time to develop. In patients with thalassemia intermedia, the prevalence is approximately 20% to 24%. This entity is rare in the first decade of life and in thalassemia major patients due to regular blood transfusions [11, 23]. EMH may cause cord compression leading to complications including low back pain, lower extremity pain, paresthesia, proprioception disorders, paraplegia, urinary or bowel incontinence, spastic gate, paraparesis, exaggerated deep tendon reflexes, and cauda equina syndrome. With that said, most patients stay asymptomatic and the masses are incidentally discovered on radiographs, CT, or MRI [24].
It is worth noting that when paraspinal masses are encountered on imaging, a broad differential diagnosis should be considered. These can include neurogenic tumors (schwannoma, neurofibroma, etc.), lymphoma, esophageal neoplasms, metastasis, paraspinal phlegmon/abscess, hematoma, or lymphangioma. Radiography and CT are usually sufficient for diagnosis of EMH in the appropriate clinical context, but MRI is the modality of choice in patients with neurologic symptoms or when there is doubt about these findings (Figs. 6, 7, and 8). MRI accurately characterizes the extent of the lesions and localizes the exact site of involvement for radiotherapy or surgery [21]. On MRI, inactive old lesions have high signal intensity on both T1- and T2-weighted sequences due to internal fat content. Meanwhile, active lesions manifest as intermediate T1 and T2 signal intensity with subtle or no enhancement. Iron deposition makes for low signal intensity on both T1- and T2-weighted MR images compared to the paraspinal muscles [25]. If findings are inconclusive, heat damaged 99m-Tc red blood cell scan or 99m-Tc-sulfur colloid scan can be used to confirm the diagnosis. Tissue sampling can be performed as a last resort in patients with high suspicion for metastatic diseases and indeterminate radiologic findings [26]. Management of spinal EMH differs based on the size and location of the mass, patient comorbidities, and prior treatments. Therapeutic options include increased frequency of blood transfusions, surgical decompression, radiotherapy, and fetal hemoglobin induction with Hydroxy-Carbamide or a combination of these treatments [24].
Appendicular skeleton
Tubular bone abnormalities include medullary expansion, cortical thinning, trabecular coarsening, decreased bone density, and pathologic fractures [27]. These changes are not seen in the first 6 months of life due to the presence of fetal hemoglobin but generally become apparent during the second year of life. The primary manifestations become notable in the metacarpal, metatarsal, and phalangeal bones where the medullary expansion results in squared or sausage-shaped appearance of the bones. Since this hyper-proliferated marrow needs more blood supply, the nutrient foramina dimensions increase, creating round or oval lucencies about the mid-diaphysis of these bones on radiographs [28].
The long bones lose their normal concave shape and become convex as a result of marrow expansion. This mostly occurs at the humerus and femur. In addition, the medullary space becomes more reticular in appearance secondary to trabecular thickening. Trabecular coarsening is accompanied by multiple cyst-like lucencies within the bone, making a characteristic appearance called the “worm eaten” bone [27]. Severely widened metaphysis and epiphysis cause the so-called Erlenmeyer flask deformity (Fig. 10) [3], which can be seen in other hemoglobinopathies and deposition diseases such as Gaucher disease, sickle cell disease, and bone dysplasia. Irregular hyperdense transverse lines within the metaphysis can be seen on radiographs. These are referred to as “Harris lines”, implying episodes of growth arrest which are not necessarily pathognomonic for thalassemia. In the pelvis, the main changes include marrow expansion, cortical thinning, and coarsening of the remaining trabeculae, which results in a “cob-web” appearance [4].

A 14-year-old male with β-thalassemia major. a AP radiograph of the right knee in a shows early fusion of the physis with widening of epiphysis and metaphysis resulting in an early “Erlenmeyer” deformity. b AP hand radiograph in another patient shows decreased bone mineralization and a squared first metacarpal (arrow). c Elbow radiograph shows cortical thinning, decreased bone density, and epiphyseal widening
As patients become older, the marrow proliferation migrates to the axial skeleton so the appendicular skeleton manifestations regress or completely disappear in most of the cases. Some degree of trabecular blurring and medullary canal expansion can remain, especially if the treatment regimen is sub-optimal. For instance, the first metacarpal bone can stay squared in adult patients despite treatment [3].
Early fusion of epiphysis
Another common musculoskeletal finding affecting tubular bones is early fusion of the epiphysis, especially in patients who started blood transfusions at a later age. This finding was initially reported in 1964 by Currarino and Elrandson on radiographs of thalassemia patients [29]. This abnormality is mainly seen in children older than 10 years of age. The overall prevalence is thought to be 10–15% in patients with thalassemia major and approximately 23% in patients greater than 10 years old. The most common site for premature fusion of epiphysis is the proximal humeral physis. Distal femur, tibia, fibula, and proximal femur are the next common sites of early fusion [4, 13].
Unilateral or bilateral epiphyseal fusion leads to growth retardation and subsequent short stature, short limb length, and limb discrepancy (Fig. 11). It also results in skeletal deformity secondary to irregular fusion of growth plates and subsequent angulation and deviation of the epiphyses. The most common deformity is the varus deformity of the humerus [20]. In patients with thalassemia major and open growth plates, irregularity across the physis is a marker of early fusion followed by bony bridges across the physis [4].

A 12-year-old male with β-thalassemia major. a Lateral leg radiograph shows early fusion of the epiphysis at the distal tibia. b Lateral knee radiograph in the same patient demonstrates epiphyseal fusion of the distal femur and proximal tibia. Note the diffuse demineralization and thinning of the cortices
Skeletal changes secondary to hyper-transfusion
High serum iron and uric acid levels secondary to hyper-transfusion cause synovial and articular cartilage abnormalities in thalassemic patients. Iron deposition and secondary hemochromatosis in these patients cause articular disorders, which usually affect the big joints, though not as commonly seen in comparison to patients with primary hemochromatosis [30]. Symmetric articular space narrowing, synovitis, subchondral cysts, osteophyte formation, and chondrocalcinosis are the main radiologic manifestations seen in patients with thalassemia major [31]. Uncommonly, gouty arthritis occurs in thalassemic patients due to high red blood cell turnover and hyperuricemia. Involvement of uncommon articular spaces such as the sacroiliac joint has been reported. Sacroiliac arthritis manifests as articular subchondral erosions and reactive sclerosis [6].
Blood transfusions substantially increase the life expectancy of patients suffering from thalassemia; however, this life-saving therapy could be associated with numerous complications that make close follow up vital in these patients. According to the thalassemia longitudinal cohort study guidelines [32], annual bone mineral densitometry should be performed in these patients after 10 years of age. Additionally, iron deposition is toxic to the heart, liver, and endocrine organ parenchyma which are affected in over 90% of patients with thalassemia. This iron overload occurs 1–2 years after initiation of regular blood transfusions. Iron overload monitoring is feasible by usage of T2*-based MRI sequences. The presence of growth retardation, osteoporosis, kyphosis, and fracture indicate the late and severe stages of the disease. According to treatment guidelines, routine radiological workups are not recommended except for MRI for monitoring of iron deposition and annual bone densitometry. Imaging is particularly helpful in evaluation of treatment related complications such as avascular necrosis, skeletal changes secondary to hyper-transfusion, and secondary complications due to decreased bone density such as fractures, and to detect other related musculoskeletal manifestations including myopathy and spinal cord compression due to extra-medullary hematopoiesis. Knowledge of musculoskeletal manifestations of thalassemia is crucial for clinicians to guide treatment and initiate supportive strategies to address these specific manifestations.
Skeletal changes secondary to iron-chelation therapy
Deferoxamine-induced bone dysplasia
The first use of iron chelators for management of patients with thalassemia major dates back to the late 1970s [4]. Since then, specific skeletal abnormalities have come to notice in patients using iron-chelator drugs such as deferoxamine (DFX). It is believed that DFX impedes fibroblast proliferation, collagen synthesis, and osteoblast activity in the metaphyseal region of the bones, particularly in fast-growing long bones and spine. This results in decreased growth rate, growth arrest, pain, and skeletal deformities [3, 33]. The dysplastic changes are usually bilateral and are mainly seen in the distal femur, proximal tibia, and distal ulna. Other affected sites include the proximal femur, proximal humerus, distal radius, and distal tibia [3]. These changes commonly occur in patients receiving high therapeutic doses (50 mg/kg/day) of iron-chelators initiated below the age of three [34].
The most common findings on imaging are irregularly splayed epiphyseal/metaphyseal junction and physeal widening. Additionally, progressive changes include coarsening of the metaphysis associated with cystic-like lucencies, cupping and fraying appearance, which can resemble rickets dysplasia. Metaphyseal defects have low signal intensity on T1-weighted images and intermediate-high signal intensity on T2-weighted MR images [35]. Genu-varus or -valgus deformities and gait abnormalities due to severe epiphyseal dysplasia of the lower extremities can also be seen [36]. Spinal involvement may cause platyspondyly, kyphosis, scoliosis, degenerative disc disease, and disproportionate short trunk. Platyspondyly is the most noticeable spinal finding and manifests as anterior elongation and flattening of the vertebral bodies. Its severity depends on the age at which iron-chelator therapy was started. The H-shaped vertebrae is rarely seen in thalassemia patients [4]. Patients with thalassemia major are at higher risk for degenerative disc disease [37]. This is mainly caused by iron deposition or DFX therapy [1]. Scoliosis may present following DFX therapy or iron deposition. Its incidence correlates with duration of DFX therapy, hematocrit, and ferritin levels. Scoliosis is typically S-shaped (right-sided thoracic and left-sided lumbar). Most cases do not require interventional procedures and may even resolve spontaneously [38].
Early diagnosis of DFX-induced osseous dysplasia is essential as dose reduction is proved to ameliorate skeletal dysplasia and improve growth. Annual hand radiography is helpful in monitoring for early diagnosis of DFX-induced dysplasia [39].
Deferiprone-induced arthropathy
Deferiprone (DFP) is an oral iron-chelator medication, which can cause arthropathy, especially at high doses. It mainly affects the knee joints, but other appendicular joints can be involved. DFP-induced arthropathy manifests as cartilage and synovial thickening and subchondral flattening of the femoral and tibia condyles. Severe changes are not usually reversible even after DFP termination; however, the symptoms usually resolve. Muscle stiffness, arthralgia, joint swelling, effusion, and reduced range of motion are other common complications [4, 40]. MRI may reveal various degrees of effusion, irregular thickening of the cartilage, and subchondral erosions. Intense synovial enhancement after injection of gadolinium-based intravenous contrast can be observed in some cases, indicating severe synovitis [40].
Deferasirox-induced myopathy
Deferasirox is a new iron-chelating agent known to increase bone mineral density. It is believed that deferasirox can cause muscular weakness and atrophy. The increase in muscle echogenicity and atrophy can be detected by ultrasound. These findings are more prominent in proximal muscles of the lower limbs. MRI reveals muscle atrophy. Findings suggesting myositis including intramuscular contrast enhancement and edema are typically absent [41, 42].
Other musculoskeletal manifestations of beta-thalassemia
Osteoarthrosis
In patients with thalassemia major, early osteoarthritis may occur. This is attributed to multiple factors such as bone marrow expansion, iron deposition, iron-chelation therapy, hypermetabolic state, and endocrine abnormalities (i.e., hypoparathyroidism). The most common site for osteoarthritis in thalassemia major patients is the ankle joint. Nowadays, early osteoarthrosis is not prevalent in thalassemia patients due to early treatment with new effective iron chelating agents [6, 12].
Avascular necrosis
There are rare cases of avascular necrosis (AVN) of the femoral head reported in literature in patients with thalassemia major [43, 44]. This occurs secondary to multiple microfractures, osteonecrosis, medullary expansion, and subsequent arterial compression. Radiography reveals manifestations of AVN including subchondral lucency, sclerosis, and subarticular plate collapse and flattening. MRI plays an important role in detecting AVN at an early stage when radiographic examination is unremarkable. MRI reveals characteristic findings of marrow infarction including a serpentine low-intermediate signal intensity line on T1-weighted images and a high signal intensity line on fluid-sensitive sequences resulting in the subarticular double-line pattern [44].
Bone demineralization
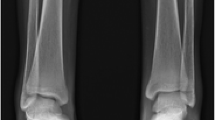
Despite optimal transfusion, initiation of iron-chelation therapy, and hormonal and mineral replacement, osteoporosis remains a common skeletal finding in thalassemia patients. Osteoporosis is a major health concern in patients with thalassemia major and is one of the most common causes of morbidity. The prevalence is reported at approximately 26% to 89% in various studies [6,7,8,9]. Delayed diagnosis and management of this debilitating problem results in various complications such as pain, fractures (Figs. 12 and 13), disability, and spinal deformities [45].

A 23-year-old female with poorly controlled β-thalassemia. AP (a) and lateral (b) radiographs of the thoracolumbar spine demonstrate diffuse demineralization of the bones secondary to osteoporosis. Additionally, there is cortical thinning and trabecular coarsening of the thoracolumbar spine. There is a compression fracture of the T9 and L1 vertebral bodies with anterior wedging and height loss, resulting in a kyphotic alignment

A 12-year-old male with β-thalassemia major. a AP radiograph shows epiphyseal irregularity and sclerosis of the distal humerus (arrow). There is pseudo-arthrosis as a result of pathologic fracture due to decreased bone density. b AP foot radiograph in a different patient shows fracture of the first proximal phalanx (arrow). c Sagittal and d axial STIR images demonstrate significant marrow and soft tissue edema surrounding the low signal intensity fracture line
The etiology is multifactorial and includes bone turn over imbalance, iron deposition, iron-chelator toxicity, and endocrine diseases (i.e., hypogonadism, thyroid or parathyroid disorders, diabetes, vitamin and mineral deficiency), resulting in generalized demineralization of both cancellous and tubular bones [46]. Osteoporosis predisposes thalassemia patients to insufficiency fractures. Prevalence of fractures has been reported at 15 to 50% in different studies [47, 48]. The most common site of fracture is the thoracolumbar junction where multiple microfractures cause short stature and kyphosis [10].
Differential diagnosis of beta-thalassemia
The radiological appearance of thalassemia may be detectable in other hemoglobinopathies or storage diseases. Although, the imaging features can help with the proper diagnosis, additional clinical data and laboratory findings are crucial for definitive diagnosis. Many systemic diseases can involve the craniofacial bones.
Bone marrow expansion and cortical thinning occur secondary to severe anemia from hematologic disorders such as thalassemia, sickle cell disease, or even from heavy smoking. The hair-on-end appearance is a well-documented but rarely seen radiologic appearance that is secondary to skull bone marrow expansion with an abrupt stop at the occipital bone. It is usually found in later stages of thalassemia major and less frequently in patients with thalassemia intermedia. It is also seen in patients with sickle cell disease, hereditary spherocytosis, and severe iron deficiency anemia.
Lymphoma can affect the skeletal system with or without nodal involvement. Extensive marrow infiltration with mild cortical bone destruction and “floating tooth appearance” can be seen when lymphoma affects the maxillofacial bones. Lower T2 signal intensity with marrow infiltration and adjacent soft tissue expansion on MRI suggests the diagnosis of lymphoma over other differential diagnoses such as thalassemia.
Paget disease manifests with coarsened trabecula, cortical thickening, and extensive bone formation. It affects skull bones in 25–65% of cases. Imaging appearance varies with the stage of the disease. The early stage is typically the destructive phase and commonly manifests with the “osteoporosis circumscripta” appearance. In the intermediate phase, there is mixed osteoblastic and osteolytic activity leading to the “cotton-wool” appearance. The last stage is the sclerotic phase which shows increased cortical and trabecular thickening and enlargement of affected the bones. Early in the disease, large areas of radiolucency are detectable within the frontal and occipital bones unlike thalassemia which spares the occipital bone.
The “bone within bone” appearance of spine occurs in various conditions including chronic diseases or sever systemic illnesses such as osteogenesis imperfecta, which could have similar musculoskeletal manifestations to thalassemia. The main radiographic appearance of osteogenesis imperfecta are bone deformities, osteopenia, and bone fractures. “Bone within bone” appearance is seen in these patients as a result of bisphosphonate therapy. The “bone within bone” is also seen in other conditions such as metastatic disease or leukemia as a result of subchondral osteopenia, which is in contrast to thalassemia where osteoporosis or generalized decreased in bone mass is detected.
Fibrous dysplasia is pathologic fibro-osseous tissue replacement of normal bone that can be monostotic or polyostotic. In the axial skeleton, it typically appears as areas of homogenous ground glass matrix with no periosteal reaction. Expansile endosteal scalloping can also be seen with cortical contour preservation and a thick layer of sclerotic margin. Progressive bowing and coxa varus deformity of the proximal femur secondary to fibrous dysplasia shape the shepherd’s crook deformity which is distinct from the Erlenmeyer deformity of thalassemia. Erlenmeyer deformity is defined as abnormal cortical thinning, lack of remodeling, and concavity of dia-metaphysis of long bones that occurs in various disorders that lead to marrow infiltration including Gaucher disease, hemoglobinopathies, and skeletal dysplasia. Presence of other musculoskeletal manifestations and clinical data help in differentiating these entities.
Conclusion
Beta-thalassemia is an inherited multisystem disease that leads to various musculoskeletal abnormalities. The main underlying reason for these abnormalities is bone marrow hyperplasia. Radiography is the most commonly used modality for diagnosis of these musculoskeletal manifestations but CT and MRI can provide additional value to help troubleshoot indeterminate findings. While MSK manifestations of thalassemia do not directly guide management (primarily done via monitoring hemoglobin levels, serum ferritin, liver iron concentration, or cardiac T2*), they are most helpful in early detection of complications (myopathy, avascular necrosis, spinal cord compression due to extra-medullary hematopoiesis) and surgical planning. Familiarity of radiologists with the imaging features of beta-thalassemia is crucial in diagnosis and timely management of thalassemia and its complications.
References
Haidar R, Mhaidli H, Musallam KM, Taher AT. The spine in β-thalassemia syndromes. Spine. 2012;37(4):334–9.
Rund D, Rachmilewitz E. Beta-Thalassemia. N Engl J Med. 2005;353(11):35–46.
Adamopoulos SG, Petrocheilou GM. Skeletal radiological findings in thalassemia major. J Res Pract Musculoskelet Syst. 2019. (accepted article). Available from: http://www.jrpms.eu. Accessed 10/29/2020
Tyler PA, Madani G, Chaudhuri R, Wilson LF, Dick EA. The radiological appearances of thalassaemia. ClinRadiol. 2006;61(1):40–52.
Hollar M. The hair-on-end sign. Radiology. 2001;221(2):347–8.
Noureldine MHA, Taher AT, Haydar AA, Berjawi A, Khamashta MA, Uthman I. Rheumatological complications of beta-thalassaemia: an overview. Rheumatology (Oxford). 2018;57(1):19–27.
Karimi M, Ghiam AF, Hashemi A, et al. Bone mineral density in beta-thalassemia major and intermedia. Indian Pediatr. 2007;44(1):29–32.
Toumba M, Skordis N. Osteoporosis syndrome in thalassaemia major: an overview. Review article. J Osteoporos. 2010;2010:537673.
Gaudio A, Morabito N, Catalano A, Rapisarda R, Xourafa A, Lasco A. Pathogenesis of thalassemia major-associated osteoporosis: A review with insights from clinical experience. J Clin Res Pediatr Endocrinol. 2018;11(2):110–117. https://doi.org/10.4274/jcrpe.galenos.2018.2018.0074.
Vogiatzi M, Macklin E, Fung E, et al. Prevalence of fractures among the thalassemia syndromes in North America. Bone. 2006;38:571–5.
Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006;37(1):12–20.
Hughes M, Akram Q, Rees DC, Kenneth Peter Jones A. Haemoglobinopathies and the rheumatologist. Rheumatology. 2016;55:2109–18.
Lawson JP (2018) Thalassemia imaging. Blood article (serial online). Available from https://emedicine.medscape.com/article/396792-overview. Accessed 10/29/2020
Carter R, Anslow P. Imaging of the calvarium. Semin Ultrasound CT MR. 2009;30(6):465–91.
Jyothi A, Rallabandi M, Mule A, Tirupathi P, Thota E, Alluri K. Radiological features of thalassaemia—an update. J Clin Diagn Res. 2019;13(3):ZE01–4.
Karakas S, Tellioglu AM, Bilgin M, Omurlu IK, Caliskan S, Coskun S. Craniofacial characteristics of thalassemia major patients. Eurasian J Med. 2016;48(3):204–8.
Takriti M, Dashash M. Craniofacial parameters of Syrian children with β-thalassemia major. J Investig Clin Dent. 2011;2(2):135–43.
Toman HA, Nasir A, Hassan R, Hassan R. Skeletal, dentoalveolar, and soft tissue cephalometric measurements of Malay transfusion-dependent thalassaemia patients. Eur J Orthod. 2011;33(6):700–4.
Hattab FN. Periodontal condition and orofacial changes in patients with thalassemia major: a clinical and radiographic overview. J Clin Pediatr Dent. 2012;36(3):301–7.
Hassanzadeh M. Images in clinical medicine. Extramedullary hematopoiesis in thalassemia. N Engl J Med. 2013;369(13):1252.
Ghieda U, Elshimy M, El Beltagi AH. Progressive spinal cord compression due to epidural extramedullary hematopoiesis in thalassemia intermedia: a case report and literature review. NeuroradiolJ. 2013;26(1):111–7.
Martinoli C, Bacigalupo L, Forni GL, Balocco M, Garlaschi G, Tagliafico A. Musculoskeletal manifestations of chronic anemias. SeminMusculoskeletRadiol. 2011;15(3):269–80.
Soman S, Rosenfeld DL, Roychowdhury S, Drachtman RS, Cohler A. Cord compression due to extramedullary hematopoiesis in an adolescent with known beta thalassemia major. J Radiol Case Rep. 2009;3(1):17–22. https://doi.org/10.3941/jrcr.v3i1.83.
Haidar R, Mhaidli H, Taher AT. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur Spine J. 2010;19(6):871–8.
Papanastasiou DA, Ellina A, Baikousis A, Pastromas B, Iliopoulos P, Korovessis P. Natural history of untreated scoliosis in beta-thalassemia. Spine (Phila Pa 1976). 2002;27(11):1186–90.
Ileri T, Azik F, Ertem M, Uysal Z, Gozdasoglu S. Extramedullary hematopoiesis with spinal cord compression in a child with thalassemia intermedia. J Pediatr Hematol Oncol. 2009;31(9):681–3.
Haidar R, Musallam KM, Taher AT. Bone disease and skeletal complications in patients with β thalassemia major. Bone. 2011;48(3):425–32.
Dinan D, Epelman M, Guimaraes CV, Donnelly LF, Nagasubramanian R, Chauvin NA. The current state of imaging pediatric hemoglobinopathies. Semin Ultrasound CT MR. 2013;34(6):493–515.
Currarino G, Erlandson ME. Premature fusion of the epiphyses in Cooley's anemia. Radiology. 1964;83:656–64.
Haines D, Martin M, Carson S, et al. Pain in thalassaemia: the effects of age on pain frequency and severity. Br J Haematol. 2013;160(5):680–7.
Resnick D. Diagnosis of bone and joint disorders, vol. 3. 4th ed. Philadelphia: Saunders; 2002. p. 2167–76.
Tubman VN, Fung EB, Vogiatzi M, Thompson AA, Rogers ZR, Neufeld EJ, et al. Thalassemia clinical research network. Guidelines for the standard monitoring of patients with thalassemia: report of the thalassemia longitudinal cohort. J Pediatr Hematol Oncol. 2015;37(3):e162.
Chan YL, Pang LM, Chick KW, Cheng JC, Li CK. Patterns of bone diseases in transfusion-dependent thalassemia major: predominance of osteoporosis and desferrioxamine-induced bone dysplasia. PaediatrRadiol. 2002;32(7):492–7.
Bedair EM, Helmy AN, Yakout K, Soliman AT. Review of radiologic skeletal changes in thalassemia. Pediatr Endocrinol Rev. 2008;6(Suppl 1):123–6.
Drakonaki EE, Maris TG, Maragaki S, Klironomos V, Papadakis A, Karantanas AH. Deferoxamine versus combined therapy for chelating liver, spleen and bone marrow iron in beta-thalassemic patients: a quantitative magnetic resonance imaging study. Hemoglobin. 2010;34(1):95–106.
De Sanctis V, Soliman AT, Elsefdy H, et al. Bone disease in β-thalassemia patients: past, present and future perspectives. Metabolism. 2018;80:66–79.
Desigan S, Hall-Craggs MA, Ho CP, Eliahoo J, Porter JB. Degenerative disc disease as a cause of back pain in the thalassemic population: a case-control study using MRI and plain radiographs. Skelet Radiol. 2006;35(2):95–102.
Hall-Craggs MA, Porter J, Gatehouse PD, Bydder GM. Ultrashort echo time (UTE) MRI of the spine in thalassemia. Br J Radiol. 2004;77(914):104–10.
Chan YL, Li CK, Pang LM, Chik KW. Desferrioxamine-induced long bone changes in thalassaemic patients-radiographic features, prevalence and relations with growth. Clin Radiol. 2000;55(8):610–4.
Kellenberger CJ, Schmugge M, Saurenmann T, et al. Radiographic and MRI features of deferiprone-related arthropathy of the knees in patients with beta-thalassemia. AJR Am J Roentgenol. 2004;183(4):989–94.
Vill K, Müller-Felber W, Teusch V, et al. Proximal muscular atrophy and weakness: an unusual adverse effect of deferasirox iron chelation therapy. NeuromusculDisord. 2016;26(4–5):322–5.
Poggi M, Sorrentino F, Pugliese P, et al. Longitudinal changes of endocrine and bone disease in adults with b-thalassemia major receiving different iron chelators over 5 years. Ann Hematol. 2016;95(5):757–63.
Al-Zahrani H, Malhan H, Al-Shaalan M. Recurrent avascular necrosis of the femoral head and intramedullary bone infarcts in thalassaemia major. J Appl Hematol. 2012;3:127–8.
Kanthawang T, Pattamapaspong N, Louthrenoo W. Acute bone infarction: a rare complication in thalassemia. Skelet Radiol. 2016;45(7):1013–6.
Shamshirsaz AA, Bekheirnia MR, Kamgar M, et al. Bone mineral density in Iranian adolescents and young adults with beta-thalassemia major. Pediatr Hematol Oncol. 2007;24(7):469–79.
De Sanctis V, Soliman AT, Elsedfy H, et al. Osteoporosis in thalassemia major: an update and the I-CET 2013 recommendations for surveillance and treatment. Pediatr Endocrinol Rev. 2013;11(2):167–80.
Fung EB, Harmatz PR, Milet M, et al. Multi-center iron overload study group. Fracture prevalence and relationship to endocrinopathy in iron overloaded patients with sickle cell disease and thalassemia. Bone. 2008;43(1):162–8.
Sutipornpalangkul W, Janechetsadatham Y, Siritanaratkul N, Harnroongroj T. Prevalence of fractures among Thais with thalassaemia syndromes. Singap Med J. 2010;51(10):817–21.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
Majid Chalian: Medical Advisor for Imagen Technologies Ltd.
Other authors have nothing to disclose.
Conflict of interest
None.
The authors declare that they had full access to all the data in this study and the authors take complete responsibility for the integrity of the data and the accuracy of data analysis.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hajimoradi, M., Haseli, S., Abadi, A. et al. Musculoskeletal imaging manifestations of beta-thalassemia. Skeletal Radiol 50, 1749–1762 (2021). https://doi.org/10.1007/s00256-021-03732-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00256-021-03732-9