
Abstract
Obtaining a sucrose isomerase (SIase) with high catalytic performance is of great importance in industrial production of isomaltulose (a reducing sugar). In order to obtain such SIase mutant, a high-throughput screening system in microtiter plate format was developed based on a widely used 2,4-dinitrosalicylic acid (DNS) method for determination of reducing sugar. An SIase from Erwinia sp. Ejp617 (ErSIase) was selected to improve its catalytic efficiency. After screening of ~ 8000 mutants from a random mutagenesis library, Q209 and R456 were identified as beneficial positions. Saturation mutagenesis of the two positions resulted in a double-site mutant ErSIase_Q209S-R456H that showed the highest catalytic efficiency, and its specific activity reached 684 U/mg that is 17.5-fold higher than that of the wild-type ErSIase. By employing the lyophilized Escherichia coli (E. coli) cells harboring ErSIase_Q209S-R456H, a high space–time yield (STY = 3.9 kg/(L·d)) was achieved toward 600 g/L sucrose. Furthermore, the in silico analysis suggested that the hydrogen bond network was improved and steric hindrance was reduced due to the beneficial substitutions.
Key points
• A sucrose isomerase mutant with high catalytic efficiency was obtained.
• The highest space–time yield was achieved toward high-concentration sucrose.
• The optimized H-bond network contributed to the enhanced catalytic efficiency.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Isomaltulose (6-O-α-D-glucopyranosyl-D-fructofuranose) is a reducing disaccharide found in natural honey (Low and Sporns 1988) and natural sugarcane juice (Takazoe, 1985) in extremely little amounts. Isomaltulose is a structural isomer of sucrose, and they both share similar organoleptic and physical properties (Hamada 2002; Mu et al. 2014). Due to its non-carcinogenic nature and low calorific value, isomaltulose is considered as an ideal sugar substitute in the food market (Pilak et al. 2020). Isomaltulose is now used as an alternative industrial precursor to isomaltitol, which is obtainable from isomaltulose via catalytic hydrogenation (Wu et al 2015; Zhang et al. 2019a, b).
Sucrose isomerase (SIase, EC 5.4.99.11), also known as isomaltulose synthase, is widely used to produce isomaltulose and trehalulose. Several microorganisms, including Serratia plymuthica ATCC 15,928 (Véronèse and Perlot 1999), Klebsiella sp. LX3 (Li et al. 2003), Protaminobacter rubrum CBS547.77 (Lee et al. 2008), Enterobacter sp. FMB-1 (Cha et al. 2009), Pantoea dispersa UQ68J (Wu and Birch 2004), Pseudomonas mesoacidophila MX-45 (Nagai et al. 1994) and Erwinia rhapontici (Cheetham 1984), enable conversion of sucrose to trehalulose and isomaltulose. During the isomerization of sucrose to trehalulose and isomaltulose, a number of by-products such as fructose and glucose are produced (Véronèse and Perlot 1999; Zhang et al. 2002; Wu and Birch 2005). The different product ingredients mainly depend on the bacterial strain used. Recently, we have isolated SIase-encoding genes from Erwinia sp. Ejp617, heterologously expressed in E. coli and name the enzyme as ErSIase. However, the activity and catalytic efficiency of wild-type ErSIase (ErSIase WT) were still low, which is the main drawback toward large-scale isomaltulose production.
Directed evolution as a powerful tool has been widely used to improve enzymes activity, stability and enantioselectivity (Cheng et al. 2015), but random screening is still a cost-effective procedure, although it is a tedious and time-consuming process (Cheng et al. 2018). Therefore, an efficient screening method is necessary to achieve the goal within a limited time period. In this study, a simple and rapid screening method for SIase was developed by using 3,5-dinitrosalicylic acid (DNS) reagent to identify a mutant strain with improved conversion yield. Five potentially beneficial positions were selected after one round of random mutagenesis and screening (using DNS method). By employing site-saturation mutagenesis (SSM), two improved mutants were obtained. The two beneficial substitutions were combined (Q209S and R456H), which yield an improved mutant (ErSIase_Q209S-R456H) displaying the highest catalytic efficiency.
Materials and methods
Materials
All chemicals were purchased from J&K Chemical or Aladdin Co., Ltd. (Shanghai, China), all of which were of analytical-reagent grade or higher, and all other enzymes were bought from Biolabs, Fermentas and TaKaRa, unless stated otherwise. The Ni–NTA agarose resin was obtained from GE Healthcare Co., Ltd. (Shanghai, China).
Bacterial strains and culture conditions
The recombinant E. coli BL21(DE3) harboring an 1847 bp SIase gene from Erwinia sp. Ejp617 (GenBank: G37835) was constructed by our previous studies (Zhang et al. 2021). Recombinant E. coli cells were cultured in LB medium with 100 μg/mL kanamycin at 37 ℃ with agitation (180 rpm) for 10 h.
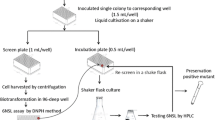
Cell cultivation and gene expression in deep 96-well plates
Single colonies were picked and inoculated in deep 96-well plates (Sangon, Shanghai, China), each well corresponded to a transformant. At the same time, E. coli BL21(DE3) harboring pET28b( +) and E. coli BL21(DE3) harboring pET28b-SIase were used as controls to inoculate in 96 deep well plates (wells H9-10 and H11-12). Each of the 96 wells contained 0.5 mL LB (contained 100 µg/mL kanamycin) and plates were further shaken under 180 rpm at 37 °C for 12 h. An aliquot of 50 μL was taken from each well and pipetted into wells of a new deep 96-well plate each well containing 950 μL LB. Then the plates were shaken at 37 °C until OD600 reached 0.6–0.8, and the induction process was started by shifting the temperature to 25 ℃ and addition of 0.1 mM isopropyl-β-D-thiogalactoside (IPTG). The gene expression process lasted for 10 h in 96 deep well plates.
High-throughput screening assay
150 μL of disodium hydrogen phosphate–citric acid buffer (100 mM, pH 6.0) containing 50 mg/mL sucrose was added to each well of microplates, and 50 μL culture from each well of deep 96-well plates was then added. The microplates were kept at 40 ℃ for 30 min, and 100 μL DNS solution was added to each of the well. Then, the microplates were heated in a microwave for 20 s. The activity of enzyme was analyzed at 540 nm using a molecular devices spectra Max M2 microplate reader (Molecular Devices, USA). The mutants showing improved activities were further determined by a reported HPLC assay (Zhang et al. 2019a, b).
Generation of random mutagenesis library by error-prone PCR (epPCR)
Random mutagenic libraries were generated based on epPCR with 0.4 mM MnCl2 using reported protocol (Markel et al. 2021). The epPCR was performed in a 50 μL reaction mixture containing 60 ng plasmid (pET28b-SIase) as the template, 60 pmol of each mutagenic primer (E2-F, E2-R, Table S1), 0.4 mM MnCl2, 2 × Taq PCR StarMix (TaKaRa, Dalian, China). The amplification process of gene encoding SIase was: 94 ℃ for 2 min followed by 30 cycles of 94 ℃ for 30 s, 60 ℃ for 30 s and 72 ℃ for 2 min and finally 72 ℃ for 10 min. Then, PCR products were digested using QuickCut Dpn I at 37 ℃ for 3 h to remove the template. Then, the library gene fragments were purified using MiniBEST DNA Fragment Purification Kit (TaKaRa, Dalian, China). The purified gene fragments were inserted into Nco I and Xho I sites of pET28b to form the recombinant plasmids pET28b-SIaseMUT. The plasmids were then transformed into E. coli BL21(DE3) and all transformed clones were grown overnight.
Site-directed/saturation mutagenesis
The site-directed/saturation mutagenesis at positions 209 and 465 was performed as previously described (Cheng et al. 2021). Primer sequences are listed in Table S2. The 50 μL reaction mixture consisted of 100 ng plasmid DNA (pET28b-SIase), 10 pmol of forward and reverse primers, 25 μL PrimeSTAR Max Premix (TaKaRa, Dalian, China). The amplification process of gene encoding SIase was: 98 ℃ for 2 min followed by 30 cycles of 98 ℃ for 10 s, 60 ℃ for 5 s and 72 ℃ for 30 s and finally 72 ℃ for 10 min. PCR products were digested using QuickCut™ Dpn I at 37 ℃ for 3 h to remove the template. Then, the library gene fragments were purified using MiniBEST Plasmid Purification Kit. The resulting plasmids were transformed into E. coli DH(5α) and then retransformed into E. coli BL21(DE3) competent cells. The best mutants were screened and sequenced.
Enzyme purification
The E. coli BL21(DE3) containing pET28b-ErSIase were cultured in 50 mL LB medium containing kanamycin (100 μg/mL) at 37 ℃ for 12–16 h. The culture was transferred to a fresh LB broth containing the same concentration of kanamycin using 2% inoculum. The cells were cultured at 37 ℃ until OD600 reached 0.6–0.8 and the protein SIase was induced by 0.1 mM isopropyl-β-D-thiogalactoside (IPTG) at 25 ℃ for 10 h. The cell pellet was carried out by centrifugation (10,000 rpm for 10 min at 4 ℃) and washed twice with 50 mM Tris–HCl (pH 7.0). Cell precipitation was resuspended in lysis buffer (pH 7.0) containing 50 mM NaH2PO4, 300 mM NaCl and 10 mM imidazole. The suspension was ultrasonically broken at 300 W for 30 min in an ice bath and then centrifuged at 12,000 rpm for 20 min (4 ℃). The supernatant was collected as crude protein solution.
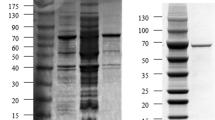
The crude SIase sample was isolated using nickel-nitrilotriacetic acid (Ni–NTA) affinity column chromatography (Qiagen). The molecular mass of SIase was investigated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) (Tripathi et al. 2011). The protein concentration was measured by the Bradford method (Bradford, 1976). The final enzyme solution was further used in the experiments after calculating the protein concentration and enzyme activity.
Determination of enzyme activity
The SIase activity was determined in a 1 mL mixture (100 mM disodium hydrogen phosphate–citric acid buffer, pH 6.0) with 10 μg enzyme solution and 100 mg/mL sucrose. The obtained solution was incubated at 40 oC for 15 min. Then the reaction solution was boiled for 7 min to terminate the reaction. Finally, the reaction mixture centrifuged at 12,000 rpm. Quantification of sugar composition was carried out by High-performance liquid chromatograph (HPLC) equipped with a refractive index detector (Waters 2414, Waters Ltd., Milford, MA, USA) using a ZORBAX carbohydrate analysis column (Agilent Technologies Ltd., California, USA), mobile phase acetonitrile: water = 75:25 (v/v), flow rate 1.0 mL/min at 30 °C. One unit of sucrose isomerase activity was defined as the amount of enzyme producing 1 μmol isomaltulose per min under the above conditions.
Determination of kinetic parameters
In order to determine the kinetic parameters, SIase activity was measured at different sucrose concentrations (50–250 mM) at pH 6.0 and 40 °C. Values of maximum reaction rate (Vmax) and Michaelis–Menten constant (Km) were calculated using nonlinear regression (Zhang et al 2019a, b). The kcat was calculated from the ratio of Vmax to the enzyme concentration (Cheng et al. 2022).
Effects of pH and temperature on ErSIase activities
Assay reactions were performed at different pHs from 4.5 to 10.0 in 100 mM disodium hydrogen phosphate–citric acid buffer (pH 4.5–7.5), 100 mM Tris–HCl buffer (pH 7.5–8.5) and 100 mM Gly-NaOH buffer (pH 8.5–10.0). The pH stability was confirmed by determining the residual activity of pre-incubated SIase at various pHs ranging from 3.0 to 11.0. The optimum temperature of SIase was investigated by measuring enzyme activity at different temperatures (4–80 °C). Thermal stability was assessed by detecting residual enzyme activity of pre-incubated SIase at 4, 20, 30, 40, 50, 60, 70 and 80 ℃ for different time spans (0 − 1 h).
Effect of detergents on wild type and mutants SIase activities
The effects of detergents (SDS, EDTA, Urea, Tween 20, NP 40, Tween 80 and Triton-X 100) were investigated in 100 mM disodium hydrogen phosphate–citric acid buffer (pH 6.0) at 40 ℃. The enzyme was incubated in detergents (various detergents were in a concentration of 0.1%) for 2 h at 20 ℃. The low temperature was chosen to decrease the effects of temperature on SIase activity. The enzymatic activity with untreated SIase was set as 100%. Assays were carried out in triplicate.
Time course of isomaltulose production
The reaction was carried out in 10 mL mimic biotransformation system containing 100 mM disodium hydrogen phosphate–citric acid buffer (pH 6.0). The substrate concentration was 600 g/L. The enzyme concentration (wild-type and best mutants) was set as 0.1 mg/mL. The reaction temperature was 40 ℃ for specified time and sample taken with a time interval of 30 min. The production of isomaltulose was determined after the reaction was terminated by boiling for 7 min.
Homology modeling and docking analysis
Homology models of ErSIase and its mutants were generated using the SWISS-MODEL (Arnold et al. 2006). The crystal structures of sucrose isomerase (PmSI from Pseudomonas mesoacidophila, PDB: 2PWE, resolution: 2.0 Å) were selected from the Protein Date Bank database. The sequence alignment of ErSIase toward PmSI was generated ClustalW program (Larkin et al. 2007). The QMEAN value and Z score were used to evaluate theoretical protein models. The Ramachandran plot was constructed to visualize the energetically allowed regions for backbone dihedral angles φ against ψ of the amino acid residues in ErSIase structure. Docking was performed by Autodock 4.0.
Results
Directed evolution of ErSIase for increased catalytic performance
Firstly, a directed evolution campaign was performed by employing a colorimetric HT screening assay. Firstly, a random mutagenesis library of ErSIase gene was generated by error-prone PCR (epPCR) using ErSIase_WT as template ([Mn2+] = 0.1 mM). After screening ~ 8000 ErSIase mutants toward 50 g/L sucrose, two ErSIase mutants (ErSIase_Q209N: an abbreviation of the ErSIase mutant harboring the substitution of Glu to Asp at position 209; ErSIase_R456K) were selected (Fig. 1) and were purified by His-tag chromatography (Fig. 2). The catalytic performance of ErSIase_WT and its mutants were quantitatively characterized by HPLC-based method. The specific activity of ErSIase WT, ErSIase_Q209N, ErSIase_R456K were 39 U/mg, 59 U/mg, 165 U/mg, respectively (Table 1). The specific activity of ErSIase_Q209N and ErSIase_R456K were improved by 1.5- and 4.2-fold.

A typic result of high-throughput screening assay in microtitle plate format. The colorimetric DNS method is used for detection of reducing sugar. The color ranges from yellow to brown. The darker in color indicated the higher enzyme activity

SDS–PAGE analysis of wild-type ErSIase (ErSIase_WT) and its mutants. Lane M, molecular mass markers; Lane WT, ErSIase_WT; Lane 1, WT_Q209N; Lane 2, WT_R456K; Lane 3, WT_Q209S; Lane 4, WT_R456H; Lane 5, WT_Q209S-R456H
Saturation of identified beneficial positions of 209 and 456
The two beneficial residue positions (209 and 456) were identified by random mutagenesis. Then the site-saturation mutagenesis was employed to the creation of two libraries and each of them included 100 clones (library I for position 209 and library II for position 456). These clones were screened for higher activity toward 100 g/L sucrose. The ErSIase activity was increased by introducing residues with poplar uncharged sidechains (Asn and Ser) at position 209. The “best” substitution was Q209S, and the ErSIase_Q209S showed about 6 times higher than that of WT. Toward library II, only three residues (His, Arg and Lys) at position 456 showed activity, while other mutants were inactive, which indicated that the positive charged residues are necessary at position 456. The “best” substitution at this position was R456H, and ErSIase_R456H displayed ~ tenfold higher than that of WT in crude enzyme format.
Kinetic analysis of ErSIase WT and its mutants
The ErSIase WT and its mutants were purified by His-tag chromatography (Fig. 2) and characterized in terms of specific activity and kinetic characteristics (Table 1). The specific activity of ErSIase_Q209S and ErSIase_R456H SIases were improved by 7.3- and 11.5-fold compared to ErSIase_WT. The introduction of Ser at position 209 reduced Km value by 30% compared to ErSIase_WT. And the kcat values of ErSIase_R456K and ErSIase_R456H were increased to 370.6 s−1 and to 528.2 s−1, respectively. The catalytic efficiency of ErSIase_Q209S and ErSIase_R456H were improved by 2.4- and 6.0-fold. Furthermore, the specific activity of double mutant ErSIase_Q209S-R456H reached 684 U/mg and the catalytic efficiency of 22.67 s−1 mM−1, which is more than 16-times higher than that of ErSIase_WT.
Effect of detergents on wild type and mutants SIase activities
In order to further investigate the effect of 0.1% (w/v) detergents on the activity of ErSIase_WT and its mutants, seven detergents were selected. The activity with untreated ErSIase was set as 100%. The results indicated that the ErSIase_Q209S-R456H mutants ErSIase activities could be significantly increased compared with wild type in the presence of 0.1% (w/v) Tween 20, NP 40, Tween 80 and Triton-X 100, especially NP 40 was increased to 159.8% (Table 2). Nevertheless, SDS (6.5%) and urea (67.5%) had rapid inhibitory effects on ErSIase activity.
Effect of pH and temperature on SIase activity
Using sucrose as the substrate, the enzyme activity was measured at 30 °C and different pH conditions. Unlike the previous SIases with narrow pH spectrum, such as SIase from Enterobacter sp. (Cha et al. 2009), SIase from K. pneumonia (Aroonnual et al. 2007) and SIase from Erwinia sp. SIase (Kawaguti et al. 2010), the ErSIase_WT and ErSIase_Q209S-R456H displayed relatively wide pH spectrum (Fig. 2a and b). The optimum pH of ErSIase_WT and ErSIase_Q209S-R456H were 6.5 and 6.0 (Fig. 3b and d), respectively. Moreover, when pH is higher than 9.0, the relative SIase activity was decreased sharply, and nearly no enzyme activity when pH is 10, which was close to SIase from P. dispersa (Wu and Birch 2005) and Erwinia rhapontici NX-5 (Ren et al. 2011). The ErSIase_WT and ErSIase_Q209S-R456H showed high pH stability. After 1 h incubation at different pHs, the two ErSIases displayed similar activity compared to the activity before incubation (Fig. 4b and d).

The optimum temperature and pH of ErSIase_WT and ErSIase_Q209S-R456H. (A) The optimum temperature of ErSIase_WT, (B) the optimum pH of ErSIase_WT, (C) the optimum temperature of ErSIase_Q209S-R456H and (D) the optimum pH of ErSIase_Q209S-R456H

The thermal stability and pH stability of ErSIase_WT and ErSIase_Q209S-R456H. (A) The thermal stability of ErSIase_WT, (B) the pH stability of ErSIase_WT, (C) the thermal stability of ErSIase_Q209S-R456H and (D) the pH stability of ErSIase_Q209S-R456H
In order to further optimize the temperature, the enzyme activity was measured in the temperature range of 20 to 55 ℃. As shown in Fig. 3a and c, purified ErSIase_WT and ErSIase_Q209S-R456H both had highest enzyme activity at about 40 °C, which was similar to that of SIase from Pseudomonas mesoacidophila (Nagai et al. 1994). The optimum temperature was higher than that of SIase from Erwinia rhapontici, Pantoea dispersa and Erwinia sp. strain (Contesini et al. 2013; Li et al. 2011, 2017), which indicated that ErSIase_WT and ErSIase_Q209S-R456H had a good thermostability. The thermostability of the ErSIase_Q209S-R456H was overall slightly higher than that of the ErSIase_WT. However, when the reaction temperature reached above 40 ℃, the activity of both decreased gradually (Fig. 4a and c).
Time course of isomaltulose production
Biotransformation of sucrose to isomaltulose was carried out by ErSIase_WT and its mutants. The results are shown in Figs. 5 and 6. ErSIase_Q209S-R456H mutant SIase exhibited the “best” performance, by which the yield of isomaltulose reached 75.2% within 60 min, and finally reached up to 93.6% when the reaction time was extended to 180 min. Compared with the ErSIase_WT, ErSIase_Q209S and ErSIase_R456H, also displayed better performances, affording 59.6% and 69.3% isomaltulose yields in just 60 min, and 76.6% and 80.7% yields after 180 min, respectively (Fig. 5). Therefore, the ErSIase_Q209S-R456H was selected for a bioconversion against different concentrations of sucrose (100 − 600 g/L), in which a high space–time yield (STY = 3.9 kg/(L·d) was achieved (Fig. 6).

Time courses of sucrose conversion by ErSIase_WT and its mutants toward 600 g/L sucrose

Time courses of isomaltulose production by ErSIase_Q209S-R456H toward different concentrations of sucrose
Discussion
Obtaining an SIase with high catalytic efficiency toward high concentration of sucrose is of great importance in industrial production of isomaltulose. However, the direct rational design of SIase is challenging, because the SIase-catalyzed reaction is complicated and we know little about the structure and reaction mechanism of SIase. Thus, directed evolution of enzymes was applied to tune the enzyme properties of isomerases such as substrate specificity of an isomerase from Geobacillus stearothermophilus toward isomerization of l-arabinose (Laksmi et al. 2018). The DNS method is widely used to detect reducing sugars by color differences. Different concentrations of reducing sugars yield different color intensities with the principle of oxidation of the aldehyde or ketone functional groups of reducing sugars. In this work, based on DNS method, a high-throughput screening system was developed in microtiter plate format. After screening of ~ 8000 mutants, two beneficial residues Q209 and R456 were identified and then saturated, which resulted in an improved mutant ErSIase_Q209S-R456H that showed the highest catalytic efficiency of 22.67 s−1 mM−1, which is more than 16-times higher than that of ErSIase_WT, and comparable with that of ErSIases from Ps. Mesoacidophila MX-45 (8.6 s−1 mM−1) (Watzlawick and Mattes 2009) and P. dispersa UQ68J (17.9 s−1 mM−1) (Wu and Birch 2005).
To better understanding the beneficial substitutions of Q209S and R456H, the ErSIase_WT model was built on the crystal structure of PmSIase (an SIase from Pseudomonas mesoacidophila, PDB: 2PWE, resolution = 2.0 Å). The Q-mean value of this model is 0.52, which indicates its quality is satisfied. Sucrose molecule was docked into the substrate-binding pocket of SIase by docking under semiempirical force field. The “best” conformation was selected, which displayed the strongest binding with lowest binding energy. The active site is surrounded by loops that formed an arginine-rich substrate-binding pocket (Arg239, Arg325, Arg456 and Arg460) (Fig. 7a). The model of mutant ErSIase_Q209S-R456H was built by FoldX software, which showed that the substitutions Q209S and R456H were located at the substrate-binding pocket (Fig. 7b). The R456H is a beneficial substitution that contributed to the improved catalytic efficiency and lower optimum pH (ErSIase_WT: 6.5 and ErSIase_Q209S-R456H: 6.0). The possible reason is that hydrogen bonding interaction can be formed between the oxygen of Asp457 (carboxyl group), the imidazolium ring of H456 in protonated state and the substrate (Fig. 7b). On the other hand, substrate-binding pocket was increased from 121 Å3 to 124 Å3 by introducing the beneficial substitution Q209S, which may contribute to the reduced Km value (ErSIase_WT: 69.28 mM; ErSIase_Q209S: 48.21 mM).

The docked conformation of SIase with sucrose. (A) The sucrose docked into the arginine-rich substrate-binding pocket of ErSIase_WT. The residues of R239, R325, R456 and R460 were shown in stick form; (B) the ErSIase_Q209S-R456H with sucrose. The residues of S209 and R456 were shown in stick form
In the batch biotransformation, by employing the lyophilized E. coli cells harboring ErSIase_Q209S-R456H exhibited the “best” performance, by which the yield of isomaltulose reached 93.6% after 180 min biotransformation toward 600 g/L sucrose. Finally, a high space–time yield (STY = 3.9 kg/(L·d)) was achieved. To the best of our knowledge, it is the highest STY toward high concentration sucrose compared to all the reports.
References
Aroonnual A, Nihira T, Seki T, Panbangred W (2007) Role of several key residues in the catalytic activity of sucrose isomerase from Klebsiella pneumoniae NK33-98-8. Enzyme Microb Technol 40:1221–1227
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
Cha J, Jung JH, Park SE, Cho MH, Seo DH, Ha SJ, Yoon JW, Lee OH, Kim YC, Park CS (2009) Molecular cloning and functional characterization of a sucrose isomerase (isomaltulose synthase) gene from Enterobacter sp. FMB-1. J Appl Microbiol 107:1119–1130
Cheetham PSJ (1984) The extraction and mechanism of a novel isomaltulose-synthesizing enzyme from Erwinia rhapontici. J Biochem 220:213–220
Cheng F, Chen Y, Qiu S, Zhai QY, Liu HT, Li SF, Weng CY, Wang YJ, Zheng YG (2021) Controlling stereopreferences of carbonyl reductases for enantioselective synthesis of atorvastatin precursor. ACS Catal 11:2572–2582
Cheng F, Tang XL, Kardashliev T (2018) Transcription Factor-Based Biosensors in High-Throughput Screening: Advances and Applications. Biotechnol J. 13(7):e1700648
Cheng F, Zhu L, Schwaneberg U (2015) Directed evolution 2.0: improving and deciphering enzyme properties. Chem Commun 51:9760–9772
Cheng F, Zhang JM, Jiang ZT, Wu XH, Xue YP, Zheng YG (2022) Development of an NAD(H)-driven biocatalytic system for asymmetric synthesis of chiral amino acids. Adv Synth Catal. https://doi.org/10.1002/adsc.202101441
Hamada S (2002) Role of sweeteners in the etiology and prevention of dental caries. Pure Appl Chem 74(7):1293–1300
Kawaguti HY, Celestino EM, Moraes ALL, Yim DK, Yamamoto LK, Sato HH (2010) Characterization of a glucosyltransferase from Erwinia sp. D12 and the conversion of sucrose into isomaltulose by immobilized cells. Biochem Eng J 48:211–217
Laksmi FA, Arai S, Tsurumaru H, Nakamura Y, Saksono B, Tokunaga M, Ishibashi M (2018) Improved substrate specificity for D-galactose of L-arabinose isomerase for industrial application. Biochim Biophys Acta Proteins Proteom 11:1084–1091
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) ClustalW and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lee HC, Kim JH, Kim SY, Lee JK (2008) Isomaltose production by modification of the fructose-binding site on the basis of the predicted structure of sucrose isomerase from “Protaminobacter rubrum”. Appl Environ Microbiol 74:5183–5194
Li X, Zhao C, An Q, Zhang D (2003) Substrate induction of isomaltulose synthase in a newly isolated Klebsiella sp. LX3. J Appl Microbiol 95:521–527
Low NH, Sporns P (1988) Analysis and quantitation of minor di- and trisaccharides in honey, using capillary gas chromatography. J Food Sci 53(2):558–561
Markel U, Lanvers P, Sauer DF, Wittwer M, Dhoke GV, Davari MD, Schiffels J, Schwaneberg U (2021) A Photoclick-Based High-Throughput Screening for the Directed Evolution of Decarboxylase OleT. Chemistry 27(3):954–958
Mu W, Li W, Wang X, Zhang T, Jiang B (2014) Current studies on sucrose isomerase and biological isomaltulose production using sucrose isomerase. Appl Microbiol Biotechnol 98(15):6569–6582
Nagai Y, Sugitani T, Tsuyuki K (1994) Characterization of alpha-glucosyltransferase from Pseudomonas mesoacidophila MX-45. Biosci Biotechnol Biochem 58:1789–1793
Pilak P, Schiefner A, Seiboth J, Oehrlein J, Skerra A (2020) Engineering a Highly Active Sucrose Isomerase for Enhanced Product Specificity by Using a “Battleship” Strategy. ChemBioChem 21(15):2161–2169
Ren B, Li S, Xu H, Feng XH, Cai H, Ye Q (2011) Purification and characterization of a highly selective sucrose isomerase from Erwinia rhapontici NX-5. Bioprocess Biosyst Eng 34:629–637
Takazoe I (1985) New trends on sweeteners in Japan. Int Dent J 35:58–65
Véronèse T, Perlot P (1999) Mechanism of sucrose conversion by the sucrose isomerase of Serratia plymuthica ATCC 15928. Enzyme Microb Technol 24:263–269
Wu L, Birch RG (2005) Characterization of the highly efficient sucrose isomerase from Pantoea dispersa UQ68J and cloning of the sucrose isomerase gene. Appl Environ Microb 71(3):1581–1590
Wu L, Liu Y, Chi B, Xu Z, Feng X, Li S, Xu H (2015) An innovative method for immobilizing sucrose isomerase on epsilon-poly-L-lysine modified mesoporous TiO2. Food Chem 187:182–188
Wu L, Birch RG (2004) Characterization of Pantoea dispersa UQ68J: producer of a highly efficient sucrose isomerase for isomaltulose biosynthesis. J Appl Microbiol 97:93–103
Watzlawick H, Mattes R (2009) Gene cloning, protein characterization, and alteration of product selectivity for the trehalulose hydrolase and trehalulose synthase from “Pseudomonas mesoacidophila” MX-45. Appl Environ Microbiol 75:7026–7036
Zhang D, Li X, Zhang L-H (2002) Isomaltulose synthase from Klebsiella sp. strain LX3: gene cloning and characterization and engineering of thermostability. Appl Environ Microb 68(6):2676–2682
Zhang F, Cheng F, Jia DX, Gu YH, Liu ZQ, Zheng YG (2021) Characterization of a recombinant sucrose isomerase and its application to enzymatic production of isomaltulose. Biotechnol Lett 43(1):261–269
Zhang P, Wang ZP, Liu S, Wang YL, Zhang ZF, Liu XM, Du YM, Yuan XL (2019a) Overexpression of secreted sucrose isomerase in Yarrowia lipolytica and its application in isomaltulose production after immobilization. Int J Biol Macromol 121:97–103
Zhang YJ, Chen CS, Liu HT, Chen JL, Xia Y, Wu SJ (2019b) Purification, identification and characterization of an esterase with high enantioselectivity to (S)-ethyl indoline-2-carboxylate. Biotechnol Lett 41(10):1223–1232
Acknowledgements
This work was financially supported by the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang, P. R. China (2018R01014) and the Zhejiang Provincial Qianjiang Talent Project.
Author information
Authors and Affiliations
Contributions
FZ and FC conceived and designed research. ZL and YZ supervised the project. FZ conducted experiments. FZ, DJ and QL analyzed data. FZ, FC and ZL wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, F., Cheng, F., Jia, DX. et al. Tuning the catalytic performances of a sucrose isomerase for production of isomaltulose with high concentration. Appl Microbiol Biotechnol 106, 2493–2501 (2022). https://doi.org/10.1007/s00253-022-11891-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-11891-5