
Abstract
Pseudomonas stutzeri strain XL-2 exhibited significant performance on biofilm formation. Extracellular polymeric substances (EPS) secreted by strain XL-2 were characterized by colorimetry and Fourier transform infrared (FT-IR) spectroscopy. The biofilm growth showed a strong positive correlation (rP=0.96, P<0.01) to extracellular protein content, but no correlation to exopolysaccharide content. Hydrolyzing the biofilm with proteinase K caused a significant decrease in biofilm growth (t=3.7, P<0.05), whereas the changes in biofilm growth were not significant when the biofilm was hydrolyzed by α-amylase and β-amylase, implying that proteins rather than polysaccharides played the dominant role in biofilm formation. More specifically, confocal laser scanning microscopy (CLSM) revealed that the extracellular proteins were tightly bound to the cells, resulting in the cells with EPS presenting more biofilm promotion protein secondary structures, such as three-turn helices, β-sheet, and α-helices, than cells without EPS. Both bio-assays and quantitative analysis demonstrated that strain XL-2 produced signal molecules of N-acylhomoserine lactones (AHLs) during biofilm formation process. The concentrations of C6-HLS and C6-oxo-HLS were both significantly positively correlated with protein contents (P<0.05). Dosing exogenous C6-HLS and C6-oxo-HLS also resulted in the increase in protein content. Therefore, it was speculated that C6-HLS and C6-oxo-HLS released by strain XL-2 could up-regulate the secretion of proteins in EPS, and thus promote the formation of biofilm.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biofilm is a highly structured community of bacterial cells enclosed in a self-produced extracellular polymeric substances (EPS) matrix attached to a surface (Costerton et al. 1999; O'Toole et al. 2000). Biofilm has been widely studied because they represent a fascinating example of microbial development and also because they can be problematic or beneficial in natural and artificial surfaces. Biofilms are formed from planktonic (free-swimming) cells in a complex and multiple-step process (Flemming and Wingender 2010). In general, planktonic cells that transitioned into a biofilm lifestyle are thought to initiate interactions with a surface reversibly, followed by irreversible attachment of cells, which thereafter form micro-colonies. Progressively, the micro-colonies differentiate into EPS-encased, mature biofilms (O'Toole and Kolter 1998; Rasamiravaka et al. 2015; Huang et al. 2019).
EPS, mostly excreted by microorganisms themselves, is essential for the biofilm formation (O'Toole et al. 2000; Flemming and Wingender 2010; Zhang et al. 2015). Proteins and polysaccharides are the major compositions of EPS, with low concentrations of extracellular DNA, lipids, humic acids, and other substances. Polysaccharides have been reported to play an important role in cell surface interactions during biofilm formation and serve as structural compositions of the biofilm matrix (Jennings et al. 2015; Skariyachan et al. 2018). Extracellular proteins, including various enzymes and structural proteins, also play a crucial role in the formation of biofilm (Karunakaran and Biggs 2011; Lv et al. 2014; Dube and Guiot 2019). Certain protein secondary structures play an important role in promoting adhesion, aggregation, and biofilm formation (Beech et al. 1999; Tretinnikov and Tamada 2001; Badireddy et al. 2010; Wang et al. 2018b). However, mixtures of extracellular enzymes for the dispersal of biofilms were also described (Flemming and Wingender 2010). To date, it is still controversial whether polysaccharides or proteins play a major role in promoting biofilm formation. Therefore, the potential links between specific compositions of EPS and biofilm formation need to be further discussed, as it may vary in different bacteria.
Furthermore, the secretion of EPS might be regulated by quorum sensing (Gilbert et al. 2010; Inhülsen et al. 2012). Quorum sensing occurs through signal molecules, which are microbial metabolic products and released to gauge microbial population density. When the microbial population density in the environment reaches the threshold, the concentration of signal molecules reaches a certain level, thus inducing the expression of certain genes. N-Acylhomoserine lactones (AHLs), one of the important signal molecules in Gram-negative bacteria, have been reported to be involved in EPS biosynthesis and thus affect the formation of biofilm (Davies et al. 1998; Tan et al. 2014; Wang et al. 2018a). However, much more investigations are needed to further clarify whether signal molecules promote biofilm formation by mainly regulating proteins or polysaccharides in EPS. Also, this may vary in different bacteria.
Among the strategies to clean up pollutants, biofilm process presents an increasing need for application in the upcycling of wastewater treatment plants all around the world in recent years (Huang et al. 2019). The biofilm provides structural integrity, bacterial protection, and nutrient capturing and consumption, and thus is vitally important to the performance of biofilm reactors. The formation of biofilm on carriers is a base of successful start-up and stable operation. However, there are still many controversies and unknowns about the role of EPS in biofilm formation and the signal regulation of EPS secretion. Previously, a biofilm formation bacterium Pseudomonas stutzeri strain XL-2 has been isolated from activated sludge in a municipal wastewater treatment plant (Zhao et al. 2018). Exploring the role of EPS in biofilm formation by P. stutzeri strain XL-2 may provide a further understanding of biofilm formation mechanisms, and may also lead to an application of this strain in the wastewater biofilm treatment process. Therefore, the present study is focused on elucidating the role of EPS in biofilm formation by P. stutzeri strain XL-2. To achieve this purpose, the EPS secretion from strain XL-2 was characterized by colorimetry and Fourier transform infrared (FT-IR) spectroscopy. The contribution of proteins and polysaccharides in EPS to biofilm formation was revealed by Pearson correlation analysis, enzymatic hydrolysis, confocal laser scanning microscopy (CLSM) and protein secondary structures analysis. Furthermore, the signal regulation of EPS secretion was explored by bio-assays and quantitative analysis of AHLs and exogenous AHLs dosing experiment.
Materials and methods
Strain
The strain used in this study, Gram-negative Pseudomonas stutzeri XL-2 (GenBank No. KY435925), was isolated from activated sludge in the secondary sedimentation tank after anaerobic–anoxic–oxic (A2/O) treatment unit in Jiguanshi wastewater treatment plant (Chongqing, China) (Zhao et al. 2018). Strain XL-2 has been deposited into the China General Microbiological Culture Collection Center (CGMCC) (Beijing, China) (accession number CGMCC No. 13252). The cells of strain XL-2 were stocked in a 25% glycerol solution at −80°C.
Media
Luria–Bertani broth (LB) was used to enrich strain XL-2. The stock solution (1 ml) of strain XL-2 was inoculated in a 250-ml flask containing 100 ml LB and cultivated for about 24 h at 30°C and 120 rpm. This was used as a pre-culture.
Basal medium (BM) contained (per liter) NH4Cl 0.19 g, KNO3 0.14 g, CH3COONa 2.39 g, NaCl 4 g, MgSO4 1.2 g, NaH2PO4·2H2O 1.83 g, Na2HPO4·12H2O 4.37 g, and trace element solution 3 ml.
The trace element solution contained (per liter) MnSO4 3 g, ZnSO4·7H2O 3 g, H3BO3 1.1 g, FeSO4·7H2O 0.3 g, and CaCl2·2H2O 0.6 g.
Determination of biofilm growth
Biofilm formation of P. stutzeri strain XL-2 was determined in a 96-well microtiter plate according to O'Toole and Kolter (1998) and modified. The pre-culture (1 ml) was inoculated into a 250-ml flask containing 100 ml BM and incubated at 30°C and 120 rpm for 24 h. Then cells were harvested by centrifugation (5000×g, 10 min, 4°C) and washed twice with PBS (pH ~7.0). The cell pellet was suspended with 0.9% NaCl solution and the final optical density (OD) at 600 nm (OD600) was adjusted to 1.0. This solution was named as cell suspension. The cell suspension (20 μl) was pipetted into a polystyrene, flat-bottomed 96-well microtiter plate (Corning, Costar, USA) containing 180 μl BM. Cells grown in the microtiter plate at 30°C for a desired period of time, and then the wells were washed twice with PBS (pH ~7.0) to remove planktonic bacteria. Subsequently, the microtiter plate was dried at 45°C and the cells were stained with 0.1% (w/v) crystal violet (200 μl) for 5 min. After that, the plate was washed twice and dried again at 45°C. Finally, 95% ethanol (200 μl) was used to dissolve the cell-bound dye. After 30 min, OD was measured at 570 nm by a microplate reader (Thermo Fisher, USA) to determine the biofilm growth. All the experiments were performed in triplicates.
EPS extraction
The EPS of strain XL-2 was extracted by using a modified method according to Liang et al. (2010). Biofilms in six-well microtiter plates (Corning, Costar, USA) at different cultivation times were dissolved in 0.9% NaCl solution to prepare suspension. Then the suspension was centrifuged (5000×g, 10 min, 4°C) and the cell pellet was washed twice with PBS (pH ~7.0). The re-suspension was treated by ultrasonic disruption (20 kHz, 40 W) and centrifuged (5000×g, 10 min, 4°C). After that, the cell pellet was re-suspended to a volume of 30 ml with PBS (pH ~7.0) and 70 g/g cell dry weight of cation exchange resin (CER) was added. The mixture was shaken at 600 rpm and 4°C for 1 h and remained static to settle CER. Then the suspension was centrifuged (10,000×g, 15 min, 4°C). The supernatant was collected as EPS and the pellet was cells without EPS. The EPS was filtered by a 0.45-μm filter membrane before further analysis. The concentration of polysaccharides was measured by anthrone colorimetry (Gaudy 1962), and the concentration of proteins was determined by Lowry method (Frølund et al. 1996). The DNA content was measured by the diphenylamine colorimetry using calf thymus DNA as the standard. The cell dry weight was estimated by freeze-drying method (Du and Yu 2002). To evaluate the cell lysis during the EPS extraction process, the activity of glucose-6-phosphate-dehydrogenase (G6PDH), an intracellular enzyme, was measured in the EPS sample using G6PDH assay kit (BC0260; Solarbio) (Liang et al. 2010). The specific enzyme activity (U/l) was defined as the amount of enzyme units that catalyzed the generation of 1 nmol NADPH per minute by the liquid in the system in liters. The total content of EPS was the sum of polysaccharides, proteins, and DNA.
Fourier transform infrared (FT-IR)
FT-IR (Nicolet iS50; Thermo Fisher, China) was used to determine the functional groups at 4000–400 cm−1 (mid-infrared) in EPS sample. EPS sample was ground with IR-grade KBr powder and molded into a disc. To determine the protein secondary structures, IR analysis was performed using Peakfit V4 software. The second derivatives of the amide I region at 1700–1600 cm−1 were generated and smoothed with 15% smoothing (Pickering et al. 2012). A Gaussian line shape was chosen as quantitative analysis, and the fitting process was performed with a correlation (r2) ≥0.999 in all of the fitted spectra.
Enzymatic hydrolysis
The enzymatic hydrolysis was aimed to evaluate the contribution of proteins and polysaccharides to biofilm formation. Biofilm was cultivated in six-well microtiter plates (Corning, Costar, USA) for 24 h. Proteinase K was an endolytic protease and utilized to hydrolyze proteins in EPS. α-Amylase and β-amylase were used to break down (α1-4) glycosidic bonds and β-linkages in polysaccharides in EPS, respectively (Adav et al. 2008). The enzyme activity of proteinase K was about 500 U/μl in PBS at pH 8.0. Before addition, the proteinase K was incubated at 58°C for 2 h. α-amylase and β-amylase were dissolved in PBS at pH 6.9 and pH 5.0, respectively, with the enzyme activity of about 200 U/μl. The biofilms in microtiter plates were digested by 2 μl of proteinase K, α-amylase, and β-amylase, respectively, and incubated at 30°C for 60 min in a shaker at 20 rpm. The biofilm without hydrolysis (2 μl of PBS was added to maintain the same volume) was used as a control. Then OD570 was measured by a microplate reader (Thermo Fisher, USA) to indicate the biofilm growth. Then the biofilms treated with different enzymes were dissolved in 0.9% NaCl solution. The EPS was extracted and determined according to the section of EPS extraction. All the experiments were conducted with six parallel samples.
Confocal laser scanning microscopy (CLSM) analysis
The biofilm of strain XL-2 was incubated on coverslips in a six-well microtiter plate (Corning, Costar, USA) for 24 h. Cells, proteins, and polysaccharides of the biofilm were stained by SYTO63 stain (Thermo, USA), fluorescein isothiocyanate (FITC) (Sigma, USA), and calcofluor white (Sigma, USA), respectively. The staining process was conducted according to Wang et al. (2018b). The stained biofilm was observed by a CLSM (TCS SP5; Leica, Inc., Germany). The entire staining process should be carried out in the dark to avoid fluorescence quenching.
Extraction of AHLs
Biofilm of P. stutzeri strain XL-2 was cultivated in six-well microtiter plates (Corning, Costar, USA) at 30°C for a desired period of time. The extraction method for signal molecules was modified according to Feng et al. (2014). Biofilm and culture in the wells were collected at different culture times and 0.9% NaCl solution was added to prepare the suspension. The suspension was then centrifuged (8000×g, 5 min, 4°C) to collect the supernatant. The pellet was re-suspended in 0.9% NaCl solution and then the supernatant was collected by centrifugation again. These operations were repeated three times. All the supernatants were collected and adjusted to 50 ml by 0.9% NaCl solution, and this was referred to as washing water phase. The remaining pellet was also adjusted to 50 ml by 0.9% NaCl solution, and this was referred to as sludge phase. Ethyl acetate was added to the washing water phase and sludge phase to extract organic phase three times. Then, an appropriate amount of anhydrous sodium acetate was added to the organic phase to remove water. The organic phase was evaporated to about 1 ml by rotary evaporator and then completely dried to solid by nitrogen. The solid was re-dissolved in 1 ml 50% acetonitrile solution and filtrated by a 0.45-μm filter membrane. Then the AHL extracts from washing water phase and sludge phase were obtained and stored at −80°C for subsequent determination of AHLs.
Protein secretion induced by exogenous signal molecules
One milliliter of pre-culture was inoculated into flasks containing 100 ml BM. Meanwhile, C6-HLS and C6-oxo-HLS were added to the flasks with concentrations of 0, 10−4, 10−5, 10−6, 10−7, 10−8, and 10−9 g/l, respectively. After 24 h of cultivation at 30°C and 120 rpm, bacterial samples were collected and the EPS was extracted. Then the concentrations of proteins in EPS were determined.
Determination of AHLs
Bio-indicator methods were applied for semi-quantitative analysis of AHLs. Chromobacterium violaceum CV026 was used to indicate the presence of short and medium acyl chain AHLs (n=4–7) by determining the production of purple pigment violacein (Blosser and Gray 2000). Then 1% (v/v) strain CV026 was inoculated into LB containing 1% (w/v) kanamycin, and 100 μl of AHL extract from washing water phase or sludge phase was added. The culture was incubated at 30°C and 120 rpm overnight and OD600 was determined. To estimate violacein, 1 ml of this overnight bacterial culture was lysed by adding an equal volume of 10% (w/v) sodium dodecyl sulfate solution (SDS), vortexed for 0.5 min, and incubated in a water bath at 37°C for 5 min. Then, 2.7 ml of water-saturated butanol was added and vortexed for 0.5 min. The organic phase was separated by centrifugation (20,000×g, 5 min, 4°C) and was used to measure the absorbance at 585 nm (OD585) to calculate the violacein content. The content of violacein corresponds to the concentration of short and medium acyl chain AHLs. The violacein unit was calculated as (OD585/OD600)×1000.
The inducement of β-galactosidase by Agrobacterium tumefaciens JZA1 was used to indicate the presence of long acyl chain AHLs (n=8–18). Then 1% (v/v) strain JZA1 was inoculated into LB containing 1% (w/v) kanamycin, and 100 μl of AHL extract from washing water phase or sludge phase was added. The culture was incubated at 28°C and 120 rpm overnight and OD600 was determined. To estimate the activity of β-galactosidase, 100 μl of this overnight bacterial culture was mixed with 0.9 ml pre-heated Z-buffer solution (0.06 mol/l Na2HPO4, 0.04 mol/l NaH2PO4, 0.01 mol/l KCl, 0.001 mol/l MgSO4, 0.05 mol/l β-mercaptoethanol). The cells were then lysed by adding 100 μl of CHCl3 and 50 μl of 1% (w/v) SDS, vortexed for 10 s, and incubated in water bath at 28°C for 3 min. Then, 200 μl of o-nitrophenyl-β-d-galactopyranoside (4 mg/ml) was added to the mixture, and the time of addition was recorded. After the appearance of sufficient color, the reaction was stopped by adding 500 μl of Na2CO3 (1 mol/l) and the stop time was recorded. The mixture was centrifuged (20,000×g, 5 min, 4°C) to remove the cell fractions. Then the absorbance of the mixture was measured at 420 and 550 nm, and the activity of β-galactosidase was calculated as (OD420−1.75×OD550)/(100 μl×reaction time×OD600)×1000. The level of β-galactosidase activity corresponds to the concentration of long acyl chain AHLs.
A high-performance liquid chromatography– tandem mass spectrometry (LC-MS/MS) system (LCMS-8060; Shimadzu, Japan) equipped with C-18 column (2.0 μm particle size, 2.1×150 mm) was used in an electrospray ionization interface (ESI) positive mode for quantitative analysis of AHLs. Before injection, the AHL extract from washing water phase or sludge phase was passed through a 0.22-μm filter membrane. AHLs were eluted with 50% acetonitrile solution at a flow rate of 0.3 ml/min. The column temperature was 30 °C and the mass range was 80–1200 m/z. Synthetic AHLs (Sigma-Aldrich, Germany) were used as a standard. The retention time of C6-HLS, C6-oxo-HLS, and C8-HLS was 0.88, 0.75, and 1.398 min, respectively.
Statistical analysis
Statistical analysis was conducted by using the soft IBM SPSS 24.0 (SPSS Inc., USA). Pearson correlation test and one-way analysis of variance (ANOVA) followed by least-significant difference multiple comparison test (LSD-t) were applied to evaluate the statistical significance. The P <0.05 level indicates significant and the P <0.01 level indicates highly significant. The closer the absolute value of Pearson correlation coefficient (rP) is to 1, the stronger the correlation is.
Results
Biofilm formation capability of P. stutzeri strain XL-2
Biofilm formation of P. stutzeri strain XL-2 within 60 h on the surface of microwells was determined (Fig. 1). The biofilm growth was measured in terms of the absorbance of the dissolved crystal violet dye from microtiter plates. Initially, the biofilm growth (OD570) was not significant and then a rapid increase in biofilm growth was detected in 1–12 h. The growth of biofilm gradually slowed down after 12 h and reached the maximum at 24 h with an OD570 of 2.14±0.19. After that, the biofilm growth was decreased. These results demonstrate that P. stutzeri strain XL-2 has the capability of biofilm formation.

Biofilm growth (OD570) of P. stutzeri strain XL-2 with different incubation time. Error bars are shown as ±SD
Characteristics of EPS from P. stutzeri strain XL-2
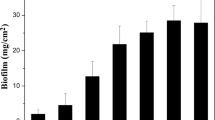
The contents and compositions of EPS from P. stutzeri strain XL-2 during growth period of 60 h are presented in Fig. 2. The lower activity of G6PDH indicates the noticeable cell lysis (Aguilera et al. 2008; Liang et al. 2010). It was obviously showed that the main composition of EPS from strain XL-2 was proteins, followed by polysaccharides. The content of DNA was extremely low. In the first hour, only a low content of proteins and polysaccharides was observed. Then for proteins, a dramatic increase was observed from 1 to 12 h, and then the increase in proteins content slowed down. The maximum protein content was observed at 24 h with 313±5.5 mg/g cell dry weight and then gradually decreased. This trend was similar to that of biofilm growth in Fig. 1. For polysaccharides, a continuous increase was observed from 1 to 48 h and then a decrease appeared.

Contents and compositions of EPS from P. stutzeri strain XL-2 during growth period. Error bars are shown as ±SD
The FT-IR spectra were used to further analyze the compositions of EPS secreted from strain XL-2 (Fig. 3). The bands observed at 3435–3120 cm−1 were assigned to the N–H and O–H stretching vibrations in the amide groups (Saleh 2011; Zhou et al. 2013; Saleh et al. 2016). The band at 2367 cm−1 belonged to the interference peak caused by CO2 in the air (Ando 2005). The bands observed at 1648 cm−1 corresponded to the C=O and C=C stretching vibration of amide I in proteins (Badireddy et al. 2010; Yuan et al. 2011). And the peak at about 1241 cm−1 belonged to the C–N stretching vibrations of amide III (Badireddy et al. 2010; Yang et al. 2015; Wang et al. 2018b). The asymmetrical band at 1115 cm−1 corresponded to the P–O, C–O–C, C–O–P of phosphodiesters and polysaccharides (Badireddy et al. 2008; Merroun and Selenska-Pobell 2008; Yang et al. 2015). And the band at 1049 cm−1 was associated with the C–O–C and C–H deformation vibrations of polysaccharide groups (Yuan et al. 2011; Zhang et al. 2014). The band at 987 cm−1 corresponded to diverse functional groups in carbohydrates, such as C–O and C–C stretching vibrations (Barth and Zscherp 2002). Besides, the peak at 949 cm−1 was associated with O–P–O stretches in nucleic acids (Badireddy et al. 2008). The bands observed at 860–814 cm−1 and 610 cm−1 were known as the fingerprint region (900–600 cm−1) and attributed to the ring vibrations associated with C–C and C–OH, which were reported from aromatic amino acids and nucleotides (Quiroz et al. 2006; Badireddy et al. 2010). Therefore, the main bands in the EPS components from strain XL-2 were several absorption peaks in the mid-infrared region: (1) 3435–3120 cm−1, 1700–1600 cm−1 (amide I region), and 1300–1200 cm−1 (amide III region) were all associated with proteins; and (2) 1200–900 cm−1 and 900–600 cm−1 (fingerprint region) were associated with polysaccharides and nucleic acids. Thus, EPS of strain XL-2 mainly included proteins and polysaccharides, and might have a few nucleotides.

Fourier-transform infrared spectroscopy spectra of EPS from P. stutzeri strain XL-2
Contribution of proteins and polysaccharides in biofilm formation
The relationship between biofilm growth and EPS compositions was investigated by statistical correlation analysis (Fig. 4). By comparing the P value and Pearson correlation coefficient (rP), there was a strong positive correlation (rP=0.96, P<0.01) between biofilm growth and proteins content. However, the polysaccharide content showed no correlation (rP=0.61, P>0.05) to biofilm growth. This result indicates that the proteins rather than polysaccharides in the EPS might play a more important role in biofilm formation of strain XL-2.

(a) Pearson correlation analysis between proteins and biofilm growth (rp=0.96, P<0.01); (b) Pearson correlation analysis between polysaccharides and biofilm growth (rp=0.61, P>0.05). Error bars are shown as ±SD
Selective enzymatic hydrolysis of proteins, α-polysaccharides, and β-polysaccharides in the EPS was conducted to further confirm the role of proteins and polysaccharides in biofilm formation of strain XL-2. It can be seen from Table 1 that each enzyme effectively hydrolyzed specific compositions in the EPS. After a hydrolysis by proteinase K, the proteins in EPS decreased from 312±7.5 mg/g cell dry weight to 98.8±5.8 mg/g cell dry weight (t=37.81, P<0.05). The polysaccharide content after hydrolysis with α-amylase and β-amylase remained at 30.2±4.2 mg/g cell dry weight (t=11.14, P<0.05) and 27.5±3.8 mg/g cell dry weight (t=11.89, P<0.05), respectively. The biofilm hydrolysis by proteinase K exhibited a significant decrease in biofilm growth (t=3.7, P<0.05), whereas the changes in biofilm growth were not significant when the biofilm was hydrolyzed by α-amylase and β-amylase. These results further demonstrated that proteins rather than polysaccharides in the EPS played the dominant role in biofilm formation.
The protein secondary structures
To further understand the role of proteins in EPS, the protein secondary structures of cells with EPS and cells without EPS were analyzed according to the curve fittings in amide I region (1700–1600 cm−1) (Badireddy et al. 2010; Wang et al. 2018b). The conformations of aggregated strands, α-helix, β-sheet, random coil, three-turn helices, and antiparallel β-sheets/aggregated strands were observed at 1620–1610 cm−1, 1660–1650 cm−1, 1640–1620 cm−1, 1650–1640 cm−1, 1680–1660 cm−1, and 1695–1680 cm−1, respectively. The assigned sub-peak spectra of each sample were determined in Fig. 5 and the contents of each protein secondary structure by the assigned sub-peak spectra are shown in Table 2.

The sub-peak spectra from original spectra by curve-fitted amide I region (1700–1600 cm−1) of cells with EPS (a) and cells without EPS (b)
In the sample of cells with EPS, the secondary structures of three-turn helices and β-sheets presented 34.5% and 34.3%, respectively. Therefore, the sum of these two contents occupied more than 50% of all compositions. Following the three-turn helices and β-sheets were α-helices with a percentage of 21.5%, while the random coil structures were not observed. Interestingly, the α-helix structures were not detected after EPS extraction. The dominant secondary structures in the sample of cells without EPS were determined as random coil structures (28.1%) and antiparallel β-sheets/aggregated strands (27.2%), followed by the three-turn helices (22.5%).
CLSM analysis
CLSM analysis was conducted to understand the distribution of proteins, polysaccharides, and cells in the biofilm. The CLSM showed that the trend of the fluorescence intensity curves of proteins and cells were similar, while the fluorescence intensity curve of polysaccharides fluctuated (Fig. 6). This phenomenon indicates that proteins were evenly distributed and tightly bound to the cells, whereas the distribution of polysaccharides was relatively inhomogeneous in the biofilm.

The fluorescence intensity curves of cells, proteins, and polysaccharides
Release of AHLs by P. stutzeri strain XL-2
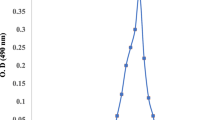
The release of AHLs by P. stutzeri strain XL-2 was initially detected by bioassay. All the AHLs were detected in the washing water phase, but not in the sludge phase. The violacein production by strain CV026 induced by AHLs from strain XL-2 was used to indicate the presence of short and medium acyl chain AHLs. The result in Fig. 7a suggests that no short and medium acyl chain AHLs was released before 12 h. With further development of biofilm, the release of short and medium acyl chain AHLs increased and reached the maximum at 24 h, and then gradually decreased after 30 h. The induction of β-galactosidase in strain JZA1 was used to indicate the presence of long acyl chain AHLs. The result in Fig. 7b suggests that long acyl chain AHLs were not released at the first 24 h, and began to appear at 30 h with low content, and disappeared at 48 h.

Semi-quantitative analysis of short and medium acyl chain AHLs (a) and long acyl chain AHLs (b) by bio-assays. Quantitative analysis of C6-HLS, C6-oxo-HLS and C8-HLS (c) by LC-MS/MS. Pearson correlation analysis between C6-HLS and proteins (rp=0.86, P<0.05) (d), and between C6-oxo-HLS and proteins (rp=0.89, P<0.05) (e). Error bars are shown as ±SD
Furthermore, the concentrations of C6-HLS, C6-oxo-HLS, and C8-HLS in the culture during the biofilm formation process of P. stutzeri strain XL-2 were quantitatively detected by LC-MS/MS. As shown in Fig. 7c, the concentrations of C6-HLS and C6-oxo-HLS began to be detectable at 12 h, and achieved the maximum at 24 h with 0.63 μg/l and 0.45 μg/l, respectively. After that, the concentrations of C6-HLS and C6-oxo-HLS decreased gradually. However, C8-HSL was only detected within 36–48 h with low concentrations of 0.02–0.05 μg/l. These quantitative results were consistent with the bio-assay results. The correlations between the concentrations of C6-HLS/C6-oxo-HLS and extracellular proteins/extracellular polysaccharides at corresponding culture time were calculated. No correlations between C6-HLS/C6-oxo-HLS and polysaccharides were observed (data not shown). However, there was a significant positive correlation (rP=0.86, P<0.05) between C6-HLS and proteins (Fig. 7d). Also, a significant correlation (rP=0.89, P<0.05) was observed between C6-oxo-HLS and proteins (Fig. 7e). These results suggest that C6-HLS and C6-oxo-HLS might stimulate the secretion of extracellular proteins.
Protein secretion induced by exogenous signal molecules
To further determine whether the extracellular proteins of P. stutzeri strain XL-2 were stimulated by C6-HLS and C6-oxo-HLS, exogenous signal molecules of C6-HLS and C6-oxo-HLS were added to BM for the cultivation of strain XL-2. Cultivation system without the addition of exogenous signal molecules was used as a control. As shown in Fig. 8a, there was no obvious difference in the content of proteins from strain XL-2 in response to the addition of 10−9–10−8 g/l of C6-HLS compared with control group. When exogenous C6-HLS increased to 10−7–10−4 g/l, an increase in protein content was observed. Also, compared with the control group, no obvious difference in the content of proteins in response to the addition of 10−9–10−7 g/l of exogenous C6-oxo-HLS was observed (Fig. 8b). However, 10−6–10−4 g/l of exogenous C6-oxo-HLS led to an increase in protein content. These results further suggest that C6-HLS and C6-oxo-HLS could stimulate the secretion of proteins in EPS.

The content of proteins in EPS from P. stutzeri strain XL-2 stimulated by different concentration of exogenous C6-HLS (a) and C6-oxo-HLS (b). Error bars are shown as ±SD
Discussion
P. stutzeri strain XL-2 exhibited a rapid biofilm growth (OD570) in microtiter plate (Fig. 1), suggesting that strain XL-2 presented significant performance on biofilm formation. In most biofilms, the microorganisms account for less than 10% of the dry mass, whereas the EPS that forms the biofilm matrix can account for over 90% (Flemming and Wingender 2010). The production of copious quantities of EPS was thought to be one prominent feature for the maturation of biofilms (Flemming and Wingender 2010; Zhang et al. 2015). Obviously, as shown in Fig. 2, EPS was secreted from strain XL-2 with biofilm growth. According to FT-IR spectra and quantitative analysis (Fig. 2 and Fig. 3), the main composition of EPS from strain XL-2 was proteins, followed by polysaccharides. This was consistent with the compositions of EPS in many reports (Adav et al. 2008; Wang et al. 2018b).
Moreover, the trend of proteins content with cultivation time (Fig. 2) was quite similar to that of biofilm growth (Fig. 1). The statistical correlation analysis further indicates that there was a strong positive correlation (rP=0.96, P<0.01) between biofilm growth and proteins content, but no correlation (rP=0.61, P>0.05) between biofilm growth and polysaccharides content (Fig. 4). Moreover, a significant decrease in biofilm growth (t=3.7, P<0.05) was observed when proteins in EPS were hydrolyzed by proteinase K. However, the changes in biofilm growth were not significant when α-polysaccharides and β-polysaccharides in EPS were hydrolyzed by α-amylase and β-amylase respectively (Table 1). All these results demonstrated that proteins rather than polysaccharides in the EPS from strain XL-2 played the dominant role in biofilm formation. Similarly, a dramatic reduction in biofilm formation by Streptococcus pneumoniae was observed when proteins were inhibited (Moscoso et al. 2006). For Bacillus cereus, extracellular proteins functioned as non-specific adhesions during biofilm formation (Karunakaran and Biggs 2011). The EPS of Shewanella sp. HRCR-1 contained biofilm-promoting protein BpfA and surface-associated serine protease, which were crucial functional components that promoted cell surface attachment and biofilm development (Cao et al. 2011). Extracellular proteins have also been reported to play important roles in the development of “aerobic granular activated sludge biofilm” (Lv et al. 2014). Contrarily, the polysaccharides in EPS from Bacillus megaterium TF10, which have the structure of the long backbone with active side chains, were the active constituents for the high bioflocculation activity (Yuan et al. 2011). Therefore, the potential links between specific compositions of EPS and biofilm formation varied in different bacteria and bacterial communities.
To further understand the role of proteins in biofilm formation by strain XL-2, the protein secondary structures were analyzed (Fig. 5), as it related to microbial adhesion, aggregation/flocculation, and biofilm formation (Beech et al. 1999; Tretinnikov and Tamada 2001; Omoike and Chorover 2004; Badireddy et al. 2010; Wang et al. 2018b). Microbes appeared to aggregate and form biofilm when the contents of biofilm positive protein secondary structures, such as β-sheets, α-helices, three-turn helices, and aggregated strands, were high. However, the aggregation and biofilm formation ability would be inhibited when the contents of biofilm negative protein secondary structures, such as random coils and antiparallel β-sheets/aggregated strands, were high. When EPS was extracted from cells, the sum of biofilm positive protein secondary structures transformed from 95.8% to 36.7%, while the sum of biofilm negative protein secondary structures increased from 2.9% to 55.3% (Table 2). This result implies that the proteins in EPS from strain XL-2 contained more biofilm positive protein secondary structures. The proteins in B. megaterium TF10 were found to play a minor role in bio-flocculation and the low percentage of α-helices was considered as one of the main factors (Yuan et al. 2011). The EPS in Enterobacter sp. strain FL, which exhibited significant capability of auto-aggregation, occupied more than 50% of secondary structures that stimulated aggregation (β-sheets, α-helices, and three-turn helices) (Wang et al. 2018b). Therefore, protein secondary structures might play a crucial role in biofilm formation by strain XL-2. The CLSM analysis (Fig. 6) indicates that proteins were tightly bound to the cells, which would be beneficial to alter the protein secondary structures of cells’ surface, and thus increased the adhesion and aggregation ability of strain XL-2 to form large colonies, further accelerating the formation of biofilm.
Furthermore, both bio-assays and quantitative analysis demonstrated that P. stutzeri strain XL-2 could produce signal molecules such as AHLs (Fig. 7a–c). Moreover, short and medium acyl chain AHLs presented relatively higher concentrations compared with long acyl chain AHLs. In combination with Fig. 1, it can be seen that signal molecules mainly appeared during the period of high biofilm growth. When the biofilm growth was maximum, the concentration of short and medium acyl chain AHLs also reached the maximum, suggesting that the AHLs from strain XL-2 might regulate the biofilm formation (Davies et al. 1998; De Kievit et al. 2001). Investigations have shown that signal molecules could regulate the secretion of EPS and thus affect the formation of biofilm (Gilbert et al. 2010; Inhülsen et al. 2012). For strain XL-2, the concentrations of C6-HLS and C6-oxo-HLS were both significantly positively correlated with proteins contents (P<0.05) (Fig. 7d, e). Moreover, when the exogenous C6-HLS reached the concentration of 10−7 g/l, or the exogenous C6-oxo-HLS reached the concentration of 10−6 g/l, an increase in protein content was observed (Fig. 8). These results provided evidence that C6-HLS and C6-oxo-HLS could stimulate the secretion of proteins in EPS from strain XL-2. Meanwhile, Fig. 4 and Table 1 demonstrated that proteins in the EPS from strain XL-2 played the dominant role in biofilm formation. Therefore, it was speculated that C6-HLS and C6-oxo-HLS released by strain XL-2 could up-regulate the secretion of proteins in EPS, and thus promote the formation of biofilm.
P. stutzeri strain XL-2 presented significant performance on biofilm formation due to the secretion of EPS. In the wastewater biofilm treatment processes, the formation of biofilm on carriers is a base of successful start-up and stable operation of biofilm reactors. Supplementation of strain XL-2 in biofilm reactors might enhance initial adhesion and EPS production. This would be a desirable bio-augmentation for biofilm formation of biofilm reactors. Therefore, further investigations are undergoing to evaluate the bio-augmentation by strain XL-2 for reducing start-up time and maintaining high treatment performance of biofilm reactors.
References
Adav SS, Lee DJ, Tay JH (2008) Extracellular polymeric substances and structural stability of aerobic granule. Water Res 42:1644–1650. https://doi.org/10.1016/j.watres.2007.10.013
Aguilera A, Souza-Egipsy V, San Martin-Uriz P, Amils R (2008) Extraction of extracellular polymeric substances from extreme acidic microbial biofilms. Appl Microbiol Biotechnol 78:1079–1088. https://doi.org/10.1007/s00253-008-1390-9
Ando DJ (2005) Infrared spectroscopy: fundamentals and applications. Wiley, Chichester
Badireddy AR, Korpol BR, Chellam S, Gassman PL, Engelhard MH, Lea S, Rosso KM (2008) Spectroscopic characterization of extracellular polymeric substances from Escherichia coli and Serratia marcescens: suppression using sub-inhibitory concentrations of bismuth thiols. Biomacromolecules 9:3079–3089
Badireddy AR, Chellam S, Gassman PL, Engelhard MH, Lea AS, Rosso KM (2010) Role of extracellular polymeric substances in bioflocculation of activated sludge microorganisms under glucose-controlled conditions. Water Res 44:4505–4516. https://doi.org/10.1016/j.watres.2010.06.024
Barth A, Zscherp C (2002) What vibrations tell us about proteins. Q Rev Biophys 35:369–430
Beech I, Hanjagsit L, Kalaji M, Neal AL, Zinkevich V (1999) Chemical and structural characterization of exopolymers produced by Pseudomonas sp. NCIMB 2021 in continuous culture. Microbiology 145:1491–1497. https://doi.org/10.1099/13500872-145-6-1491
Blosser RS, Gray KM (2000) Extraction of violacein from Chromobacterium violaceum provides a new quantitative bioassay for N-acyl homoserine lactone autoinducers. J Microbiol Methods 40:47–55
Cao B, Shi L, Brown RN, Xiong Y, Fredrickson JK, Romine MF, Marshall MJ, Lipton MS, Beyenal H (2011) Extracellular polymeric substances from Shewanella sp. HRCR-1 biofilms: characterization by infrared spectroscopy and proteomics. Environ Microbiol 13:1018–1031. https://doi.org/10.1111/j.1462-2920.2010.02407.x
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilm: a common cause of persistent infections. Science 284:1318–1322
Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP (1998) The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295–298
De Kievit TR, Gillis R, Marx S, Brown C, Iglewski BH (2001) Quorum-sensing genes in Pseudomonas aeruginosa biofilms: their role and expression patterns. Appl Environ Microbiol 67:1865–1873
Du GC, Yu J (2002) Green technology for conversion of food scraps to biodegradable thermoplastic polyhydroxyalkanoates. Environ Sci Technol 36:5511–5516. https://doi.org/10.1021/es011110o
Dube CD, Guiot SR (2019) Characterization of the protein fraction of the extracellular polymeric substances of three anaerobic granular sludges. AMB Express 9:23. https://doi.org/10.1186/s13568-019-0746-0
Feng H, Ding Y, Wang M, Zhou G, Zheng X, He H, Zhang X, Shen D, Shentu J (2014) Where are signal molecules likely to be located in anaerobic granular sludge? Water Res 50:1–9. https://doi.org/10.1016/j.watres.2013.11.021
Flemming HC, Wingender J (2010) The biofilm matrix. Nat Rev Microbiol 8:623–633. https://doi.org/10.1038/nrmicro2415
Frølund B, Palmgren R, Keiding K, Nielsen PH (1996) Extraction of extracellular polymers from activated sludge using a cation exchange resin. Water Res 30:1749–1758. https://doi.org/10.1016/0043-1354(95)00323-1
Gaudy AF (1962) Colorimetric determination of protein and carbohydrate. Ind Water Wastes 7:17–22
Gilbert KB, Kim TH, Gupta R, Greenberg EP, Schuster M (2009) Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol Microbiol 73:1072–1085
Huang H, Peng C, Peng P, Lin Y, Zhang X, Ren H (2019) Towards the biofilm characterization and regulation in biological wastewater treatment. Appl Microbiol Biotechnol 103:1115–1129. https://doi.org/10.1007/s00253-018-9511-6
Inhülsen S, Aguilar C, Schmid N, Suppiger A, Riedel K, Eberl L (2012) Identification of functions linking quorum sensing with biofilm formation in Burkholderia cenocepacia H111. Microbiologyopen 1:225–242
Jennings LK, Storek KM, Ledvina HE, Coulon C, Marmont LS, Sadovskaya I, Secor PR, Tseng BS, Scian M, Filloux A, Wozniak DJ, Howell PL, Parsek MR (2015) Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc Natl Acad Sci USA 112:11353–11358. https://doi.org/10.1073/pnas.1503058112
Karunakaran E, Biggs CA (2011) Mechanisms of Bacillus cereus biofilm formation: an investigation of the physicochemical characteristics of cell surfaces and extracellular proteins. Appl Microbiol Biotechnol 89:1161–1175
Liang Z, Li W, Yang S, Du P (2010) Extraction and structural characteristics of extracellular polymeric substances (EPS), pellets in autotrophic nitrifying biofilm and activated sludge. Chemosphere 81:626–632. https://doi.org/10.1016/j.chemosphere.2010.03.043
Lv J, Wang Y, Zhong C, Li Y, Hao W, Zhu J (2014) The effect of quorum sensing and extracellular proteins on the microbial attachment of aerobic granular activated sludge. Bioresour Technol 152:53–58. https://doi.org/10.1016/j.biortech.2013.10.097
Merroun ML, Selenska-Pobell S (2008) Bacterial interactions with uranium: an environmental perspective. J Contam Hydrol 102:285–295
Moscoso M, Garcia E, Lopez R (2006) Biofilm formation by Streptococcus pneumoniae: role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J Bacteriol 188:7785–7795. https://doi.org/10.1128/JB.00673-06
Omoike A, Chorover J (2004) Spectroscopic study of extracellular polymeric substances from Bacillus subtilis: aqueous chemistry and adsorption effects. Biomacromolecules 5:1219–1230. https://doi.org/10.1021/bm034461z
O'Toole G, Kolter R (1998) Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304
O'Toole G, Kaplan HB, kolter R (2000) Biofilm formation as microbial development. Annu Rev Microbiol 54:49–79
Pickering KL, Verbeek CJR, Viljoen C (2012) The effect of aqueous urea on the processing, structure and properties of CGM. J Polym Environ 20:335–343
Quiroz NGA, Hung CC, Santschi PH (2006) Binding of thorium(IV) to carboxylate, phosphate and sulfate functional groups from marine exopolymeric substances (EPS). Mar Chem 100:337–353
Rasamiravaka T, Labtani Q, Duez P, El Jaziri M (2015) The formation of biofilms by Pseudomonas aeruginosa: a review of the natural and synthetic compounds interfering with control mechanisms. Biomed Res Int 2015:759348. https://doi.org/10.1155/2015/759348
Saleh TA (2011) The influence of treatment temperature on the acidity of MWCNT oxidized by HNO3 or a mixture of HNO3 /H2SO4. Appl Surf Sci 257:7746–7751
Saleh TA, Al-Shalalfeh MM, Al-Saadi AA (2016) Graphene dendrimer-stabilized silver nanoparticles for detection of methimazole using surface-enhanced Raman scattering with computational assignment. Sci Rep 6:1–12
Skariyachan S, Sridhar VS, Packirisamy S, Kumargowda ST, Challapilli SB (2018) Recent perspectives on the molecular basis of biofilm formation by Pseudomonas aeruginosa and approaches for treatment and biofilm dispersal. Folia Microbiol 63:413–432. https://doi.org/10.1007/s12223-018-0585-4
Tan CH, Koh KS, Xie C, Tay M, Zhou Y, Williams R, Ng WJ, Rice SA, Kjelleberg S (2014) The role of quorum sensing signalling in EPS production and the assembly of a sludge community into aerobic granules. Isme J 8:1186–1197
Tretinnikov ON, Tamada Y (2001) Influence of casting temperature on the near-surface structure and wettability of cast silk fibroin films. Langmuir 17:7406–7413. https://doi.org/10.1021/la010791y
Wang J, Ding L, Li K, Huang H, Hu H, Geng J, Xu K, Ren H (2018a) Estimation of spatial distribution of quorum sensing signaling in sequencing batch biofilm reactor (SBBR) biofilms. Sci Total Environ 612:405–414
Wang X, An Q, Zhao B, Guo JS, Huang YS, Tian M (2018b) Auto-aggregation properties of a novel aerobic denitrifier Enterobacter sp. strain FL. Appl Microbiol Biotechnol 102:2019–2030. https://doi.org/10.1007/s00253-017-8720-8
Yang L, Han DH, Lee BM, Hur J (2015) Characterizing treated wastewaters of different industries using clustered fluorescence EEM-PARAFAC and FT-IR spectroscopy: implications for downstream impact and source identification. Chemosphere 127:222–228
Yuan SJ, Sun M, Sheng GP, Li Y, Li WW, Yao RS, Yu HQ (2011) Identification of key constituents and structure of the extracellular polymeric substances excreted by Bacillus megaterium TF10 for their flocculation capacity. Environ Sci Technol 45:1152–1157
Zhang P, Fang F, Chen YP, Shen Y, Zhang W, Yang JX, Li C, Guo JS, Liu SY, Huang Y, Li S, Gao X, Yan P (2014) Composition of EPS fractions from suspended sludge and biofilm and their roles in microbial cell aggregation. Chemosphere 117:59–65
Zhang Y, Wang F, Zhu X, Zeng J, Zhao Q, Jiang X (2015) Extracellular polymeric substances govern the development of biofilm and mass transfer of polycyclic aromatic hydrocarbons for improved biodegradation. Bioresour Technol 193:274–280. https://doi.org/10.1016/j.biortech.2015.06.110
Zhao B, Cheng DY, Tan P, An Q, Guo JS (2018) Characterization of an aerobic denitrifier Pseudomonas stutzeri strain XL-2 to achieve efficient nitrate removal. Bioresour Technol 250:564–573. https://doi.org/10.1016/j.biortech.2017.11.038
Zhou W, Zhang HO, Ma Y, Zhou J, Zhang Y (2013) Bio-removal of cadmium by growing deep-sea bacterium Pseudoalteromonas sp. SCSE709-6. Extremophiles 17:723–731
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 51208534) and Technical Innovation and Application Demonstration Project of CQ CSTC (grant no. cstc2018jscx-msybX0308).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animal performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ding, X.S., Zhao, B., An, Q. et al. Role of extracellular polymeric substances in biofilm formation by Pseudomonas stutzeri strain XL-2. Appl Microbiol Biotechnol 103, 9169–9180 (2019). https://doi.org/10.1007/s00253-019-10188-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-10188-4