
Abstract
Alishewanella sp. WH16-1 is a heavy metal-resistant bacterium. Previously, a putative YebC family regulator gene, designated ruvR, was associated with Cr(VI) resistance. In this study, comprehensive analyses were performed to study the role of ruvR and its adjunct putative DNA repairing genes, ruvCAB, in the heavy metal resistance of Alishewanella sp. WH16-1. RT-PCR analysis showed that ruvR is cotranscribed with ruvCAB. Gene mutation and complementation experiments indicated that ruvRCAB contributes to Cr(VI), As(III), Sb(III), and Cd(II) resistance in vivo. Random amplification of polymorphic DNA-PCR revealed that ruvCAB is associated with DNA repair mediated by these metals, and the presence of the metals in the cells was confirmed by elemental mapping and energy-dispersive X-ray spectrograph analysis. In addition, qRT-PCR, reporter gene assay, and in vivo and in vitro protein-DNA interaction experiments indicated that RuvR positively regulates the transcription of ruvCAB and is induced by Cr(VI). Finally, site-directed mutagenesis demonstrated that Asp103 is essential for the DNA binding ability of RuvR. The above results suggest that RuvR is involved in Cr(VI) resistance and resistance to other metals and that RuvR positively regulates the expression of ruvCAB. Based on our study and literatures, a model of RuvRCAB detailing bacterial heavy metal resistance is proposed. The RuvRCAB system plays an important role in the ability of Alishewanella sp. WH16-1 to survive in environments with heavy metals.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
With the rapid development of industrialization, heavy metal contamination has been a serious problem to the environment, and it has caused deleterious effects in living organisms (Meena et al. 2017). Chromium (Cr) is one of the most toxic heavy metals, and it is widespread in the environment. Chromate [Cr(VI)] and chromite [Cr(III)] are the most common forms of chromium in the environment (Dhal et al. 2013). Trace Cr(III) is essential for human and other organisms (Pechova 2007) since it contributes to lipid, sugar, and amino acid metabolism (Bai et al. 2015). However, Cr(VI) is highly toxic, mutagenic, and carcinogenic (He et al. 2010, 2011). Cr(VI) can cross cell membranes through sulfate transporters (Ramírez-Díaz et al. 2008), and it can be subsequently reduced by glutathione, cysteine, or reductases to Cr(III) (Valko et al. 2006). During the reduction process, free radicals and reactive oxygen species (ROS) may be produced (O'Brien et al. 2003), and as the amount of Cr(VI) increases (Ramírez-Díaz et al. 2008), DNA may be damaged by alteration, enzyme inhibition, or lipid and protein oxidation (Arslan et al. 1987; Kadiiska et al. 1994). Arsenic (As), antimony (Sb), and cadmium (Cd) are also common environmental metal(loid) pollutants, and they act as inducers of peroxidation to stimulate the production of excess ROS (De et al. 2003; Li et al. 2015; Sinha et al. 2008; Xia et al. 2016a).
ROS may lead to DNA-protein crosslinks, gene mutations, loss of nucleotides, base oxidation, or single- and double-strand DNA breaks (Cooke et al. 2003; O’Brien et al. 2003). Among all of the types of DNA damage that cells cope with, double-strand breaks are the most harmful. Several studies have demonstrated that heavy metals, such as Cr (Xie et al. 2005), Cu, Cd (Haldsrud and Krokje 2009), and As (Xu et al. 2016), can induce double-strand breaks in eukaryotes. Homologous recombination is a fundamental way to repair bacterial DNA double-strand breaks. In homologous recombination, a four-way DNA intermediate, called Holliday junction, is formed. Following its formation, it must migrate and be resolved. In E. coli, the proteins RuvA, RuvB, and RuvC form a tripartite complex, RuvCAB, to displace and resolve the Holliday junction (Grove et al. 2008). RuvA and RuvB bind to the DNA and drive the branch migration of the Holliday junction in the presence of ATP (Parsons et al. 1992; Tsaneva et al. 1992), while RuvC resolves the Holliday junction (Connolly et al. 1991).
Previously, a gene encoding a putative YebC family regulator protein, designated ruvR, was found to be associated with Cr(VI) resistance by means of Tn5 transposon mutagenesis (Xia et al. 2018a). The YebC family regulator proteins are widespread and conserved in many bacteria. The X-ray structure of a YebC family protein in Aquifex aeolicus showed a negatively charged cavity on the surface, and it was predicted to be a DNA binding domain (Shin et al. 2002). The YebC family protein PmpR negatively regulates the quorum-sensing response regulator pqsR of the quinolone signal system (PQS) in Pseudomonas aeruginosa PAO1 (Liang et al. 2008). YebC is also a potential transcriptional regulator for the acid tolerance response in Lactobacillus delbrueckii subsp. bulgaricus CAUH1 (Zhai et al. 2014) and is involved in the regulation of the proteolytic system in L. delbrueckii subsp. lactis CRL 581 (Brown et al. 2017). Interestingly, yebC-like genes were found to be adjacent to ruvCAB genes in various microorganisms (Yan et al. 2012) indicating that the YebC family protein may also regulate the expression of ruvCAB. However, no direct evidence has been provided for the regulation mechanism of ruvCAB to date.
Alishewanella sp. WH16-1 (CCTCC M201507) was isolated from the soil of a copper and iron mine. The strain is resistant to multiple heavy metals, including Cr(VI), As(III), Sb(III), Cd(II), and Cu(II), and the minimal inhibitory concentrations (MICs) for these metals are 45, 1, 1, 0.1, and 1 mmol/L, respectively (Xia et al. 2016b; Zhou et al. 2016). Moreover, it can reduce Cr(VI), Se(VI), and S(VI) to Cr(III), Se(0), and S(-II), respectively (Xia et al. 2016b, 2018b; Zhou et al. 2016). The microreagent prepared with the fermentation of strain WH16-1 exhibited an enhanced ability to remediate Pd and Cd contamination in pot experiments (Shi et al. 2018; Zhou et al. 2016) (Chinese patent number: CN201710787339). Strain WH16-1 exhibited a particularly high Cr(VI) resistance level and Cr(VI) reduction ability (Xia et al. 2018b). In the genome of strain WH16-1, ruvR is located adjacent to ruvCAB, indicating that ruvR may possess a function related to the transcription of ruvCAB. To explore whether RuvCAB is associated with Cr(VI) resistance and the resistance to other metal(loid) and to determine whether RuvCAB is regulated by RuvR, bioinformatics, genetics, biochemical, microscopical, and chemical analyses were performed in this study.
Materials and methods
Strains, plasmids, and growth conditions
The strains and plasmids used in this study are listed in Table S1, and the primers are listed in Table S2. Alishewanella sp. WH16-1 and the related strains were grown in Luria-Bertani (LB) broth at 37 °C with shaking at 150 rpm unless otherwise noted. Rifampin (Rif, 50 mg/mL), kanamycin (Km, 50 mg/mL), chloramphenicol (Cm, 25 mg/mL), tetracycline (Tet, 5 mg/mL), K2CrO4 [Cr(VI)], NaAsO2 [As(III)], C8H4K2O12Sb2S3(H2O) [Sb(III)], CdCl2 [Cd(II)], and CuSO4 [Cu(II)] were added when required.
Gene arrangement and cotranscriptional analysis
To analyze the gene arrangement of ruvR and ruvCAB, different homologous operons from various microbial genomes were selected (Alishewanella sp. WH16-1 (LCWL00000000), Alishewanella agri BL06T (NZ_AKKU01000001), Alishewanella aestuarii B11T (NZ_ALAB01000001), Rheinheimera pacifica DSM 17616T (NZ_FNXF01000046), Rheinheimera salexigens KH87T (NZ_MKEK01000001), Escherichia coli K-12 (NC_000913), Shewanella oneidensis MR-1 (NC_004347), Shewanella fidelis ATCC BAA-318 (NZ_KI912458), Shewanella woodyi ATCC 51908 (NR_074846.1), Pseudomonas formosensis JCM 18415T (NZ_FOYD01000028), Pseudomonas luteola NBRC 103146 (NZ_BDAE01000001), Pseudomonas aeruginosa PAO1 (NC_002516), Kyrpidia tusciae DSM 2912T (NC_014098), and Nocardioides gansuensis WSJ-1T (QDGZ00000000)). Phylogenetic analysis based on ruvRCAB operon sequences was carried out using MEGA 6.0 (Tamura et al. 2013) and applying the neighbor-joining algorithm (Saitou and Nei 1987) with 1000 bootstrap repetitions to calculate the reliability.
For the cotranscriptional analysis, strain WH16-1 was cultured in 5-mL LB broth with 5 mmol/L Cr(VI) until the OD600 reached approximately 0.3. RNA was extracted with Trizol reagent (Invitrogen), and the residual DNA was removed by digestion with DNase I (Takara). Reverse transcription was carried out using the TRUEscript RT MasterMix (Aidlab) with 300 ng RNA. The obtained cDNA was used as the template in RT-PCR using primers designed between the genes in the ruvRCAB operon (Table S2). Genomic DNA was used as a positive control. RNA and ddH2O were used as negative controls.
Construction of the ruvR mutant and its complemented strain
The ruvR mutant strain was constructed with the suicide allelic exchange vector pCM184 (Marx and Lidstrom 2002). The upstream and downstream sequences of ruvR were PCR amplified and cloned into the AatII-BsrGI site and the ApaI-SacI site of pCM184, respectively. Then, the construction and selection of the mutant strain WH16-1-∆ruvR was performed as described by Xia et al. (2018b). For ∆ruvR complementation, the ruvRCAB sequence was obtained by PCR amplication, and the fragment was cloned into the BamHI-SacI site of the pCT-Zori plasmid (Chen et al. 2015). The generated complemented plasmid was transformed into WH16-1-∆ruvR via conjugation with E. coli S17-1 to generate WH16-1-∆ruvR-C.
Reporter gene assays
The putative promoter and promoter-ruvR sequences were amplified and cloned into the EcoRI-BamHI site of the pLSP kt2lacZ plasmid to produce pLSP-promoter and pLSP-promoter-ruvR, respectively. Then, the pLSP-promoter and pLSP-promoter-ruvR plasmids were each introduced into WH16-1, and the pLSP-promoter was introduced into WH16-1-∆ruvR via biparental conjugation. The strains were each incubated in LB broth overnight, and fresh LB broth was inoculated with 1% of each culture and incubated for another 6 h. The β-galactosidase reporter gene assay was performed as described by Miller (1972). In addition, strain WH16-1 pLSP-promoter-ruvR was cultured to an OD600 of approximately 0.3 and 0, 0.5, 1, and 3 mmol/L Cr(VI) were each added to the broth. The cultures were collected then used for the reporter gene assay after an additional 4-h incubation.
Metal(loid) resistance tests
For the Cr(VI) resistance tests, cultures containing 100-mL LB broth and 0 mmol/L or 5 mmol/L Cr(VI) were each inoculated with 1% of WH16-1, WH16-1-∆ruvR, or WH16-1-∆ruvR-C strains overnight culture. At designated times, the OD600 of the cultures were measured by a spectrophotometer (DU800, Beckman). In addition, LB plates with and without 5 mmol/L Cr(VI) were used to evaluate Cr(VI) sensitivity, and they were incubated for 24 h.
To determine the resistance levels to Cr(VI), As(III), Sb(III), Cd(II), and Cu(II), the above three strains were each transferred to 5-mL LB broth with different concentrations of Cr(VI) (1, 3, 5, and 7 mmol/L), As(III) (200, 400, 600, and 800 μmol/L), Sb(III) (100, 200, 300, and 400 μmol/L), Cd(II) (20, 40, 60, and 80 μmol/L), and Cu(II) (0.5 and 1 mol/L). After incubating for 24 h, the OD600 of each culture was measured using a spectrophotometer.
Elemental mapping and energy disperse X-ray spectroscopy analysis
Strain WH16-1 was cultured in 100-mL LB broth until the OD600 reached approximately 0.3, and 5 mmol/L Cr(VI), 600 μmol/L As(III), 400 μmol/L Sb(III), and 80 μmol/L Cd(II) were added. After 24 h of cultivation, the cell pellets were harvested by centrifugation at 8000 rpm for 5 min. The pellets were washed with 0.85% NaCl solution and were fixed in 1-mL 2.5% glutaraldehyde overnight. Then, ultrathin sectioning was performed. Elemental mapping and EDX analysis were performed according to our previous description (Xia et al. 2018c). In addition, the cell pellets were collected after 24 h of cultivation, and the intracellular concentrations of Cr(VI) and Cd(II) were measured and compared with the Cr(VI) and Cd(II) concentrations initially added. The cell pellets were washed with ddH2O three times to fully remove extracellular metals, and they were lysed using an ultrasonic cell disruptor (Ningbo Xinzhi Instruments, China). After filtering twice (0.22 μM), the intracellular Cr(VI) and Cd(II) concentrations were measured using a spectrophotometer (DU800, Beckman) and an atomic absorption spectrometer (AAS; 986A, Bejing Puxi General Instrument Co., Beijing, China), respectively.
Random amplification of polymorphic DNA-PCR
WH16-1, WH16-1-∆ruvR, and WH16-1-∆ruvR-C strains were each incubated in 100-mL LB broth until the OD600 reached 0.3, and 5 mmol/L Cr(VI), 600 μmol/L As(III), 400 μmol/L Sb(III), and 80 μmol/L Cd(II) were added. At designated times, aliquots of the cultures were collected for RAPD-PCR. RAPD-PCR generally uses several oligonucleotide primers to randomly amplify genomic DNA fragments, and each primer can be used as a forward and reverse primer in PCR. The differences in amplified fragment numbers and sizes indicate the DNA nucleotide changes (Williams et al. 1990).
Genomic DNA from WH16-1, WH16-1-∆ruvR, and WH16-1-∆ruvR-C strains were extracted and purified using a conventional phenol/chloroform method (De Los Reyes-Gavilán et al. 1992). The DNA concentration was assessed by a spectrophotometer (NanoDrop 2000, Thermo). The RAPD-PCR condition was prepared as previously described (Tofalo and Corsetti 2017) with some changes. Briefly, the 25-μL reaction consisted of 2.5-μL 10 × PCR buffer (Mg2+ plus), 3.0 μL dNTP (2.5 mmol/L), 3 μL primer (10 μmol/L), 1 μL rTaq (2 U/μL), 300 ng DNA, and ddH2O. PCR amplification was performed with an initial denaturation step at 95 °C for 5 min followed by 45 cycles consisting of 45 s at 95 °C, 45 s at 45 °C, 2 min at 72 °C, and a final extension of 10 min at 72 °C. The amplified products were electrophoresed in 2.0% agarose gels stained with ethidium bromide.
Bacterial one-hybrid system assay
The interaction between RuvR and the promoter region of the ruvRCAB operon was tested in vivo using a bacterial one-hybrid system (Guo et al. 2009). The ruvR gene and its promoter region were amplified and inserted into the BamHI-EcoRI site of the pTRG vector and the XcmI site of the pBXcmT vector to yield pTRG-RuvR and pBXcmT-PruvR, respectively. Then, the two recombinant plasmids were cotransformed into the reporter strain E. coli XL1-Blue MRF’ Kan. The growth of this strain was tested on a selective screening medium plate containing 20 mmol/L 3-amino-1,2,4-triazole (3-AT), 16 μg/mL Str, 15 μg/mL Tet, 34 μg/mL Cm, and 50 μg/mL Km for 4 days. In addition, E. coli XL1-Blue MRF’ Kan containing the vectors pTRG-Rv3133c and pBXcmT-Mt2031p was used as the positive control, while the strain containing pTRG and pBXcmT was used as a negative control.
Overexpression and purification of RuvR
The RuvR coding sequence was amplified and cloned into the BamHI-HindIII site of the pET32a to generate pET32a-ruvR, which was confirmed by PCR and sequencing. The plasmid was then introduced into E. coli BL21 (DE3). The expression strain was cultured in LB broth at 37 °C, and 0.2 mmol/L IPTG was added when the OD600 reached 0.3. The strain was cultured for 8 h at 28 °C, and then, the cells were harvested by centrifugation and washed twice with 50 mmol/L Tris-HC1 (pH 8.0). The washed cells were lysed via French Press (JN-02C, JNBIO, China) at 120 MPa and centrifuged to remove cell debris. The supernatant was mixed with 1-mL pre-equilibrated Ni NTA Beads 6FF (Smart-lifesciences) in a 10-mL gravity-flow column and agitated at 4 °C for 1 h. Then, 6 mL Tris-HCl with 100 mmol/L imidazole was used to wash miscellaneous proteins, and 2 mL Tris-HCl with 500 mmol/L imidazole was used to elute the target protein. The quality and quantity of the protein were assessed by a spectrophotometer (NanoDrop 2000, Thermo) and SDS-PAGE.
Electrophoretic mobility shift assay
The interaction between RuvR and the promoter region of ruvRCAB was tested in vitro using EMSA. The promoter region was amplified using the primers YP-F/YP-R (listed in Table S2). EMSA was performed with 100 ng FAM-labeled probe (generated using the primer pair YP-F/YP-R FAM) and RuvR (0, 3, 6, and 9 μM), while the heat-inactivated protein was used as a negative control. To verify the specificity of the probe, competition assays were performed with different concentrations of unlabeled probes (400, 600, and 800 ng) in a mixture containing 9 μM RuvR and 100 ng labeled probe. To determine whether Cr(VI) or H2O2 are ligands for RuvR, ligand-dependent EMSA experiments between RuvR and DNA were performed as described above with the addition of different concentrations of Cr(VI) (0, 5, 10, 50, 100, 300, 500, and 700 μmol/L) and H2O2 (0, 0.01, 0.05, 0.1, 0.5, 1, 5, and 10 mmol/L). The reaction mixtures used for EMSA were incubated at 37 °C for 30 min in binding buffer [20 mmol/L Tris-HC1, pH 7.0; 50 mmol/L NaCl; 1 mmol/L dithiothreitol (DTT); 10 mmol/L MgCl2; 100 μg/mL bovine serum (BSA)]. The incubated samples were loaded onto an 8% native PAGE gel that had been prerunning for 30 min at 80 V. The gel was then exposed to a phosphor imaging system (Fujifilm FAL-500) after running at 100 V for 1 h in 0.5× TBE buffer.
Sequence alignment and site-directed mutagenesis of RuvR
The amino acid sequences of YebC family proteins were obtained from NCBI. Multiple alignments were constructed by ClustalW and Espript 3.0 as described elsewhere (Robert and Gouet 2014; Thompson et al. 1994).
Site-directed mutagenesis was performed using the Easy Mutagenesis System Kit (Beijing TransGen Biotech, China) according to the instruction manual. The pET32a-ruvR plasmid was used as a PCR template to amplify the mutant plasmids. After sequencing confirmation, the mutant plasmids were each transformed into E. coli BL21 (DE3). The expression and purification of the mutant protein and ESMA were performed as described above.
Results
The putative regulator gene ruvR was cotranscribed with the DNA repairing genes ruvCAB
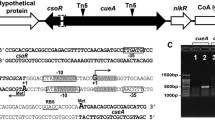
The RuvR (YebC family protein) is a conserved protein in many strains, and the gene arrangement between ruvR and ruvCAB is similar in Alishewanella sp. WH16-1 and other bacterial strains (Fig. 1a). The gene ruvR (AAY72_05130) is located in contig 25 and is directly adjacent to ruvC (AAY72_05135), followed by ruvA (AAY72_05140) and ruvB (AAY72_05145) in strain WH16-1. The cotranscription of these four genes was verified by RT-PCR. The DNA fragments were successfully amplified using primers across any two genes and cDNA as the template. The results indicated that the four genes (ruvR/ruvC, ruvC/ruvA, and ruvA/ruvB) were cotranscribed as an operon (Fig. 1b). The typical RuvRCAB arrangement was found in several Gram-negative bacteria, such as Alishewanella, Rheinheimera, Pseudomonas, and Shewanella spp., while in E. coli and some Gram-positive bacteria (Kyrpidia and Nocardioides spp.), the RuvRCAB arrangement is different to the typical RuvRCAB (Fig. 1), indicating the presence of various DNA repairing mechanisms mediated by RuvR. In addition, the RuvRCAB ortholog was not found in databases of Cyanobacteria (taxid:1117), archaea [Crenarchaeota (taxid: 28889) and Euryarchaeota (taxid:28890) database], fungi [Saccharomyces (taxid:4930)], plants [Arabidopsis (taxid:3701)] or animals [Mus (taxid:10088) and Drosophila (taxid:7215)].

Analysis of the ruvRCAB operon and verification of ruvR mutation and complementation. a The NJ phylogenetic tree based on RuvRCAB amino acid sequences (left) and the gene arrangement of the ruvRCAB operon and its adjacent genes (right). b The cotranscriptional validation of ruvR, ruvC, ruvA, and ruvB. c Verification of ruvR mutation and complementation. M: Trans 2K Plus II DNA marker (TransGen Biotech); lane 4: PCR mixture that lacks DNA
To verify the function of the ruvRCAB operon in Cr(VI) resistance and the resistance to other metal(loid)s, gene knockout and complementation experiments were performed. The completed ruvRCAB operon was complemented in WH16-1-∆ruvR and confirmed by PCR (Fig. 1c) and DNA sequencing (data not shown).
RuvR was induced by Cr(VI) and positively regulated the expression of ruvCAB
To further investigate the roles of RuvR, lacZ reporter gene assays were performed using the strategy shown in Fig. S1. The β-galactosidase activity of E. coli DH5α pLSP-promoter-ruvR was significantly higher than that of DH5α pLSP-promoter (Fig. 2a), and the results for strains WH16-1 and WH16-1-∆ruvR were consistent with that for E. coli DH5α (Fig. 2b). These results suggested that RuvR positively regulates the expression of ruvCAB. Furthermore, we found that the β-galactosidase activity in strain WH16-1 pLSP-promoter-ruvR was highly upregulated when Cr(VI) was added (Fig. 2c). This result was in agreement with the qRT-PCR analysis (Fig. 2d). These results indicate that RuvR is induced by Cr(VI).

The expression of RuvCAB was positively regulated by RuvR and induced by Cr(VI). aβ-galactosidase activities in DH5α pLSP-promoter and DH5α pLSP-promoter-ruvR. bβ-galactosidase activities in WH16-1 pLSP-promoter, WH16-1 pLSP-promoter-ruvR, and WH16-1-∆ruvR pLSP-promoter. c Effects of Cr(VI) on the expression of RuvCAB. β-galactosidase activities were increased following the addition of Cr(VI). d Transcription of ruvR and ruvC detected by qRT-PCR in strain WH16-1 cultivated in LB broth with or without 5 mmol/L of Cr(VI). Data are shown as the mean of three replicates with the error bars representing the standard deviation
The ruvRCAB contributed to Cr(VI) resistance and the resistance to other metal(loid)s
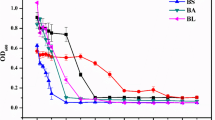
The growth and resistance of WH16-1 (wild-type), WH16-1-∆ruvR (mutant strain), and WH16-1-∆ruvR-C (complemented strain) were tested in LB broth and LB plates with or without Cr(VI). The growth curves of the wild-type, mutant, and complemented strains were similar in the absence of Cr(VI) (Fig. 3a). In contrast, the growth of the mutant strain was significantly inhibited with the addition of Cr(VI) compared to that of the wild-type and complemented strains (Fig. 3b, c). These results indicate that ruvRCAB is associated with Cr(VI) resistance in Alishewanella sp. WH16-1.

Effects of the ruvRCAB operon on Cr(VI) resistance and the resistance to other metals. The growth curves of WH16-1 (wild-type), WH16-1-∆ruvR (mutant strain), and WH16-1-∆ruvR-C (complemented strain) with 0 (a) and 5 mmol/L Cr(VI) (b) in LB broth, and the growth of these strains with 0 and 5 mmol/L Cr(VI) on LB medium plates (c). OD600 values of these strains were measured following the addition of different concentrations of Cr(VI) (d), As(III) (e), Sb(III) (f), and Cd(II) (g). The data are the means of three replicates
It is well known that RuvCAB is a protein complex involved in the migration and resolution of Holliday junctions, which are generated via the homologous recombination repair system. Similar DNA-damaging effects may exist for other heavy metals. Therefore, the growth of WH16-1, WH16-1-∆ruvR, and WH16-1-∆ruvR-C strains in different concentrations of Cr(VI), As(III), Sb(III), Cd(II), and Cu(II) were examined. WH16-1-∆ruvR exhibited increased sensitivity to Cr(VI), As(III), Sb(III), and Cd(II) (Fig. 3d–g), but it was insensitive to Cu(II) (data not shown). Complemented WH16-1-∆ruvR-C exhibited similar metal(loid)-resistant levels compared to the wild-type strain. The above results revealed that the ruvRCAB operon plays an important role in Cr(VI), As(III), Sb(III), and Cd(II) resistance.
Characterization of intracellular metallic elements
To determine the intracellular metallic elements in strain WH16-1 following its exposure to metal(loid)s, elemental mapping and energy disperse X-ray (EDX) spectroscopy analyses were carried out (Fig. 4). The EDX spectrum (Fig. 4c) and TEM elemental mapping (Fig. 4d–g) results revealed that Cr(VI) was present intracellularly in strain WH16-1. After treatment with As(III), Sb(III), and Cd(II), EDX also revealed the presence of the metal(loid)s inside the cells (data not shown). Meanwhile, the intracellular concentrations of Cr(VI) and Cd(II) were also measured, and there were approximately 4.80 nmol Cr(VI)/mg dry weight (± 0.5, n = 3) and 7.88 nmol Cd(II)/mg dry weight (± 0.4, n = 3) accumulated in the cells. The intracellular Cr(VI) content accounted for 0.06% of the added Cr(VI), which was far less than the initial concentration. However, the intracellular Cd(II) content accounted for up to 8.8% of the added Cd(II), indicating that Cd(II) is very easily entering cells and causing the highly toxicity (Fig. S2).

Characterization of intracellular metal content after treated with Cr(VI). TEM image (a), high angle annular dark filed (HAADF) image (b), EDX spectrum (c) and Cr, C, N, and O elemental mapping (d–g) images of cells of strain WH16-1
The effect of metal(loid)s on the RAPD-PCR profiles of WH16-1, WH16-1-ΔruvR, and WH16-1-ΔruvR-C strains
To explore the effects of Cr(VI), As(III), Sb(III), and Cd(II) on the DNA arrangement in WH16-1, WH16-1-∆ruvR, and WH16-1-∆ruvR-C strains, five oligonucleotide random primers were used in RAPD-PCR analysis (Fig. 5). Using primers AK, AM1, AM2, LA1, and M13, the RAPD profiles of strain WH16-1-∆ruvR exposed to Cr(VI), As(III), Sb(III), and Cd(II) exhibited significant changes compared to that of strain WH16-1, with visible changes in the amplified DNA fragment numbers and sizes. In addition, the complemented strain WH16-1-∆ruvR-C exhibited similar RAPD profiles compared to WH16-1. The RAPD profiles amplified using other primers showed no DNA polymorphism among the wild-type, mutant, and complemented strains, indicating that the RAPD-PCR is reliable (Fig. S3). These results suggest that the ruvRCAB operon plays an important role in repairing DNA rearrangements caused by Cr(VI), As(III), Sb(III), and Cd(II).

RAPD-PCR profiles of DNA for WH16-1, WH16-1-∆ruvR, and WH16-1-∆ruvR-C strains treated with Cr(VI) (a), As(III) (b), Sb(III) (c), and Cd(II) (d). Lane M, 1, 2, and 3 show Trans 2K Plus II DNA marker (TransGen Biotech), WH16-1, WH16-1-∆ruvR, and WH16-1-∆ruvR-C, respectively; M13, LA1, AM1, AM2, and AK are primer names. The red line indicates one primer
RuvR interacts with the promoter DNA of ruvRCAB
Bacterial one-hybrid assays were carried out to examine the interaction between RuvR and its promoter region in vivo (Guo et al. 2009). Detection of the protein-DNA interaction is based on the transcriptional activation of His3 (imidazoleglycerol-phosphate dehydratase coding gene involved in histidine biosynthesis) and addA (streptomycin resistance gene). The reporter strain containing pTRG-RuvR and pBXcmT-PruvR exhibited similar growth to the positive control on the screening plate, while the negative control strain did not grow. These results suggest that RuvR interacts with the ruvRCAB promoter in vivo (Fig. 6a).

Bacterial one-hybrid assay and EMSA analysis for the interaction between RuvR and the promoter of the ruvRCAB operon. a Bacterial one-hybrid assay. The reporter strain containing pBX-PruvR/pTRG-RuvR was an experimental strain, and strains containing pBX-Mt2031p/pTRG-Rv3133c and the empty vector pBXcmT/pTRG were used as the positive and negative control strains, respectively. Overnight LB broth cultures (5 mL each) were inoculated with the experimental, positive, or negative control strains. Then, 2 μL of each culture was spotted onto His-selective plate (+ 3 AT, +Strr) and LB plate (− 3 AT, −Strr). b Purification of RuvR. c EMSA assay. Lanes 1–4, the band shift was enhanced by increasing the amount of RuvR. Lane 5, heat-inactivated protein was used as a negative control. Lanes 6–8, FAM-labeled DNA was outcompeted by unlabeled DNA. d The predicted structural model of Alishewanella sp. WH16-1 RuvR created using SWISS-MODEL. Conserved residues mutated in this study are indicated in red (Asp103) and blue (Thr86, Asn104, and Val129). e The functional verification of the site mutants. The Asp103 mutant lost its ability to bind DNA, while the other mutants retained their ability to interact with DNA
The binding ability of RuvR was then detected in vitro by EMSA using the purified His6-tag RuvR (Fig. 6b) and the promoter DNA of the ruvRCAB operon. With increasing amounts of RuvR, the free DNA substrates gradually disappeared, and the strength of the DNA-protein complex increased (Fig. 6c). In addition, the unlabeled DNA probe competed with the labeled probe for the DNA-RuvR interaction (Fig. 6c). To confirm whether Cr(VI) and H2O2 affect the binding, ligand-dependent analyses revealed that the free DNA did not change with increasing amounts of Cr(VI) and H2O2 (Fig. S4), indicating that Cr(VI) and H2O2 are not direct ligands of RuvR. These results suggest that RuvR interacts with the promoter region of ruvRCAB in vitro.
The amino acid alignments of RuvR with its homologs revealed that there are 14 conserved residues (Fig. S5). To determine the functional sites of RuvR, a structural model of RuvR from Alishewanella sp. WH16-1 was predicted using SWISS-MODEL (Arnold et al. 2006; Guex et al. 2009; Kiefer et al. 2009), and the conserved residues Asp103, Thr86, Asn104, and Val129 (Fig. 6d) were each mutated. The EMSA results revealed that the Asp103 mutant lost its ability to bind the DNA, while the other mutant proteins were still able to interact with the DNA (Fig. 6e). Thus, Asp103 is essential for RuvR activity.
Discussion
Based on the genetics, in vivo and in vitro protein-DNA interactions and chemical analysis in Alishewanella sp. WH16-1, we found that RuvCAB plays a role in the resistance to several metal(loid)s by association with DNA repair and RuvR has a positive regulatory effect on the expression of RuvCAB. Based on our results and literatures, we propose a model representing bacterial Cr(VI), As(III), Sb(III), and Cd(II) resistance mediated by RuvRCAB. (1) Cr(VI), As(III), Sb(III), and Cd(II) enter cells through transporter systems; (2) the metal(loid)s cause DNA rearrangement; (3) RuvR positively regulates RuvCAB to repair DNA rearrangement (Fig. 7). Bioinformatics analysis implicates that the DNA repairing mechanisms mediated by RuvRCAB may be mainly distributed in some Gram-negative bacteria.

A model representing the heavy metal resistance mechanisms of WH16-1. (1) Cr(VI), As(III), Sb(III), and Cd(II) enter cells through transporter systems; (2) the metal(loid)s cause DNA rearrangement; (3) RuvR positively regulates RuvCAB’s expression to repair the DNA rearrangements
Using transposon mutagenesis, RuvB was previously found to be associated with Cr(VI) resistance in P. aeruginosa PAO1 and Ochrobactrum tritici 5bvl1 (Miranda et al. 2005; Morais et al. 2011). Santoyo et al. (2015) reported that Cr(VI) induces the expression of RuvB in Rhizobium etli. Joe et al. (2011) found that RuvCAB was induced by Cd(II) using a whole-genome DNA microarray, but Cd(II) resistance was not studied. Our results are in agreement with these findings, but in addition to the above studies, here we provide comprehensive genetic evidence for the necessity of RuvCAB in the bacterial resistance to metal(loid)s. In our study, the synthesis of RuvCAB may have been blocked in the WH16-1-∆ruvR mutant strain; thus, the migration and resolution of four-way Holliday junctions were affected, and the repair of DNA was prevented. This may explain the higher Cr(VI) susceptibility of this mutant strain. We also found that RuvCAB is involved in As(III), Sb(II), and Cd(II) resistance, but it is not involved in Cu(II) resistance. WH16-1 was isolated from Fe/Cu mine soil and exhibited extensive Cu(II) resistance. Thus, this bacterium may possess several strategies to tolerate Cu(II), and RuvCAB may not be a key player in Cu(II) tolerance in our experimental condition. In this study, we determined that certain amounts of Cr(VI) and Cd(II) were indeed present in the cytosol of strain WH16-1. Recently, it was found that approximately 2.6% and 4.4% of As(III) and Sb(III) was taken up by E. coli cells, respectively (Shi et al. 2018a). The toxicity of different metal(loid)s to bacteria is varied due to bacterial cell wall/membrane structure, uptake, efflux, redox transformation, DNA repair, and movement systems (Shi et al. 2018b).
To the best of our knowledge, we show that the YebC family protein RuvR is involved in Cr(VI), As(III), Sb(III), and Cd(II) resistance by positively regulating the expression of RuvCAB for the first time. We found that RuvR was induced by Cr(VI), but RuvR induction by As(III), Sb(III), and Cd(II) was not significant (data not shown). This may be related to the highly Cr(VI) resistance ability of strain WH16-1 compared to other metal(loid)s. In addition, the ligand-dependent EMSA analyses showed that Cr(VI) and H2O2 are not ligands that can directly bind to RuvR to regulate the transcription of downstream ruvCAB genes. It is possible that the presence of Cr(VI) in the cytosol causes other signals, such as ROS, Fenton reaction substances, or superoxide anion-related substances, to be direct ligands for the expression of RuvR.
The ruvR and ruvCAB genes were cotranscribed in strain WH16-1, and a similar gene arrangement was conserved and widely spread in various microbes indicating the functional relatedness of RuvR and RuvCAB. However, before our study, the YebC family of proteins was reported to possess other functions. For example, in P. aeruginosa PAO1, the gene arrangement of pmpR (YebC family protein gene) and ruvCAB is identical to that in WH16-1 (66% amino acid identity between PmpR and RuvR). However, PmpR regulates the PQS system by binding to the promoter region of the pqsR gene (Liang et al. 2008). YebC proteins (98% amino acid identity) from two different L. delbrueckii strains regulate acid tolerance via pyrG and clpC (Zhai et al. 2014) and the proteolytic system via prtL, oppA1, and optS (Brown et al. 2017). Recently, it was found that YebC not only regulates quorum sensing by binding to the edwR promoter region, but it also activates T3SS expression in Edwardsiella piscicida (Wei et al. 2018). These results indicate that YebC family proteins have diverse functions and that RuvR may also possess additional regulatory roles in strain WH16-1.
In addition, we found that Asp103 is essential for RuvR activity by performing site-directed mutagenesis analysis. It was reported that Asp103 forms the third ion pair network located in domain 2 with Arg108, Glu112, and Asp-230 to stabilize the entire tertiary structure of the YebC family protein in A. aeolicus (Shin et al. 2002). The protein structural analysis performed by SWISS-MODEL indicated that Asp103 is located in the coil structure at the corner that connects the beta-strand and alpha-helix; therefore, the structure of RuvR may change after the residue is mutated. This may be the reason why the Asp103 mutant of RuvR lacks the ability to bind DNA.
References
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22(2):195–201
Arslan P, Beltrame M, Tomasi A (1987) Intracellular chromium reduction. Biochim Biophys Acta 931(1):10–15
Bai J, Xun P, Morris S, Jacobs DR, Liu K, He K (2015) Chromium exposure and incidence of metabolic syndrome among American young adults over a 23-year follow-up: the CARDIA trace element study. Sci Rep 5:15606
Brown L, Villegas J, Elean M, Fadda S, Mozzi F, Saavedra L, Hebert E (2017) YebC, a putative transcriptional factor involved in the regulation of the proteolytic system of Lactobacillus. Sci Rep 7(1):8579
Chen F, Cao Y, Wei S, Li Y, Li X, Wang Q, Wang G (2015) Regulation of arsenite oxidation by the phosphate two-component system PhoBR in Halomonas sp. HAL1. Front Microbiol 6:923
Connolly B, Parsons CA, Benson FE, Dunderdale HJ, Sharples GJ, Lloyd RG, West SC (1991) Resolution of Holliday junctions in vitro requires the Escherichia coli ruvC gene product. Proc Natl Acad Sci U S A 88(14):6063–6067
Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17(10):1195–1214
De BM, Kirsch-Volders M, Lison D (2003) Cobalt and antimony: genotoxicity and carcinogenicity. Mutat Res 533(1):135–152
De Los Reyes-Gavilán CG, Limsowtin GK, Tailliez P, Séchaud L, Accolas JP (1992) A Lactobacillus helveticus-specific DNA probe detects restriction fragment length polymorphisms in this species. Appl Environ Microbiol 58(10):3429–3432
Dhal B, Thatoi HN, Das NN, Pandey BD (2013) ChemInform abstract: chemical and microbial remediation of hexavalent chromium from contaminated soil and mining/metallurgical solid waste: a review. J Hazard Mater 250–251(30):272–291
Grove JI, Harris L, Buckman C, Lloyd RG (2008) DNA double strand break repair and crossing over mediated by RuvABC resolvase and RecG translocase. DNA Repair 7(9):1517–1530
Guex N, Peitsch MC, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30(Suppl 1):S162–S173
Guo M, Feng H, Zhang J, Wang W, Wang Y, Li Y, Gao C, Chen H, Feng Y, He ZG (2009) Dissecting transcription regulatory pathways through a new bacterial one-hybrid reporter system. Genome Res 19(7):1301–1308
Haldsrud R, Krokje A (2009) Induction of DNA double-strand breaks in the H4IIE cell line exposed to environmentally relevant concentrations of copper, cadmium, and zinc, singly and in combinations. J Toxicol Environ Health A 72(3–4):155–163
He M, Li X, Guo L, Miller SJ, Rensing C, Wang G (2010) Characterization and genomic analysis of chromate resistant and reducing Bacillus cereus strain SJ1. BMC Microbiol 10:221
He M, Li X, Liu H, Miller SJ, Wang G, Rensing C (2011) Characterization and genomic analysis of a highly chromate resistant and reducing bacterial strain Lysinibacillus fusiformis ZC1. J Hazard Mater 185(2–3):682–688
Joe MH, Jung SW, Im SH, Lim SY, Song HP, Kwon O, Kim DH (2011) Genome-wide response of Deinococcus radiodurans on cadmium toxicity. J Microbiol Biotechnol 21(4):438–447
Kadiiska MB, Xiang QH, Mason RP (1994) In vivo free radical generation by chromium(VI): an electron spin resonance spin-trapping investigation. Chem Res Toxicol 7(6):800–805
Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res 37:D387–D392
Li J, Wang Q, Li M, Yang B, Shi M, Guo W, McDermott TR, Rensing C, Wang G (2015) Proteomics and genetics for identification of a bacterial antimonite oxidase in Agrobacterium tumefaciens. Environ Sci Technol 49(10):5980–5989
Liang H, Li L, Dong Z, Surette MG, Duan K (2008) The YebC family protein PA0964 negatively regulates the Pseudomonas aeruginosa quinolone signal system and pyocyanin production. J Bacteriol 190(18):6217–6227
Marx CJ, Lidstrom ME (2002) Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques 33(5):1062–1067
Meena RAA, Sathishkumar P, Ameen F, Yusoff ARM, Feng LG (2017) Heavy metal pollution in immobile and mobile components of lentic ecosystems—a review. Environ Sci Pollut Res 1–15
Miller JH (1972) Experiments in molecular genetics. 58(4):893–924
Miranda AT, Gonzalez MV, Gonzalez G, Vargas E, Campos-Garcia J, Cervantes C (2005) Involvement of DNA helicases in chromate resistance by Pseudomonas aeruginosa PAO1. Mutat Res 578(1–2):202–209
Morais PV, Branco R, Francisco R (2011) Chromium resistance strategies and toxicity: what makes Ochrobactrum tritici 5bvl1 a strain highly resistant. Biometals 24(3):401–410
O'Brien TJ, Ceryak S, Patierno SR (2003) Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res Fundam Mol 533(1–2):3–36
Parsons CA, Tsaneva I, Lloyd RG, West SC (1992) Interaction of Escherichia coli RuvA and RuvB proteins with synthetic Holliday junctions. Proc Natl Acad Sci U S A 89(12):5452–5456
Pechova A, Veterinarni a Farmaceuticka Univ, Brno (2007) Chromium as an essential nutrient: a review. Vet Med- Czech 52(1):1–18
Ramírez-Díaz MI, Díaz-Pérez C, Vargas E, Riveros-Rosas H, Campos-García J, Cervantes C (2008) Mechanisms of bacterial resistance to chromium compounds. Biometals 21(3):321–332
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406
Santoyo G, Orozco-Mosqueda M, Valdez-Martinez G, Orozco-Mosqueda Mdel C (2015) Induction of the homologous recombination system by hexavalent chromium in Rhizobium etli. Microbiol Res 170:223–228
Shi X, Zhou G, Liao S, Shan S, Wang G, Guo Z (2018) Immobilization of cadmium by immobilized Alishewanella sp. WH16-1 with alginate-lotus seed pods in pot experiments of cd-contaminated paddy soil. J Hazard Mater 357:431–439
Shi K, Li C, Rensing C, Dai X, Fan X, Wang G (2018a) Efflux transporter ArsK is responsible for bacterial resistance to arsenite, antimonite, trivalent roxarsone and methylarsenite. Appl Environ Microbiol 84(24):e01842–e01818
Shi K, Wang Q, Fan X, Wang G (2018b) Proteomics and genetic analyses reveal the effects of arsenite oxidation on metabolic pathways and the roles of AioR in Agrobacterium tumefaciens, GW4. Environ Pollut 235:700–709
Shin DH, Yokota H, Kim R, Kim SH (2002) Crystal structure of conserved hypothetical protein Aq1575 from Aquifex aeolicus. Proc Natl Acad Sci U S A 99(12):7980–7985
Sinha M, Manna P, Sil PC (2008) Protective effect of arjunolic acid against arsenic-induced oxidative stress in mouse brain. J Biochem Mol Toxicol 22(1):15–26
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Tofalo R, Corsetti A (2017) RAPD-PCR as a rapid approach for the evaluation of genotoxin-induced damage to bacterial DNA. Methods Mol Biol 1644:195–201
Tsaneva IR, Müller B, West SC (1992) ATP-dependent branch migration of Holliday junctions promoted by the RuvA and RuvB proteins of E. coli. Cell 69(7):1171–1180
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160(1):1–40
Wei L, Wu Y, Qiao H, Xu W, Zhang Y, Liu X, Wang Q (2018) YebC controls virulence by activating T3SS gene expression in the pathogen Edwardsiella piscicida. FEMS Microbiol Lett 365
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18(22):6531–6535
Xia L, Chen S, Dahms HU, Ying X, Peng X (2016a) Cadmium induced oxidative damage and apoptosis in the hepatopancreas of Meretrix meretrix. Ecotoxicology 25(5):959–969
Xia X, Li J, Liao S, Zhou G, Wang H, Li L, Xu B, Wang G (2016b) Draft genomic sequence of a chromate- and sulfate-reducing Alishewanella strain with the ability to bioremediate Cr and cd contamination. Stand Genomic Sci 11:48
Xia X, Wu S, Li L, Xu B, Wang G (2018a) The cytochrome bd complex is essential for chromate and sulfide resistance and is regualted by a GbsR-type regulator, CydE, in Alishewanella sp. WH16–1. Front Microbiol 9:1849
Xia X, Wu S, Li N, Wang D, Zheng S, Wang G (2018b) Novel bacterial selenite reductase CsrF responsible for se(IV) and Cr(VI) reduction that produces nanoparticles in Alishewanella sp. WH16-1. J Hazard Mater 342:499–509
Xia X, Zhou Z, Wu S, Wang D, Zheng S, Wang G (2018c) Adsorption removal of multiple dyes using biogenic selenium nanoparticles from an Escherichia coli strain overexpressed selenite reductase CsrF. Nanomaterials 8(4):234
Xie H, Wise SS, Holmes AL, Xu B, Wakeman TP, Pelsue SC, Singh NP, Wise JP Sr (2005) Carcinogenic lead chromate induces DNA double-strand breaks in human lung cells. Mutat Res 586(2):160–172
Xu H, Zhou X, Wen X, Lauer FT, Liu KJ, Hudson LG, Aleksunes LM, Burchiel SW (2016) Environmentally relevant concentrations of arsenite induce dose-dependent differential genotoxicity through poly(ADP-ribose) polymerase inhibition and oxidative stress in mouse thymus cells. Toxicol Sci 149(1):31–41
Yan Z, Jie L, Yang G (2012) In silico identification of a multi-functional regulatory protein involved in Holliday junction resolution in bacteria. BMC Syst Biol 6(1):1–10
Zhai Z, Douillard FP, An H, Wang G, Guo X, Luo Y, Hao Y (2014) Proteomic characterization of the acid tolerance response in Lactobacillus delbrueckii subsp. bulgaricus CAUH1 and functional identification of a novel acid stress-related transcriptional regulator Ldb0677. Environ Microbiol 16(6):1524–1537
Zhou G, Xia X, Wang H, Li L, Wang G, Zheng S, Liao S (2016) Immobilization of lead by Alishewanella sp. WH16-1 in pot experiments of Pb-contaminated paddy soil. Water Air Soil Pollut 227(9)
Funding
This study was funded by the National Key Research and Development Program of China (grant number 2016YFD0800702) and National Natural Science Foundation of China (grant number 31870086).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1530 kb)
Rights and permissions
About this article

Cite this article
Wu, S., Xia, X., Wang, D. et al. Gene function and expression regulation of RuvRCAB in bacterial Cr(VI), As(III), Sb(III), and Cd(II) resistance. Appl Microbiol Biotechnol 103, 2701–2713 (2019). https://doi.org/10.1007/s00253-019-09666-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-09666-6