
Abstract
Porcine follicle-stimulating hormone (pFSH), comprising α and β subunits, is commonly used to induce superovulation in domestic animals in assisted reproduction technologies; however, the practical application of pFSH is inhibited by the limited efficiency of its production. Recombinant yeast-derived FSH offers a practical alternative; however, the heterologous expression efficiency remains disappointingly low. To improve FSH production in Pichia pastoris, a series of molecular strategies, together with fermentation optimization, were tested in the present study. By comparing clones of the Muts phenotype strain, it was observed that the yield of soluble pFSH increased by approximately 96% in clones of the Mut+ phenotype strain. The protein levels of soluble pFSHβ, which confers biological specificity, increased by approximately 143 and 22% after two kinds of codon optimization strategies, respectively. Moreover, compared with the production of soluble pFSHβ and SUMO-pFSHβ, the production of soluble protein HSA-pFSHβ was significantly improved. Furthermore, the optimum pH and methanol concentration for expressing soluble HSA-pFSH in strain H3-3 were determined as 5.0–6.0 and 1.5–2% in shake-flask, and the yield of soluble HSA-pFSH could reach 40.8 mg/l after purification. In vitro bioactivity assays showed that recombinant HSA-pFSH could efficiently stimulate cAMP synthesis in HEK293 cells expressing porcine FSHR. In conclusion, our results demonstrated that the application of phenotypic selection of aox1 mutants, combined with codon optimization, the choice of fusion partners, and fermentation optimization, considerably increased the yield of pFSH in supernatant of P. pastoris and thus provided a valuable reference for the large-scale recombinant expression of pFSH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Follicle-stimulating hormone (FSH) is a member of the glycoprotein hormone family, which share a common α subunit and a unique β subunit that confers biologic specificity (Ryan et al. 1988). FSH is widely used to induce ovulation in human clinics and superovulation in domestic animal reproductive management. However, the limited use of FSH preparations is mainly the result of the low production efficiency of FSH purified from the urine of menopausal women or the pituitary of animals.
Recombinant proteins produced in Pichia pastoris have low production costs, which could offer a practical alternative. Expression in P. pastoris not only has the advantages of prokaryotic protein expression systems, such as fast reproduction, high density fermentation, and simplified purification procedure for secreting heterologous proteins, but also has characteristics of eukaryotic expression systems, such as post-translational modification (Cregg et al. 1993; Cregg et al., 2000). The yields of many recombinant proteins are suitable for industrial production (Bar et al. 1992; Shekhar et al. 2008). Many therapeutic proteins produced by P. pastoris, such as ocriplasmin, insulin, and ecallantide, have been approved in the USA and Europe (Shekhar, 2008; Walsh et al. 2014).
However, not every heterologous protein can be efficiently expressed in P. pastoris, because of the genetic background of a P. pastoris host strain, which could affect transcription, translation efficiency, or secretion of the target protein (Macauley et al. 2005). To solve this problem, a set of strategies has been applied to improve recombinant protein expression in P. pastoris, e.g., phenotypic selection of aox1 mutants (Kim et al. 2009), codon optimization (Huang et al., 2008a; Teng et al. 2007; Elena et al. 2014), fusion partners (Liu et al. 2008; Esposito and Chatterjee, 2006; Gou et al. 2012), and culture condition optimization (Holmes et al. 2009; Jahic et al., 2003a).
P. pastoris strains can be divided according to their ability to utilize methanol: the wild-type or methanol-utilization plus phenotype (Mut+, rapid growth in methanol), the methanol-utilization slow phenotype (Muts, slow growth in methanol), and methanol-utilization minus (Mut−). The Mut− strain, disrupted both aox1 and aox2 genes, is unable to grow on methanol and thus is not suitable for high-level expression of heterologous proteins in P. pastoris. Therefore, phenotypic selection of aox1 mutants is essential to improve the production of FSH in P. pastoris.
Codon optimization could increase heterologous protein production, and many parameters of codon optimization, such as codon bias toward the host, free energy, and G + C content, can potentially affect the production of heterologous proteins in P. pastoris (Sinclair and Choy, 2002; Huang et al., 2008b). However, it remains unclear whether codon optimization would improve the efficiency of recombinant pFSH production in P. pastoris.
Additionally, fusion partners, such as small ubiquitin-related modifier (SUMO) and human serum albumin (HSA), could also increase heterologous protein expression by reducing degradation or improving protein folding and solubility. Wu et al. (2009) reported that the human keratinocyte growth factor 2 expression level could be improved by fusing it with SUMO in Escherichia coli, and the SUMO fragment could be removed by SUMO proteases (Wu et al. 2009). A similar result was observed in a eukaryotic expression system (Peroutka et al. 2008). Thus, SUMO may be a good choice as a fusion tag to increase the production of FSH in P. pastoris. Likewise, HSA fusion technology has also been widely used in P. pastoris (Bloom et al. 2003; Shen et al. 2012; Wang et al. 2013). Moreover, the production level of HSA in P. pastoris could reach 1–12 g per liter levels (Kobayashi et al. 2000; Ohya et al. 2005; Belew et al. 2008). Therefore, using HSA as a carrier protein might represent a promising strategy to improving the production of heterologous proteins (Subramanian et al. 2007).
In addition to current molecular strategies, conventional culture condition optimization (pH or methanol concentration) could also increase the production of heterologous proteins in P. pastoris. It has been reported that the yield of recombinant protein produced in P. pastoris could increase by more than 10-fold by optimizing the culture conditions (Zhao et al. 2008; Mao et al. 2015).
In the present study, we have systematically tested the effects of different molecular strategies, combined with culture condition optimization, on the yield efficiency of recombinant soluble porcine FSH (pFSH). It should be mentioned that insoluble recombinant pFSH that remained intracellularly was also collected and detected in the present study, but the biological activity of pFSH may be impaired due to misfolding and degradation (Wang et al. 2017; Idiris et al. 2010; Vanz et al. 2014).
Materials and methods
Strains, plasmids, reagents, and cell lines
E. coli strain Top10 (CWbio, Beijing, China), P. pastoris strains GS115 and KM71, and vectors pPIC9K and pPICZαA (Invitrogen, Shanghai, China) were used for cloning and heterologous expression. The HEK293 cell line was purchased from Cell Bank of Peking Union Medical College (Beijing, China). Endoglycosdase H and peptide-N-glycosidase (PNGase) F were purchased from New England Biolabs (Beijing, China). Porcine FSH was supplied by SIOUX Biochemical (Sioux Center, IA, US). The cAMP levels in cells were detected using a cAMP enzyme-linked immunosorbant assay (ELISA) kit (NewEast Bioscience Wuhan, China). All culture media used for the cells were purchased from Gibco (Invitrogen, Shanghai, China), and analytical grade chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Codon optimization of pFSHβ
The mature pFSHβ subunit cDNA (GenBank: NM_213875.1) was optimized according to the codon usage of P. pastoris (De Schutter et al. 2009). The lower frequency codons were replaced with synonymous codons with a higher frequency in P. pastoris, and the G + C content was altered, accompanied by eliminating AT-rich sequences. The secondary structure and free energy (ΔG) of the mRNA were predicted at 30 °C using an RNA structure webserver (http://rna.urmc.rochester.edu/RNAstructureWeb/). The codon adaptation index (CAI) was calculated at http://www.genscript.com/cgi-bin/tools/rare_codon_analysis. Two optimized genes (named as opt-FSHβ-A and opt-FSHβ-B, GenBank: MH249032 and MH249033, respectively), which differed mainly in their G + C contents, were used to compare the effect of different parameters of codon optimization on the production of pFSHβ. The coding sequences of pFSHβ, SUMO-pFSHβ, HSA-pFSHβ, pFSHα (Gene pFSHα also known as CGA, GenBank: MH252913), and HSA-pFSHα were optimized according to the optimal parameters determined above.
Construction of expression plasmid of pPIC9K-pFSHβ-pFSHα
The mature pFSHβ subunit cDNA bearing a sequence encoding a C-terminal 6 × His-tag and the mature pFSHα subunit cDNA were optimized according to the codon bias of P. pastoris and synthesized by Tsingke Biological Technology (Beijing, China). The PCR fragments of pFSHβ and pFSHα, amplified by primer pairs P1 + P2 and P3 + P4 (all primers are shown in Table 1), respectively, were digested with EcoRI/NotI and then ligated into corresponding site of plasmid pPIC9K to construct vectors pPIC9K-pFSHα and pPIC9K-pFSHβ.
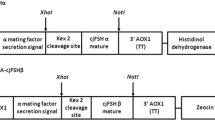
The recombinant plasmid carrying both pFSHβ and pFSHα subunit cDNAs, pPIC9K-pFSHβ-pFSHα, was constructed as described in Fig. 1a. The DNA fragment of the pFSHα expression cassette (including 5′AOX1, MF-α, pFSHα, 3′AOX1 TT) was amplified from vector pPIC9K-pFSHα using primer pairs P5 + P6 (underlined XbaI site) and then digested by XbaI, followed by insertion into the linearized plasmid pPIC9K-pFSHβ cut with XbaI. The vector pPIC9K-pFSHβ-pFSHα was used to screen the optimal Mut phenotype of P. pastoris strains for expressing pFSH. The Mut phenotype of the GS115 recombinant strain was confirmed in MM (1.34% yeast nitrogen base (YNB), 0.00004% biotin, 0.5% methanol, 1.5% Agar), and MD (1.34% YNB, 0.00004% biotin, 2% dextrose, 1.5% Agar) plates.

Schematic diagram of the pPIC9K construct. a Construction of expression plasmid pPIC9K-pFSHβ-pFSHα. b Schematic representation of the pFSHβ, SUMO-pFSHβ, and HSA-pFSHβ expression cassettes in vector of pPIC9K. 5′AOX1, promoter of an alcohol oxidase 1; MFα, an α-factor secretion signal; 3′AOX1 TT, transcription termination of an alcohol oxidase 1; MCS, multiple cloning site; GGGGS, a flexible peptide linker
Construction of fusion protein expression plasmids pPIC9K-pFSHβ, pPIC9K-SUMO-pFSHβ, pPIC9K-HSA-pFSHβ, and coexpression plasmid pPICZαA-HSA-pFSHα
The SUMO-encoding fragment (from vector of pY-secSUMOstar (LifeSensors, Inc.) with BglII (G > A) mutant) and truncated mature HSA fragment (GenBank: AAN17825.1, 1Asp-402Lys) were used to make N-terminal fusions with the pFSHβ sequence with a flexible linker of Gly-Gly-Gly-Gly-Ser, as shown in Fig. 1b, to form SUMO-pFSHβ and HSA-pFSHβ bearing a 6 × His-tag at the N-terminus (GenBank: MH249034 and MH249035, respectively). The DNA sequence encoding pFSHβ, SUMO-pFSHβ, and HSA-pFSHβ fragment with flanking EcoRI and NotI restriction enzymes sites were synthesized by Tsingke Biological Technology (Beijing, China). These DNA fragments and P. pastoris expression vector pPIC9K were digested by EcoRI/NotI and then ligated together to construct vectors pPIC9K-pFSHβ, pPIC9K-SUMO-pFSHβ, and pPIC9K-HSA-pFSHβ. Similarly, the HSA-pFSHα construct comprised the HSA truncated fragment, a LysArgGlu sequence as a linker, the mature sequence of pFSHα, followed by a 6 × His-tag was subcloned into P. pastoris expression vector pPICZαA to form pPICZαA-HSA-pFSHα (Fig. 1b). All fragments above were verified by DNA sequencing before transformation. All primers used in this study are shown in Table 1.
Transformation and screening recombinant strain
The pPIC9K constructs (including vectors pPIC9K-pFSHβ-pFSHα, pPIC9K-pFSHβ, pPIC9K-SUMO-pFSHβ, and pPIC9K-HSA-pFSHβ) were linearized using BglII and transformed into P. pastoris strains GS115 by the electroporation method using a Bio-Rad Gene Pulser Xcell (2 kV, 25 μF, 200 Ω). Positive transformants were incubated at 30 °C for 1 h and initially screened on MD plates. Then, 100 clones from the MD plates were selected in YPD + G418 (Invitrogen, Shanghai, China) plates (1% yeast extract, 2% peptone, 2% dextrose, 2% agar and 0.6–0.8-mg/ml G418).
For coexpression with HSA-pFSHα, the plasmid pPICZαA-HSA-pFSHα, linearized using PmeI, was introduced into recombinant GS115 strains with high production of the FSHβ fusion protein. After transformation, clones were selected directly on YPD + Zeocin (Invitrogen, Shanghai, China) plates (300 μg/ml Zeocin instead of G418) for 3–5 days at 30 °C.
Recombinant protein expression in shake flasks
Clones from YPD plates containing G418 or Zeocin were picked to inoculate into 3 ml BMGY (1% yeast extract, 2% peptone, 1.34% YNB, 0.00004% biotin, 1% glycerol, 100 mM potassium phosphate, pH 6.0) at 30 °C and shaken at 250 rpm. After 48 h of culture, cells were harvested by centrifugation at 3000 ×g for 5 min, followed by decanting all the supernatant carefully and resuspending in 1 ml of BMMY (same as BMGY but replacing glycerol with 0.5% methanol) to induce expression for 72 h. Methanol was added into the BMMY cultures every 24 h to maintain the concentration at 0.5%. After inducing, the recombinant protein in 5 μl of culture supernatant was analyzed by SDS-PAGE and western blotting.
To select the optimal pH and methanol concentration, a strain with a high yield of both FSH subunits was picked to inoculate in BMGY at 30 °C, with shaking at 250 rpm. After 48 h of growth, cells were collected and induced in BMMY at different pH values (5.0, 5.5, 6.0, 6.5, 7.0, 7.5, and 8.0) or in BMMY with different methanol concentrations (0.5, 1, 1.5, 2, 2.5, and 3%). There were three repeats for each pH or methanol concentration treatment. Every 24 h, methanol was supplied to the culture, and the supernatants were collected after 72 h for SDS-PAGE analysis.
The recombinant protein in supernatant was purified by using a BeaverBeads™ His-tag Protein Purification Kit (Bioscience, Suzhou, China) following the manufacturer’s instructions. The purified protein was obtained by elution with 500 mM imidazole and desalting by ultrafiltration.
Expression of pFSH by fed-batch fermentation
A strain with high expression of pFSH fusion proteins and the optimized culture conditions were used for fed-batch fermentation. The strain was selected from an MM plate and cultivated in 50 ml of YPD overnight, followed by continuing culture for 18 h in 600 ml YPD. The entire propagated culture was then used for fed-batch fermentation in a 15 L fermenter containing 9 L of fermentation broth. Fermentation was divided into four phase: glycerol batch, glycerol fed-batch, glycerol and methanol fed-batch, and methanol fed-batch. The medium used for high cell density fermentation was basal salt medium (BSM), as mentioned in the Pichia Fermentation Process Guidelines proposed by Invitrogen (2002). The recombinant protein in the fermentation supernatant was collected every 24 h and analyzed by SDS-PAGE and western blotting. After 144 h, all the fermentation broth was centrifuged at 8000 ×g, for 15 min at 4 °C, filtered through a 0.22 μm membrane, concentrated, and then stored at − 80 °C until analysis. For the glycosylation analysis and bioactivity assays, the concentrated supernatant was dialyzed overnight against phosphate-buffered saline (PBS) and reconstituted in FSH-binding buffer (Richard et al. 1998).
Polyacrylamide gel electrophoresis analysis and western blot
The recombinant proteins in the culture supernatant induced by methanol were analyzed on 12% SDS-PAGE and western blotting. Gels were stained by Coomassie blue R-250. For western blotting, a mouse anti-human FSHβ monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:500) was used as the primary antibody to detect FSHβ (SIOUX Biochemical, Inc., IA), SUMO-FSHβ, or HSA-FSHβ protein. A mouse anti-His tag monoclonal antibody (Abclonal Technology, Beijing, China, 1:5000) was used to detect proteins with a His-tag. The secondary antibody (Zhongshan Jinqiao Biotech, Beijing, China, diluted 1:5000–1:10000) was horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG. The immunoreactive proteins on the blots were visualized with ECL and imaged on an ImageQuant LAS 4000 instrument. The band intensities were quantified using the ImageJ software.
Mass spectrometry analysis
The protein bands on the SDS-PAGE gel stained by Coomassie blue R-250 were cut, and the protein digestion was performed using FASP method with modifications (Wiśniewski et al. 2009). Briefly, 100 μg protein was dissolved with 50 mM ABC (NH4HCO3), reduced with DTT (dithiothreitol) at 56 °C for 45 min, and alkylated with IAM (iodoacetamide) at room temperature for 30 min in the dark. The solution was transferred into a 10 K ultrafiltration tube (Vivacon 500, Satrorius), spinned at 14,000 ×g for 20 min. A 50 mM ABC solution was used to wash the protein for three times. A 2 μg trypsin in 50 μL 50 mM ABC was added and incubated at 37 °C overnight. The ultrafiltration tube was spinned at 14,000 ×g for 20 min with a new collection tube to collect digested peptides. ABC solution was added into the ultrafiltration tube to wash the digested peptide into the collection tube. The collected solution was diluted with 0.1% FA for nanoLC-MS analysis.
Raw data from the mass spectrometer were preprocessed with Mascot Distiller 2.4 for peak picking. The resulted peak lists were searched against Sus_scrofa_uniprot_pig database using Mascot 2.5 search engine. The search parameters are fixed modifications: carbamidomethyl (C), variable modifications: oxidation (M), and phosphorylation (S, T, Y). Enzyme: trypsin, maximum missed cleavages: 2, MS mass tolerance: 10 ppm, MSMS mass tolerance: 0.02 Da. Scaffold PTM was used to evaluated phosphorylation sites of the Mascot search results using Ascore algorithm.
Glycosylation analysis
The glycosylation of recombinant HSA-pFSH was analyzed using endoglycosidase H or PNGase F (NEB, Beijing, China) digestion, following the manufacturer’s instructions. In brief, recombinant HSA-pFSH or pituitary FSH was denatured by heating at 100 °C for 10 min, followed by incubation for 1 h at 37 °C in the presence or absence of either 2500 U of endoglycosidase H or PNGase F. Deglycoslated proteins were analyzed using western blotting.
Establishment of a stable HEK293-pFSHR cell line
The porcine FSHR gene was amplified from ovarian granulosa cell cDNA using specific primer pair P7 + P8 with EcoRI and XhoI restriction sites. The PCR product was digested by EcoRI/XhoI and then ligated into corresponding sites of pCAGGS-EGFP-Neo (constructed in our laboratory) to generate vector pCAGGS-Neo-EGFP-pFSHR. The vector pCAGGS-Neo-EGFP-pFSHR was used to transfect into HEK293 cells (maintained in our laboratory) using Vigofect (Vigorous Biotechnology, Beijing, China) following the manufacturer’s instructions. The stably transfected cell line was screened in DMEM medium (Gibco, 11960) containing 700 μg/ml of G418 and maintained in DMEM medium including 350 μg/ml of G418. The pFSHR expression level in HEK293-pFSHR cell line was assessed using quantitative real-time PCR (qPCR) analysis with primers P9 + P10 and P11 + P12 (human GAPDH used as reference gene).
Quantitative real-time PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen, Shanghai, Beijing), and 1 μg of total RNA was reverse transcribed using HiScript™ Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme Biotech Co., Ltd., Nanjing, China). Quantitative real-time PCR was performed using the CFX96 real-time system (Bio-Rad, Beijing, China) in a reaction mixture containing (in a total volume 10 μl): 2× SsoFast EvaGreen Supermix (Bio-Rad, Beijing, China), forward and reverse primers, cDNA, and ddH2O. PCR was performed at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 5 s, and a melting curve was constructed at the end of the amplification. Triplicate samples of each template were analyzed. The expression levels of pFSHα and pFSHβ in strains with integration of both pFSH subunit genes were determined using qPCR analysis with primers P13 + P14 and P15 + P16, respectively, and primers P17 + P18 for the reference gene ARG4 for relative quantification.
Copy number quantification of gene pFSHβ and pFSHα in strains GS115-pPIC9K-pFSHβ-pFSHα and KM71-pPIC9K-pFSHβ-pFSHα
The copy number of gene pFSHβ and pFSHα in recombinant strains were determined according to the method reported by Abad et al. (2010). Genomic DNA was isolated by using the TIANamp Yeast DNA Kit (Tiangen, Beijing, China) following the manufacturer’s instruction. Absolute real-time qPCR was used to analyze the copy number of gene pFSHα and pFSHβ in recombinant strains with primers P19 + P20 and P21 + P22, respectively, and primers P23 + P24 for reference gene GAPDH.
In vitro bioassays of recombinant pFSH
HEK293-pFSHR cells were seeded in 12-well plates at a density of 2 × 105/well. After 48 h, the cells were pretreated with 0.8 mM 1-methyl-3-isobutylxanthine (IBMX) for 15 min in new DMEM medium and after 1 h, 20 ng of recombinant HSA-pFSH or pituitary FSH (measured by ELISA) were added, and as controls, 20 μl DPBS (Gibco) or HSA (Sigma-Aldrich, MO, USA) was added. Subsequently, the cells were lysed in 0.1 M HCl. The concentration of intracellular cAMP was measured using a cAMP ELISA Kit (Neweast bioscience, Inc.) with Infinite M200 (Tecan, Sweden).
EMBL/GenBank accession numbers
The GenBank accession number of pFSHB is NM_213875.1; the GenBank accession number of opt-pFSHβ-A, opt-pFSHβ-B, and opt-CGA (pFSHα) are MH249032, MH249033, and MH252913, respectively. The GenBank accession number of SUMO-pFSHβ and HSA-pFSHβ are MH249034 and MH249035.
Results
Effect of Mut+ and Muts strains on yield efficiency of recombinant pFSH
To investigate the effect of strain phenotype (Mut+ or Muts) on the protein expression of pFSH, we compared the soluble pFSH production in supernatant of strains GS115 (Mut+) or KM71 (Muts) transformed with pPIC9K-pFSHβ-pFSHα. To obtain intact pFSH with both pFSHα and pFSHβ subunits, the expression vector pPIC9K-pFSHβ-pFSHα containing two expression cassettes (including 5′AOX1-MFα-pFSHβ/pFSHα-3′AOX1 TT) was constructed and transformed into strains GS115 (Mut+) or KM71 (Muts) (Fig. 1a). After transformation and induction, the protein levels of soluble pFSH in supernatant were determined using western blotting with an anti-His monoclonal antibody, as shown in Fig. 2a. Three protein bands were clearly observed on the western blot. The middle protein band corresponded to the pFSHβ subunit (molecular weight approximately 23 kDa, black arrow) with a C-terminal His-tag, and the upper and lower protein bands (molecular weights approximately 35 and 15 kDa, respectively) were suspected to be a dimer of pFSHβ and pFSHα and a degraded pFSH protein, respectively (Fig. 2a). To test the possible influence of integrated copy number of the heterologous genes on protein yield, we detected the copy number of pFSHβ and pFSHα in GS115-pPIC9K-pFSHβ-pFSHα (Mut+) and KM71-pPIC9K-pFSHβ-pFSHα (Muts) strains by real-time qPCR. As shown in Table 2, each strain of Mut+-1, Mut+-2, Muts-3, and Muts-4 has a single copy of pFSHα and two copies of pFSHβ integrated into the genome. Therefore, it seems that the different yields of soluble recombinant protein in the Mut+ and Muts strains are not due to the copy numbers. These data indicated that compared with protein level in supernatant of strain KM71 (Muts), the production of pFSHβ in the supernatant of strain GS115 (Mut+) was increased by about 96%, based on densitometry of the protein bands (Fig. 2b).

Effect of methanol-utilization plus (Mut+) and methanol-utilization slow (Muts) phenotypes on pFSH expression in Pichia pastoris. a The recombinant protein in supernatant from GS115-pPIC9K-pFSHβ-pFSHα (Mut+) or KM71-pPIC9K-pFSHβ-pFSHα (Muts) strains was analyzed using western blotting with a His-tag antibody. Lane M represents protein markers, lanes 1–2 represent the proteins in the supernatant of GS115-pPIC9K-pFSHβ-pFSHα strains, and lanes 3–4 represent the proteins in the supernatant of KM71-pPIC9K-pFSHβ-pFSHα strains. The middle protein band corresponds to the pFSHβ subunit with a C-terminal His-tag (marked by an arrow). b The relative amounts of FSHβ protein in each strain were estimated using ImageJ analysis software, which values were normalized to the cell density at 600 nm
Effect of codon optimization on yield efficiency of recombinant pFSHβ
Codon optimization has been widely applied to improve recombinant protein expression, but has not been used to optimize the expression of pFSH in P. pastoris. To test the possibility of improving pFSH production via codon optimization, the different coding sequences of the mature pFSHβ subunit, which mainly confers receptor specificity, were synthesized based on different parameters (named opt-pFSHβ-A and opt-pFSHβ-B). After optimization, a total 63 and 82 nucleotides were changed in the opt-pFSHβ-A and opt-pFSHβ-B sequences, respectively, which showed 80.73 and 74.92% identity with wild-type pFSHβ (wt-pFSHβ). Moreover, compared with the wt-pFSHβ sequence, the G + C content and ΔG of mRNA folding decreased from 52 to 48.3% and from − 122.4 to − 114.3 kcal mol−1 respectively, and the CAI increased from 0.67 to 0.84 in the opt-pFSHβ-A sequence (Table 3). The parameters were changed to 37.9% for G + C content, − 91.8 kcal mol−1 for ΔG, and 0.96 for the CAI in the opt-pFSHβ-B sequence (Table 3). The wild-type and optimized coding sequences were subcloned into vector pPIC9K and then expressed in strain GS115. After 72 h of induction, the yield of soluble pFSHβ in the supernatant of each strain was compared using western blotting analysis (based on the average densitometry of the protein bands; Fig. 3a). As shown in Fig. 3b, compared with the GS115-pPIC9K-wt-pFSHβ strain (the GS115 strain transformed with the wild-type pFSHβ gene), the yield of pFSHβ in GS115-pPIC9K-pFSHβ-A and GS115-pPIC9K-pFSHβ-B strains (the GS115 strain transformed by gene opt-pFSHβ-A or opt-pFSHβ-B) increased by approximately 143 and 22%, respectively, indicating that the parameters optimized in gene opt-pFSHβ-A were better for expressing pFSHβ in P. pastoris (Fig. 3c).

Comparison of the production of pFSHβ in GS115 strains transformed with different wild-type pFSHβ (wt-FSHβ) or optimized pFSHβ (opt-pFSHβ-A or opt-pFSHβ-B). a Three strains carrying each gene integration were picked and induced for 72 h; the pFSHβ yields in supernatant were determined by western blotting. Lanes 1–3: wt-pFSHβ; lanes 4–6: opt-pFSHβ-A; lanes 7–9: opt-pFSHβ-B. b The relative amounts of pFSHβ protein in each strain was estimated using ImageJ analysis software, which values were normalized to the cell density at 600 nm
Effect of SUMO or HSA fusion partners on yield efficiency of recombinant soluble pFSHβ
Next, we investigated whether the recombinant production of pFSHβ in P. pastoris could be further improved by fusion with SUMO or HSA. The expression vectors pPIC9K-pFSHβ, pPIC9K-SUMO-pFSHβ, and pPIC9K-HSA-pFSHβ were constructed and transformed into P. pastoris strain GS115 (Fig. 1b). To investigate the effect of the fusion partner on the yield of pFSHβ, four GS115-pPIC9K-pFSHβ strains, five GS115-pPIC9K-SUMO-pFSHβ strains, and four GS115-pPIC9K-HSA-pFSHβ strains were selected for the induction of pFSHβ expression. After 72 h, the protein levels of soluble pFSHβ, SUMO-pFSHβ, and HSA-pFSHβ in the supernatant of each strain were determined by SDS-PAGE with Coomassie brilliant blue staining. As shown in Fig. 4a, two protein bands for HSA-pFSHβ were clearly observed in supernatant of the GS115-pPIC9K-HSA-pFSHβ strains. The upper band and the lower band corresponded to intact HSA-pFSHβ (molecular weight approximately 68 kDa) and the truncated HSA fragment (molecular weight approximately 45 kDa), respectively, which was confirmed by western blotting (Fig. 4b) and mass spectrometry analysis (data not shown). However, no protein band of pFSHβ and SUMO-pFSHβ was detected in Coomassie brilliant blue-stained SDS-PAGE, but it was successfully expressed in supernatants of recombinant strains GS115-pPIC9K-pFSHβ or GS115-pPIC9K-SUMO-pFSHβ, respectively (Fig. 4a, b, molecular weights approximately 23 and 36 kDa). These data indicated that fusion with HSA can significantly improve the production efficiency of soluble pFSHβ.

Comparison of protein expression of pFSHβ, SUMO-pFSHβ, and HSA-pFSHβ in strains transformed with each gene. a and b SDS-PAGE (a) and western blotting (b) analysis of recombinant proteins in the supernatants from GS115-pPIC9K-pFSHβ, GS115-pPIC9K-SUMO-pFSHβ, or GS115-pPIC9K-HSA-pFSHβ strains. Lane M: protein marker, lanes 1–4, 5–9, and 10–13: culture supernatants from recombinant GS115-pPIC9K-pFSHβ, GS115-pPIC9K-SUMO-pFSHβ, and GS115-pPIC9K-HSA-pFSHβ strains, respectively
Coexpression of HSA-pFSHβ and HSA-pFSHα
A previous study showed that the bovine FSHβ subunit expressed by P. pastoris had no bioactivity to stimulate cAMP production in rat Sertoli cells, unless assembled with an ovine α subunit of luteinizing hormone to form a heterodimer (Samaddar et al. 1997). To obtain biologically active HSA-pFSH, we construct the vector pPICZαA-HSA-pFSHα, which was transformed into strain GS115-pPIC9K-pFSHβ (named as H3). Four strains, which were defined as H3-1, H3-2, H3-3, and H3-4, were selected in media containing 300 μg ml−1 Zeocin plates after transformation. At 72 h following methanol induction, the protein level in supernatants of strains H3, H3-1, H3-2, H3-3, and H3-4 was analyzed using SDS-PAGE and western blotting (using an anti-FSHβ monoclonal antibody). As shown in Fig. 5a, b, the protein band of intact HSA-pFSHβ was found in the supernatant of strains H3-1, H3-2, H3-3, and H3-4, indicating that the HSA-pFSHβ gene was not replaced by the HSA-pFSHα gene in those strains after transformation. However, the yield of HSA fragment in supernatant of those strains increased significantly compared with the yield in supernatant of strain H3. This might be related to the KRE amino acid sequence of the kex2 protease cleavage site, which was present between the HSA truncated fragment and pFSHα.

Effect of coexpression HSA-pFSHα on the production of the HSA-pFSHβ fusion protein. a and b SDS-PAGE (a) and western blotting (b) analysis of proteins in the supernatant of GS115-pPIC9K-HSA-pFSHβ strain (named as H3) transformed with pPIC9K-HSA-pFSHα. Lane M: protein marker, lane 1: supernatant from H3, lanes 2–5: supernatants from positive transformants named as H3-1, H3-2, H3-3, and H3-4, respectively. c and d Transcription level of pFSHα (c) and pFSHβ (d) at different induction times. H3-3 engineered strain was picked for induction, and intracellular pFSHα and pFSHβ expression was determined using real-time PCR at 24 h intervals
Due to the low yield of pFSHα (Fig. 5a, no protein band was detected at ~ 24 kDa) and lack of commercially available antibody against pFSHα, the expression level of gene pFSHα, as well as pFSHβ, in recombinant strain H3-3, was evaluated by real-time qPCR after induction for different times. Compared with 0 h (before being induced by methanol), the expression levels of pFSHβ and pFSHα increased significantly after induction by methanol (Fig. 5c, d), indicating that both pFSHβ and pFSHα had been successfully integrated and substantially expressed in strain H3-3.
Effect of pH and methanol concentrations on the yield efficiency of recombinant soluble pFSHβ
Based on the tested molecular strategies, we next evaluated the effect of pH (from 5.0 to 8.0) and methanol concentration (from 0.5 to 3.0%) on the production of soluble HSA-pFSH in strain H3-3. After 72 h induction, the protein levels in the supernatants were analyzed using SDS-PAGE. As shown in Fig. 6a, with increasing pH, the protein level of soluble HSA-pFSHβ in the supernatant of strain H3-3 did not change significantly; the intensities of the protein bands at approximately 180 and 100 kDa changed obviously, which may be related to the formation of multimers (marked by arrows). To decrease the formation of multimers, which may be associated with enhanced degradation, the optimum pH in the medium for soluble HSA-pFSH protein expression in strain H3-3 was set at 5.0 to 6.0 (Nordén et al. 2011). Similarly, the highest yield of soluble HSA-pFSH protein in the supernatant of strain H3-3 was obtained when the methanol concentration was between 1.5 and 2% in the medium (Fig. 6b). However, the final HSA-pFSH production decreased when the methanol concentration was more than 2.5% or less than 1.5%. These results showed that the optimum pH and methanol concentration were 5.0–6.0 and 1.5–2.0%, respectively, to express HSA-pFSH.

Expression and purification of the recombinant HSA-pFSH fusion protein. a and b Influence of pH (a) and methanol concentration (b) on the expression of HSA-pFSH in the supernatant of strain H3-3. c and d SDS-PAGE (c) and western blotting (d) analysis of purified recombinant HSA-pFSH. Lane M: protein marker; lane 1: recombinant HSA-pFSH protein purified using a His-tag protein purification magnetic bead kit; lanes 2, 3: purified HSA-pFSH detected by western blotting using a mouse monoclonal antibody against hFSHβand using a mouse anti-His-tag monoclonal antibody, respectively
Protein expression and purification in shake-flask
On the basis of the high protein yield in the supernatant of strain H3-3, we further purified the recombinant HSA-pFSH protein using a His-tag Protein Purification Kit. A total 0.94 mg of recombinant soluble HSA-pFSH was obtained from 23 ml of supernatant. The purified protein was analyzed by SDS-PAGE and western blotting. As shown in Fig. 6c, three protein bands were found on SDS-PAGE. The upper band (molecular weight approximately 68 kDa) corresponded to the HSA-pFSHβ fusion protein, which was confirmed by western blotting using a mouse monoclonal antibody against hFSHβ (Fig. 6d, lane 2). Likewise, the middle band (molecular weight approximately 45 kDa) corresponded to the truncated HSA fragment (Fig. 6d, lane 3). There is no His-tag on the end of pFSHβ when HSA-pFSHβ was disconnected, thus we speculated that the protein band at approximately 24 kDa was the pFSHα subunit with a His-tag in its C-terminus, which corresponded with the observed size of the protein band. (Fig. 6d, lane 3).
High-density fermentation of the recombinant HSA-pFSH fusion protein
To improve the concentration and yield of the fusion protein, strain H3-3 was selected for recombinant protein expression in 9 L of culture medium. The fermentation medium was sampled at 24 h intervals and the cell wet weight and total protein were measured in the medium. As a result, the cell wet weight and total protein increased with time, indicating that the cells were growing and secreting proteins (Fig. 7a). In addition, the fusion protein of soluble HSA-pFSHβ began to degrade after 48 h, and the levels of the degraded protein products of HSA-pFSHβ in the fermentation supernatant, such as HSA and pFSHβ, increased up to 120 h, indicating that the HSA-pFSH fusion protein began to disconnect after 48 h and was degraded after 120 h (Fig. 7b, c). However, when the protein band at 23 kDa was detected by mass spectrometry, we found that approximately 10% of the fraction was ubiquitin not than pFSHβ (data not shown). Thus, we speculated that a large number of proteases were generated in large-scale fermentation, which could degrade the HSA-pFSH recombinant fusion protein.

Production of recombinant protein HSA-pFSH in Fed-batch fermentation of strain H3-3. The fusion protein HSA-pFSH in the supernatant of strain H3-3 was collected every 24 h and centrifuged at 12,000 ×g, 4 °C, 3 min. a Time curve of cell wet weight and total protein concentration at 30 °C. Cell wet weight and total protein concentration were detected by a weighing test and a BCA protein assay, respectively. b and c SDS-PAGE (a) and western blotting (c) analysis of the recombinant protein in the supernatant of strain H3-3 at different induction times. The upper and lower arrows indicate the HSA-pFSHβ protein band and the pFSHβ or pFSHα protein bands, respectively. d Deglycosylation analysis of recombinant HSA-pFSH. Supernatant harvested at 144 h was concentrated 10-fold, dialyzed against PBS and reconstituted in binding buffer. Plus(+) indicates pituitary FSH or recombinant HSA-pFSH treated with PNGase F or Endo H, otherwise the data is marked as minus (−)
The N-glycosylation analysis of the recombinant HSA-pFSH protein was performed by enzymatic digestion followed by western blotting with an anti-pFSHβ antibody. As shown in Fig. 7d, recombinant soluble pFSHβ and HSA-pFSHβ protein could be cleaved by both PNGase F and Endo H enzymes, whereas pituitary FSH could only be cleaved by PNGase F. This result indicated that N-glycosylation of recombinant soluble HSA-pFSH was the high mannose-type.
In vitro bioactivity assay of recombinant soluble HSA-pFSH
FSH bioactivity can be determined by testing its ability to induce cyclic AMP production in HEK293 cells expressing FSHR (Kutteyil et al. 2015). To evaluate the bioactivity of recombinant HSA-pFSH, we constructed a cell line that constitutively expressed pFSHR. As shown in Fig. 8a, the plasmid harboring the pFSHR gene was successful transformed into HEK293 cells, as indicated by GFP expression in the cells. Moreover, compared with HEK293 cells transfected with pCAGSS, the expression level of pFSHR was significantly increased in HEK293 cells transfected with pCAGGS-pFSHR (Fig. 8b). These results indicated that pFSHR was successful expressed in HEK293.

Detection of recombinant protein HSA-pFSH’s biological activity. a Effect of the transfection of HEK293 cell with plasmid pCAGGS or pCAGGS-pFSHR. HEK293 cells were transfected using Vigofect with plasmid pCAGGS or pCAGGS-pFSHR (containing EGFP) and the transfection efficiency was determined by assaying EGFP. The HEK293-pFSHR stable cell line was screened in DMEM with 700 μg/ml G418 and maintained in DMEM with 350 μg/ml G418. Scale bars: 100 μm. b Relative mRNA expression level of pFSHR in HEK293 cells transfected by pCAGGS or pCAGGS-pFSHR. c cAMP concentration in HEK293-pFSHR cell treated with pitFSH or recombinant HSA-pFSH. Different letters above the bars indicate a significant difference at p < 0.05
The bioactivity of the purified soluble HSA-pFSH fusion protein was detected by measuring the cAMP levels in the HEK293-pFSHR cell line after treating with dialyzed supernatant or pituitary FSH for 1 h. As a result, the intracellular cAMP concentration after treatment with soluble rHSA-pFSH was dramatically increased compared with that treated with DPBS or HSA, although it was significantly lower than that in cells treated with pituitary FSH (Fig. 8c), indicating that recombinant HSA-pFSH has a satisfactory biological activity to induce FSHR signal transduction.
Discussion
FSH is essential to maintain follicular growth in females and to initiate spermatogenesis in males. To date, many bioactive FSH from different species has been successfully expressed in different protein expression systems, such as the baculovirus system (Van de Wiel et al. 1998), the Chinese hamster ovary cell expression system (Keene et al. 1989), plant systems (Dirnberger et al. 2001), P. pastoris expression systems (Richard et al. 1998), and transgenic rats or rabbits (Greenberg et al. 1991; Coulibaly et al. 2002). Until now, although recombinant FSH has been used in clinical trials, the cost per oocyte is higher than that of human chorionic gonadotropin because of the low production efficiency in recombinant expression systems (Devroey et al., 2012; Kutteyil et al. 2015; Mountford et al. 1994). To decrease the cost of FSH preparation, we applied molecular strategies, combined with culture condition optimization, to improve the production of recombinant pFSH in P. pastoris.
Different proteins are suitable for expression in different Mut phenotype P. pastoris hosts (Kim et al. 2009; Pla et al. 2006). In the current study, compared with the Muts phenotype host KM71, we found that the Mut+ phenotype of host GS115 was more suitable for expressing soluble pFSH. The codon bias and mRNA folding energy in mammals are different from those in P. pastoris, which might influence mRNA stability and translation efficiency (Kim et al. 1997). Similarly, codon optimization and altered G + C content could also increase the yield of recombinant luciferase (Sinclair and Choy, 2002). In the present study, we compared different parameters of codon optimization, especially the G + C content, for their effects of the production of soluble pFSHβ. As a result, a higher yield of soluble pFSHβ was obtained from gene opt-pFSHβ-A with 48.3% G + C content. Similarly, the protein level of truncated 1,3-1,4-β-D-glucanase also improved when the G + C content was adjusted to 48–49% in P. pastoris (Huang et al., 2008a). Moreover, the yield of a heterologous protein could be increased by altering the G + C content only, which could affect the expression of the protein by limiting the transcription level (Newman et al. 2016; Sinclair and Choy, 2002). Likewise, our results indicated that the protein level decreased when the G + C content was less than 40%. Thus, we speculated that the G + C content might be the major parameter that influences the expression of soluble pFSHβ in P. pastoris. Collectively, our data suggested that phenotypic selection of aox1 mutants and codon optimization could improve the yield of pFSHβ in P. pastoris.
The yield of recombinant bovine FSH could be improved in the baculovirus system when the expressed sequence included a major part of the FSHβ gene 3′ untranslated region (Van de Wiel et al. 1998). This provides a way to increase the expression of FSH by fusion at the N- or C-terminus of FSHβ using a fusion partner. Based on the observation that SUMO could increase the expression of recombinant proteins in yeast (Hughes et al. 2008), we fused the SUMO sequence to the N-terminus of pFSHβ and found that SUMO did not significantly increase the production of pFSHβ. Likewise, it has been reported that fusion with HSA could improve bikunin expression in P. pastoris (Gou et al. 2012). In the present study, the production of the soluble HSA-pFSHβ fusion protein was higher than that of SUMO-pFSHβ and pFSHβ. However, an HSA truncated fragment (~ 45 kDa), in addition to the intact HSA-pFSHβ protein, was detected in supernatant of strain H3, indicating that the fusion protein had been degraded. Similar degradation phenomena occurred for other HSA fusion proteins expressed in P. pastoris (Yao et al. 2009). The ~ 45 kDa fragment could be reduced or even eliminated when the FQNALLVRYTKK (403Phe-414Lys) sequence of HSA was mutanted in Saccharomyces cerevisiae (Kerry-Williams et al., 1998); however, this would not work in P. pastoris because a large part of ~ 45 kDa fragment was detected in our assay. Thus, fusion with HSA could increase the production of soluble pFSHβ, but it was accompanied by degradation. In addition to the optimization of phenotypic selection of AOX1 mutants, codon, and fusion partners, the yield of soluble HSA-pFSHβ protein in the culture supernatants can be further improved through increasing the secretion efficiency of soluble HSA-pFSHβ (Idiris et al. 2010), coexpressing with molecular chaperone (Sha et al. 2013), or increasing the copy number of gene HSA-pFSHβ (Mansur et al. 2005).
After optimization using molecular strategies, the recombinant soluble HSA-pFSH protein in the supernatant was purified and the yield reached 40.8 mg/l in the shake-flask, which was significantly higher than the 10 mg/l reported by Richard et al. (1998). Furthermore, the total protein in the supernatant from high-density fermentation reached 6 g/l; however, it included many degraded fragments of the fusion protein (Fig. 7b).
Intracellular and extracellular degradation is a prominent limitation to achieve high-efficiency recombinant protein production (Van de Wiel et al. 1998). It has been reported that intracellular degradation of recombinant bovine FSHβ might be related to hyposecretion, which can be improved by coexpression with FSHα or chaperone molecules (Mountford et al. 1994; Pajot-Augy et al. 1995). Moreover, extracellular degradation in a protease deficient strain was also observed by expressing HSA (Kang et al. 2000). In this study, HSA-pFSHβ and HSA-pFSHα fusion proteins were coexpressed in strain H3-3, which may be reduced intracellular degradation; however, the extracellular degradation of soluble HSA-pFSHβ still occurred when protein was expressed in shake-flask cultures (Fig. 5c, d), indicating that the soluble HSA-pFSH fusion protein expressed in P. pastoris was unstable. Likewise, the degradation phenomenon of recombinant HSA or cellulose-binding module and lipase B fusion protein was also reported in P. pastoris, which could be overcome by adjusting the pH of the culture medium (Kobayashi et al. 2000; Kang et al. 2000; Jahic et al., 2003b). In the present study, the effect of pH from 5.0 to 8.0 on the production of soluble HSA-pFSH was investigated in shake-flask fermentation (Damasceno et al. 2012; Dueck et al. 2004; Hiyama and Renwick, 1990). However, the production and degree of degradation of soluble fusion protein HSA-pFSH were not obviously improved when pH was varied between 5.0 and 8.0. Thus, whether the degradation of soluble HSA-pFSH produced in bioreactor cultures was associated with pH requires further study. Interestingly, a protein at approximately 180 kDa appeared when the pH was greater than 6.5, which might represent covalently-linked multimers formed among HSA-pFSHβ, pFSHα, and pFSHβ. Multimers and degradation fragments were also detected in addition to the intact protein in supernatants when aquaporins were expressed in P. pastoris, suggesting that there may be some link between multimer formation and degradation (Nordén et al. 2011). The reason for this phenomenon is not clear; however, it may be related to the culture environment, such as the pH or temperature, which may not be suitable for soluble HSA-pFSH expression in the bioreactor. Moreover, a large amount of ubiquitin related to protein degradation was found in the fermentation supernatant, indicating that proteases such as metallopeptidases might be a major factor affecting the stability of the recombinant protein (Vad et al. 2005). In addition to optimizing the pH of the culture medium, fermentation temperature optimization, and fermentation using YPS1 (encoding a protease the specifically cleaves at the C-terminal side of basic amino acids) and PEP4 (encoding proteinase A) deficient strains could also solve the degradation problem (Jahic et al., 2003a; Yao et al. 2009; Wu et al. 2013). Thus, the degradation problems associated with HSA fusion could be improved by reducing intracellular protease activity or optimizing the culture conditions.
Furthermore, CHO or HEK293 cell lines expressing different species of FSHR are widely used to detect the biological activity of recombinant FSH (Richard et al. 1998; Kim et al. 2007; Kutteyil et al. 2015). In the present study, we established an HEK293 cell line expressing porcine FSHR to detect the bioactivity of recombinant HSA-pFSH in vitro. By measuring intracellular cAMP after stimulating HEK293-pFSHR cells with recombinant HSA-pFSH, we found that recombinant soluble HSA-pFSH has biological activity; however, its activity was less potent than pituitary FSH, indicating that the bioactivity of HSA-pFSH was affected by the presence of HSA. It has been reported that fusion with HSA can decrease the protein’s bioactivity because of increased spatial interference (Bloom et al. 2003; Huang et al., 2008b), which could not be improved using different linkers between HSA and hGH177–191 (Wang et al. 2013). However, recombinant fusion proteins with HSA such as Albugon (GLP-1/albumin fusion protein) exhibited the full spectrum of biological activities in preclinical studies, albeit with reduced bioactivity in vitro (Baggio et al. 2004). Therefore, whether HSA fusion at the N-terminus of pFSH affected the bioactivity of pFSH requires further confirmation using in vivo assays. Nevertheless, the recombinant HSA-pFSH protein expressed in P. pastoris is biologically active.
In conclusion, we have systematically optimized the expression of pFSH in P. pastoris. By integrating several molecular strategies, combined with optimizing the culture condition, we achieved a satisfactory efficiency for producing recombinant pFSH. To the best of our knowledge, this is the first report of the expression of pFSH fused with HSA in P. pastoris.
References
Abad S, Kitz K, Hörmann A, Schreiner U, Hartner FS, Glieder A (2010) Real-time PCR-based determination of gene copy numbers in Pichia pastoris. Biotechnol J 5(4):413–420
Bar KA, Hopkins SA, Sreekrishna K (1992) Protocol for efficient secretion of HSA developed from Pichia pastoris. Pham Eng 12:48–51
Baggio LL, Huang Q, Brown TJ, Drucker DJ (2004) A recombinant human glucagon-like peptide (GLP)-1–albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes 53(9):2492–2500
Belew M, Li Yan M, Wei Z, Caldwell K (2008) Purification of recombinant human serum albumin (rHSA) produced by genetically modified Pichia pastoris. Sep Sci Technol 43(11–12):3134–3153
Bloom M, Block J, Duttaroy A, Grzegorzewski K, Moor P, Ou Y, Wojcik S, Zhou X, Bell A (2003) Albugon™ fusion protein: a long-acting analog of GLP-1 that provides lasting antidiabetic effect in animals. Diabetes 52:A112
Coulibaly S, Besenfelder U, Miller I, Zinovieva N, Lassnig C, Kotler T, Jameson JL, Gemeiner M, Muller M, Brem G (2002) Expression and characterization of functional recombinant bovine follicle-stimulating hormone (boFSHα/β) produced in the milk of transgenic rabbits. Mol Reprod Dev 63(3):300–308
Cregg JM, Vedvick TS, Raschke WC (1993) Recent advances in the expression of foreign genes in Pichia pastoris. Nat Biotechnol 11(8):905–910
Cregg JM, Cereghino JL, Shi J, Higgins DR (2000) Recombinant protein expression in Pichia pastoris. Mol Biotechnol 16(1):23–52
Damasceno LM, Huang CJ, Batt CA (2012) Protein secretion in Pichia pastoris and advances in protein production. Appl Microbiol Biotechnol 93(1):31–39
De Schutter K, Lin YC, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouze P, Van de Peer Y, Callewaert N (2009) Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol 27(6):561–566
Devroey P, Pellicer A, Nyboe Andersen A, Arce JC (2012) A randomized assessor-blind trial comparing highly purified hMG and recombinant FSH in a GnRH antagonist cycle with compulsory single-blastocyst transfer. Fertil Steril 97(3):561–571
Dirnberger D, Steinkellner H, Abdennebi L, Remy JJ, van de Wiel D (2001) Secretion of biologically active glycoforms of bovine follicle stimulating hormone in plants. Eur J Biochem 268(16):4570–4579
Dueck MH, Paul M, Wiesner RH, Boerner U (2004) Why does blood have a pH-value of 7.4? The theory of acid-base management. Anaesthesist 53(11):1046–1053
Elena C, Ravasi P, Castelli ME, Peirú S, Menzella HG (2014) Expression of codon optimized genes in microbial systems: current industrial applications and perspectives. Front Microbiol 5:21
Esposito D, Chatterjee DK (2006) Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol 17(4):353–358
Gou XH, Liu YY, Chen QL, Tang JJ, Liu DY, Zou L, Wu XY, Wang W (2012) High level expression of bikunin in Pichia pastoris by fusion of human serum albumin. AMB Express 2(1):14
Greenberg NM, Anderson JW, Hsueh AJ, Nishimori K, Reeves JJ, Ward DN, Rosen JM (1991) Expression of biologically active heterodimeric bovine follicle-stimulating hormone in milk of transgenic mice. Proc Natl Acad Sci 88(19):8327–8331
Hiyama J, Renwick AGC (1990) Separation of human glycoprotein hormones and their subunits by reversed-phase liquid chromatography. J Chromatogr B Biomed Sci Appl 529:33–41
Holmes WJ, Darby RAJ, Wilks MDB, Smith R, Bill RM (2009) Developing a scalable model of recombinant protein yield from Pichia pastoris: the influence of culture conditions, biomass and induction regime. Microb Cell Factories 8(1):35
Huang H, Yang P, Luo H, Tang H, Shao N, Yuan T, Wang Y, Bai Y, Yao B (2008a) High-level expression of a truncated 1,3-1,4-β-D-glucanase from Fibrobacter succinogenes in Pichia pastoris by optimization of codons and fermentation. Appl Microbiol Biotechnol 78(1):95–103
Huang YS, Chen Z, Chen YQ, Ma GC, Shan JF, Liu W, Zhou LF (2008b) Preparation and characterization of a novel exendin-4 human serum albumin fusion protein expressed in Pichia pastoris. J Pept Sci 14(5):588–595
Hughes SR, Dowd PF, Hector RE, Panavas T, Sterner DE, Qureshi N, Bischoff KM, Bang SS, Mertens JA, Johnson ET, Li XL, Jackson JS, Caughey RJ, Riedmuller SB, Bartolett S, Liu S, Rich JO, Farrelly PJ, Butt TR, Labaer J, Cotta MA (2008) Lycotoxin-1 insecticidal peptide optimized by amino acid scanning mutagenesis and expressed as a coproduct in an ethanologenic Saccharomyces cerevisiae strain. J Pept Sci 14(9):1039–1050
Idiris A, Tohda H, Kumagai H, Takegawa K (2010) Engineering of protein secretion in yeast: strategies and impact on protein production. Appl Microbiol Biotechnol 86(2):403–417
Invitrogen (2002) Pichia fermentation process guidelines. https://toolsthermofishercom/content/sfs/manuals/pichiaferm_protpdf Accessed 26 December 2016
Jahic M, Gustavsson M, Jansen AK, Martinelle M, Enfors SO (2003a) Analysis and control of proteolysis of a fusion protein in Pichia pastoris fed-batch processes. J Biotechnol 102(1):45–53
Jahic M, Wallberg F, Bollok M, Garcia P, Enfors SO (2003b) Temperature limited fed-batch technique for control of proteolysis in Pichia pastoris bioreactor cultures. Microb Cell Factories 2(1):6
Kang HA, Choi ES, Hong WK, Kim JY, Ko SM, Sohn JH, Rhee SK (2000) Proteolytic stability of recombinant human serum albumin secreted in the yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol 53(5):575–582
Keene JL, Matzuk MM, Otani T, Fauser BC, Galway AB, Hsueh AJ, Boime I (1989) Expression of biologically active human follitropin in Chinese hamster ovary cells. J Biol Chem 264(9):4769–4775
Kerry-Williams SM, Gilbert SC, Evans LR, Ballance DJ (1998) Disruption of the Saccharomyces cerevisiae YAP3 gene reduces the proteolytic degradation of secreted recombinant human albumin. Yeast 14(2):161–169
Kim CH, Oh Y, Lee TH (1997) Codon optimization for high-level expression of human erythropoietin (EPO) in mammalian cells. Gene 199(1):293–301
Kim MO, Kim SH, Lee SR, Shin MJ, Min KS, Lee DB, Lee SW, Kim KS, Kim SJ, Ryoo ZY (2007) Ectopic expression of tethered human follicle-stimulating hormone (hFSH) gene in transgenic mice. Transgenic Res 16(1):65–75
Kim SJ, Lee JA, Kim YH, Song BK (2009) Optimization of the functional expression of Coprinus cinereus peroxidase in Pichia pastoris by varying the host and promoter. J Microbiol Biotechnol 19(9):966–971
Kobayashi K, Kuwae S, Ohya T, Ohda T, Ohyama M, Ohi H, Tomomitsu K, Ohmura T (2000) High-level expression of recombinant human serum albumin from the methylotrophic yeast Pichia pastoris with minimal protease production and activation. J Biosci Bioeng 89(1):55–61
Kutteyil SS, Pathak BR, Dighe RR, Mahale SD (2015) Expression of bioactive Callithrix jacchus follicle-stimulating hormone in Pichia pastoris. Appl Biochem Biotechnol 176(2):399–411
Liu L, Spurrier J, Butt TR, Strickler JE (2008) Enhanced protein expression in the baculovirus/insect cell system using engineered SUMO fusions. Protein Expr Purif 62(1):21–28
Macauley-Patrick S, Fazenda ML, Mcneil B, Harvey LM (2005) Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270
Mansur M, Cabello C, Hernández L, País J, Varas L, Valdés J, Terrero Y, Hidalgo A, Plana L, Besada V, García L, Lamazares E, Castellanos L, Martínez E (2005) Multiple gene copy number enhances insulin precursor secretion in the yeast Pichia pastoris. Biotechnol Lett 27(5):339–345
Mao R, Teng D, Wang X, Zhang Y, Jiao J, Cao X, Wang J (2015) Optimization of expression conditions for a novel NZ2114-derived antimicrobial peptide-MP1102 under the control of the GAP promoter in Pichia pastoris X-33. BMC Microbiol 15(1):57
Mountford PS, Brandon MR, Adams TE (1994) Expression and characterization of biologically active ovine FSH from mammalian cell lines. J Mol Endocrinol 12(1):71–83
Newman ZR, Young JM, Ingolia NT, Barton GM (2016) Differences in codon bias and GC content contribute to the balanced expression of TLR7 and TLR9. Proc Natl Acad Sci 113(10):E1362–E1371
Nordén K, Agemark M, Danielson JA, Alexandersson E, Kjellbom P, Johanson U (2011) Increasing gene dosage greatly enhances recombinant expression of aquaporins in Pichia pastoris. BMC Biotechnol 11(1):47
Ohya T, Ohyama M, Kobayashi K (2005) Optimization of human serum albumin production in methylotrophic yeast Pichia pastoris by repeated fed-batch fermentation. Biotechnol Bioeng 90(7):876–887
Pajot-Augy E, Couture L, Bozon V, Remy JJ, Biache G, Severini M, Huer J-C, Pernoller J-C, Salesse R (1995) High-level expression of recombinant porcine LH receptor in baculovirus-infected insect cells or caterpillars. J Mol Endocrinol 14(1):51–66
Peroutka RJ, Elshourbagy N, Piech T, Butt TR (2008) Enhanced protein expression in mammalian cells using engineered SUMO fusions: secreted phospholipase A2. Protein Sci 17(9):1586–1595
Pla IA, Damasceno LM, Vannelli T (2006) Evaluation of Mut+ and Muts Pichia pastoris phenotypes for high level extracellular scFv expression under feedback control of the methanol concentration. Biotechnol Prog 22(3):881–888
Richard F, Robert P, Remy JJ, Martinat N, Bidart JM, Salesse R, Combarnous Y (1998) High-level secretion of biologically active recombinant porcine follicle-stimulating hormone by the methylotrophic yeast Pichia pastoris. Biochem Biophys Res Commun 245(3):847–852
Ryan RJ, Charlesworth MC, McCormick DJ, Milius RP, Keutmann HT (1988) The glycoprotein hormones: recent studies of structure-function relationships. FASEB J 2:2661–2669
Samaddar M, Catterall JF, Dighe RR (1997) Expression of biologically active β subunit of bovine follicle-stimulating hormone in the methylotrophic yeast Pichia pastoris. Protein Expr Purif 10(3):345–355
Sha C, Yu XW, Zhang M, Xu Y (2013) Efficient secretion of lipase r27RCL in Pichia pastoris by enhancing the disulfide bond formation pathway in the endoplasmic reticulum. J Ind Microbiol Biot 40(11):1241–1249
Shekhar C (2008) Pichia power: India’s biotech industry puts unconventional yeast to work. Chem Biol 15(3):201–202
Shen Q, Wu M, Wang HB, Naranmandura H, Chen SQ (2012) The effect of gene copy number and co-expression of chaperone on production of albumin fusion proteins in Pichia pastoris. Appl Microbiol Biotechnol 96(3):763–772
Sinclair G, Choy FYM (2002) Synonymous codon usage bias and the expression of human glucocerebrosidase in the methylotrophic yeast, Pichia pastoris. Protein Expr Purif 26(1):96–105
Subramanian GM, Fiscella M, Lamousé-Smith A, Zeuzem S, McHutchison JG (2007) Albinterferon α-2b: a genetic fusion protein for the treatment of chronic hepatitis C. Nat Biotechnol 25(12):1411–1419
Teng D, Fan Y, Yang Y, Tian ZG, Luo J, Wang JH (2007) Codon optimization of Bacillus licheniformis β-1,3-1,4-glucanase gene and its expression in Pichia pastoris. Appl Microbiol Biotechnol 74(5):1074–1083
Vad R, Nafstad E, Dahl LA, Gabrielsen OS (2005) Engineering of a Pichia pastoris expression system for secretion of high amounts of intact human parathyroid hormone. J Biotechnol 116(3):251–260
Van de Wiel DF, Van Rijn PA, Meloen RH, Moormann RJ (1998) High-level expression of biologically active recombinant bovine follicle stimulating hormone in a baculovirus system. J Mol Endocrinol 20(1):83–98
Vanz AL, Nimtz M, Rinas U (2014) Decrease of UPR-and ERAD-related proteins in Pichia pastoris during methanol-induced secretory insulin precursor production in controlled fed-batch cultures. Microb Cell Factories 13(1):23
Walsh G (2014) Biopharmaceutical benchmarks 2014. Nat Biotechnol 32(10):992–1000
Wang F, Wu M, Liu W, Shen Q, Sun H, Chen S (2013) Expression, purification, and lipolytic activity of recombinant human serum albumin fusion proteins with one domain of human growth hormone in Pichia pastoris. Biotechnol Appl Biochem 60(4):405–411
Wang XD, Jiang T, Yu XW, Xu Y (2017) Effects of UPR and ERAD pathway on the prolyl endopeptidase production in Pichia pastoris by controlling of nitrogen source. J Ind Microbiol Biot 44(7):1053–1063
Wiśniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6(5):359–362
Wu X, Nie C, Huang Z, Nie Y, Yan Q, Xiao Y, Su Z, Huang Y, Xiao J, Zeng Y, Tan Y, Feng W, Li X (2009) Expression and purification of human keratinocyte growth factor 2 by fusion with SUMO. Mol Biotechnol 42(1):68–74
Wu M, Shen Q, Yang Y, Zhang S, Qu W, Chen J, Sun H, Chen S (2013) Disruption of YPS1 and PEP4 genes reduces proteolytic degradation of secreted HSA/PTH in Pichia pastoris GS115. J Ind Microbiol Biotechnol 40(6):589–599
Yao XQ, Zhao HL, Xue C, Zhang W, Xiong XH, Wang ZW, Li XY, Liu ZM (2009) Degradation of HSA-AX15 (R13K) when expressed in Pichia pastoris can be reduced via the disruption of YPS1 gene in this yeast. J Biotechnol 139(2):131–136
Zhao HL, Xue C, Wang Y, Yao XQ, Liu ZM (2008) Increasing the cell viability and heterologous protein expression of Pichia pastoris mutant deficient in PMR1 gene by culture condition optimization. Appl Microbiol Biotechnol 81(2):235–241
Acknowledgments
We would like to thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript and the staff of Functional Genomic Technology Center Mass Spectrometry Laboratory for mass spectrometry analysis.
Funding
This work was supported by grants from the National Key R&D Program (2017YFD0501901; 2017YFD0501905) and the Earmarked Fund for the Innovative Teams of Beijing Swine Industrialization Research Program.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Luo, G., Tian, J., Huang, H. et al. Improving heterologous expression of porcine follicle-stimulating hormone in Pichia pastoris by integrating molecular strategies and culture condition optimization. Appl Microbiol Biotechnol 102, 8867–8882 (2018). https://doi.org/10.1007/s00253-018-9260-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9260-6