
Abstract
As a crucial factor for biocatalysts, protein thermostability often arises from a combination of factors that are often difficult to rationalize. In this work, the thermostable nature of halohydrin dehalogenase from Agrobacterium radiobacter AD1 (HheC) was systematically explored using a combinatorial directed evolution approach. For this, a mutagenesis library of HheC mutants was first constructed using error-prone PCR with low mutagenesis frequency. After screening approximately 2000 colonies, six mutants with eight mutation sites were obtained. Those mutation sites were subsequently combined by adopting several rounds of iterative saturation mutagenesis (ISM) approach. After four rounds of saturation mutagenesis, one best mutant ISM-4 with a 3400-fold improvement in half-life (t 1/2) inactivation at 65 °C, 18 °C increase in apparent T m value, and 20 °C increase in optimum temperature was obtained, compared to wild-type HheC. To the best of our knowledge, the mutant represents the most thermostable HheC variant reported up to now. Moreover, the mutant was as active as wild-type enzyme for the substrate 1,3-dichloro-2-propanol, and they remained most enantioselectivity of wild-type enzyme in the kinetic resolution of rac-2-chloro-1-phenolethanol, exhibiting a great potential for industrial applications. Our structural investigation highlights that surface loop regions are hot spots for modulating the thermostability of HheC, the residues located at these regions contribute to the thermostability of HheC in a cooperative way, and protein rigidity and oligomeric interface connections contribute to the thermostability of HheC. All of these essential factors could be used for further design of an even more thermostable HheC, which, in turn, could greatly facilitate the application of the enzyme as a biocatalyst.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rapid development of the biotechnology industry is greatly facilitated by protein engineering of enzymes to provide novel biocatalysts with desired catalytic properties (Schmidt-Dannert and Lopez-Gallego 2016; Jemli et al. 2016). Thermostability is one of the critical properties commonly considered during enzyme evolution processes as high temperature is a favorable condition in many industrial biotransformations because high temperature is usually required for increased enzyme activity and substrate solubility, and it decreases the risk of microbial contamination (Bommarius and Paye 2013). Moreover, thermostable proteins are the preferred starting points for protein evolution because of activity-stability trade-off (Bloom et al. 2006; Siddiqui 2016). However, those stable enzymes are not always available from natural sources. Therefore, the development of thermostable enzymes is of great interest for biocatalysis.
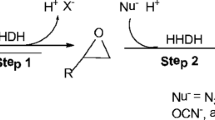
Halohydrin dehalogenases (HHDHs, EC 4.5.1.X) catalyze both the dehalogenation of vicinal halohydrins to epoxides via an intramolecular nucleophilic displacement and the reverse epoxide ring-opening reactions with a series of nucleophiles as substrates, representing valuable promiscuous biocatalysts (van Hylckama Vlieg et al. 2001; Lutje Spelberg et al. 2002; You et al. 2012). Thus, the enzymes attract much attention for the production of optically pure epoxides and a series of β-substituted alcohols that are valuable compounds in the synthesis of pharmaceuticals and fine chemicals (Fox et al. 2007; Haak et al. 2008; Majeric Elenkov et al. 2006, 2007). Compared to the other types of halohydrin dehalogenases (Schallmey et al. 2014; Xue et al. 2014; Nakamura et al. 1994), halohydrin dehalogenase from Agrobacterium radiobacter AD1 (HheC) has been studied more extensively, with aspects of X-ray structure resolvation (de Jong et al. 2003, 2005), kinetics and biochemical characterization (Tang et al. 2003a, b), and protein engineering and biocatalysis (Tang et al. 2002, 2005; Wang et al. 2015a; Guo et al. 2015; Schallmey et al. 2015; You et al. 2014). The X-ray structure of HheC demonstrates that HheC is a homotetramer and that the active site is mainly formed by four highly connected loop regions (de Jong et al. 2003). The enzyme displays a high activity and enantioselectivity toward a broad range of substrates. It exhibits a temperature optimum of 50 °C and is only active as a tetramer. Rational design and directed evolution have produced more promiscuous variants for the synthesis of highly valuable chiral compounds, particularly for the commercial preparation of ethyl (R)-4-cyano-3-hydroxybutyrate, a key building block for statin synthesis (Fox et al. 2007; You et al. 2014).
Recently, Koopmeiners et al. reported that most known halohydrin dehalogenases, including HheC, show a \( {T}_{50}^{10} \) value below 40 °C (\( {T}_{50}^{10} \) is the temperature at which half of the activity is lost after a 10-min incubation) (Koopmeiners et al. 2016), representing a drawback for industrial applications. Despite the importance of protein thermostability for biocatalysts, few publications related to the modification on the thermostability of HheC have been carried out (Tang et al. 2002; Wang et al. 2015b). Thus, the systematic investigation on the evolution of thermostable HheC mutants is of great importance. Although several sequence and structural-based computational methods have been developed for rational design of thermostable protein biocatalysts (Damborsky and Brezovsky 2014; Wijma et al. 2014; Bednar et al. 2015), many studies have demonstrated that by nature, extreme thermostability seems to be a consequence of a dramatic change in short-range and long-range interactions in proteins (Karshikoff et al. 2015), and most of the crucial interactions are beyond the range of these rational design methodologies. Thus, using a directed evolution approach to improve the thermostability of enzyme still remains a hot research field as the methodology provides the framework to explore hot spots throughout proteins.
In this study, an efficient combinatorial directed evolution strategy was adopted for the evolution of active HheC mutants with enhanced thermostability. Initially, the HheC sequence space was explored by constructing a random mutagenesis library by error-prone PCR with a low frequency of mutation, and six thermostable mutants with eight mutation sites were selected. These mutations were then combined through several rounds of iterative saturation mutagenesis (ISM), which was developed by Reetz et al. (2006). After screening 2200 colonies, four more stable mutants were compared to the parental enzyme. Subsequently, these mutant enzymes were characterized, and the results demonstrated that the most stable ISM-4 mutant was as active as wild-type HheC and exhibited a 3400-fold improvement in the half-life (t 1/2) value at 65 and 18 °C increase in apparent T m, representing the most thermostable HheC variant up to now. In addition, we also investigated the structural determinants associated with the thermostability of the ISM-4 mutant.
Materials and methods
Materials
1,3-Dicholoro-2-propanol (1,3-DCP) and racemic 2-chloro-1-phenylethanol (rac-2-CPE) were purchased from Alfa Aesar (Ward Hill, MA). Phusion high-fidelity PCR kit, restriction enzyme Dpn I, and DNA molecular weight standards were purchased from New England Biolabs (Ipswich, MA, USA). L-Arabinose was from Lancaster (Morecambe, UK). Sypro orange dye was purchased from Sigma-Aldrich (St. Louis, MO, USA). All of the other materials were purchased from local businesses.
Methods
Library construction
The DNA sequence of HheC (accession number AF397297) that was cloned in the expression vector pBAD was randomized by error-prone PCR using the plasmid-derived primers FwepC-5′GGCTAACAGGAGGAATTAACCAT3′ and RvepC-5′ATTCCCATATGGTACCAGCTGCAG3′. The random mutagenesis library used in this study was constructed as described before (Tang et al. 2013). The error-prone PCR-produced library (Ep library) was stored in a plasmid format.
Mutation integration
To accumulate all beneficial mutations, an ISM approach was adopted. ISM was started with the most thermostable mutant Ep-6 as a template, which was selected from the above constructed Ep library. All ISM libraries were constructed using a Phusion high-fidelity PCR kit (New England Biolabs, Ipswich, MA, USA), and the target sites were randomized by replacing the target codons with NNS, where N is A, G, C, or T and S is C or G. The primers used in this study were synthesized from Invitrogen (Shanghai, China) and shown in Table S1. The 20 μl reaction system contained 1× high-fidelity (HF) buffer, 200 μM (each) deoxynucleoside triphosphate (dNTP), 1 mM Mg2+, 4 ng template, 2 μM (each) primers, and 0.01 U/μl Phusion polymerase. The temperature was programmed to hold at 98 °C for 3 min, followed by 30 temperature cycles (10 s at 98 °C, 4 s at 50 °C, and 2 min at 72 °C), and finished with 10 min at 72 °C. PCR amplification products were digested with Dpn I to remove the parental templates after which the reaction mixtures were transformed into chemically competent Escherichia coli MC1061 (the strain was kindly supplied by Prof. Dick B. Janssen, Groningen University, the Netherlands). The resulting library was screened according to the library screening procedure described below. The only modification was that the tested plates were incubated at 60 °C for 1 h instead of 50 °C.
Library screening for thermostable HheC mutants
The constructed library was screened for halohydrin dehalogenase activity using the previously developed colorimetric assay in a 96-well plate format with some modifications (Tang et al. 2010). Briefly, after lysis with 0.1% Triton X-100 for 10 min, half of the cell lysate in each well was transferred into a new 96-well plate, resulting in two duplicated plates. One plate (tested plate) was then incubated at 55 °C for 1 h, while the other plate (control plate) was kept at room temperature, and then, the two plates were placed on ice for 30 min. The two plates were subsequently subjected to the activity assay. The colorimetric screening assay for activity was carried out at 30 °C using 1,3-DCP (5 mM) as a model substrate. The colonies that turned from red to yellow in both the tested and control plates were collected as positive variants for DNA sequence and further enzymatic characterization.
Protein expression and purification
The recombinant strain E. coli MC1061, carrying either pBADHheC-WT or pBADHheC-mutant gene, was incubated overnight at 37 °C in 500 ml LB medium containing 100 μg/ml ampicillin and 0.05% (w/v) L-arabinose to reach an OD600 of 1.7 to 2.0. Cells were harvested by centrifugation (5000×g, 50 min) at 4 °C and washed once with 10 mM Tris-SO4 buffer (pH 8.0). Both wild-type HheC and the mutants were purified using a Q-Sepharose column (20 ml; GE Healthcare, Chicago, IL, USA), as described before. The concentrations of purified proteins were measured using Bradford’s method.
Steady-state kinetic measurements
Steady-state kinetic parameters of wild-type and mutant HheC toward 1,3-DCP were determined in 50 mM Tris-SO4 buffer (pH = 8.0) at 30 °C by monitoring halide liberation as described elsewhere (Tang et al. 2012). All k cat values are defined as per monomer.
Stability determination
The optimum temperature of the purified wild-type HheC and variants was determined over a temperature range of 30–90 °C using the abovementioned halide liberation assay. The thermal stability of both purified wild-type HheC and HheC variants (0.5 mg/ml) was determined by incubating in Tris-SO4 buffer (50 mM, pH 8) at the indicated temperature for various period of times in a water bath (Julabo MH, Shanghai, China). Afterwards, aliquots of enzyme were taken and cooled immediately on ice for 10 min. The residual enzyme activity was determined using 5 mM of the substrate 1,3-DCP in Tris-SO4 buffer (50 mM, pH 8.0) at 30 °C. The midpoint of thermal inactivation (T m), where the activity was reduced by 50% for ISM mutants, was calculated from the plot of percent residual activity retained after incubation at temperatures ranging from 30 to 80 °C for 30 min versus temperature. The half-life (t 1/2) was calculated by determining the thermostability at 65 °C.
The thermal unfolding of the wild-type HheC and the two most stable variants, ISM-3 and ISM-4, were assessed by a thermofluor-based protein unfolding assay (Ericsson et al. 2006; Schallmey et al. 2013). The method is based on monitoring the protein unfolding process through the binding of a hydrophobic fluoroprobe, Sypro orange dye, to the exposed hydrophobic interior of unfolding proteins, resulting in an increase of the fluorescence of the dye. The fluorescence changed with increasing temperature from 30 to 95 °C at a rate of 0.5 °C/min, and it was monitored using a MyiQ real-time PCR machine (Bio-Rad, Hercules, CA, USA) with an excitation wavelength at 490 nm and an emission wavelength at 575 nm. In iQ 96-well real-time PCR plates, each well contained 17.5 μl purified protein (0.4 mg/ml) in Tris-SO4 buffer (50 mM, pH 8.0) and 7.5 μl 300-fold diluted Sypro orange dye solution, and four wells of each sample were prepared. The final fluorescence signal was the average of the four measurements. The apparent T m value was obtained by plotting the first derivative of the measured fluorescence traces versus temperature.
The chemical stability of the wild-type HheC and the most stable variant, ISM-4, was assessed by steady-state fluorescence that was measured using a F-4600 FL spectrophotometer with an excitation wavelength of 295 nm and emission wavelengths ranging from 300 to 450 nm. Protein samples (final concentration A 280 = 0.1, in 50 mM Tris-SO4 buffer, pH 8.0) were incubated with different concentrations of guanidinium hydrochloride (GdnHCl) ranging from 0 to 6 M at 25 °C for 24 h. To clearly monitor the chemical-induced protein unfolding of HheC variants, the fluorescence intensity and the emission maximum were determined. All data were analyzed, as described previously (Durão et al. 2006). Equation (1) that describes a two-state protein unfolding process was used.
Enzymatic kinetic resolution
The kinetic resolution experiments were performed using purified wild-type HheC and its variants at 30 °C as described previously (Tang et al. 2012). The catalysis of 5 mM rac-2-CPE in 18 ml Tris-SO4 buffer (200 mM, pH 8.0) was started by adding 120 μg enzyme. The enzymatic conversion was monitored by periodically taking samples (0.25 ml each) from the reaction mixture. The samples were extracted with 1 ml methyl tert-butyl ether (MTBE) containing mesitylene, which was used as an internal standard. The enantiomeric excess (e.e.) of the remaining substrates and products was determined by chiral GC under the following conditions: 100 °C for 6 min, 10 °C/min to 170 °C, and 15 min at 170 °C. All separations were carried out using a β-dex 225 chiral column (30 m, 0.25 mm, 0.25 m; Supelco).
Structure analysis
A homology-based model of the ISM-4 variant was constructed using the program Swiss model (version 8.05) with the crystal structure of HheC from A. radiobacter AD1 (PDB code 1ZMT) as a template.
Gel filtration analysis
The effect of heating on the oligomerization state of both the wild-type HheC and the variants (0.5 mg/ml) was estimated using a prepacked Superdex 200 peptide column (GE Healthcare, Chicago, IL, USA) equilibrated with 50 mM Tris-SO4 buffer containing 100 mM NaCl. Immunoglobulin G (Mr, 160 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa), and soy bean trypsin inhibitor (20.1 kDa) were used as reference proteins.
Results
Generation of thermostable Ep mutants
To obtain thermostable HheC mutants, the wild-type enzyme was first evolved using an error-prone PCR approach with a low mutagenesis frequency (approximately two to three amino acids mutated per variant). In the resulting Ep library, approximately 2000 clones were screened for increased thermostability at 55 °C using 1,3-DCP as a model substrate. Eighty colonies were initially selected based on the catalytic ability of those variants, with residual activities at least 20% higher than that of the wild-type enzyme. Those variants were subsequently expressed in 15 ml cultures and retested for both enzyme activity and thermostability. The top six mutants (namely, Ep-1, Ep-2, Ep-3, Ep-4, Ep-5, Ep-6) that demonstrated a notable improvement in thermostability without a significant decrease in activity were chosen for the following characterizations.
The six Ep mutants were purified to greater than 90% based on SDS-PAGE analysis (data not shown) and then subjected to both enzyme activity and thermostability determinations (Table 1). The results demonstrated that the wild-type HheC retained only 4.9% residual activity after heating at 55 °C for 100 min, while the six mutants retained more than 60% residual activity. Thus, the mutants gained 12.4-fold to 16.4-fold improvement in thermostability when compared with the wild-type HheC. Most importantly, two of the six mutant enzymes demonstrated a 1.5-fold higher activity than the wild-type enzyme.
Sequencing results demonstrated that the selected mutants had between one and three amino acid substitutions (Table 1). In total, eight mutation sites were obtained (namely, F12Y, K52R, Q87R, F136Y, N157I, D182E, N195D, and M252L). The two most active mutants, Ep-1 and Ep-6, contained the F12Y mutation. The F12Y mutant was also selected in our previous study for the ability to efficiently catalyze the dehalogenation of 1,3-DCP. Thus, the results implied that the tyrosine residue at the position 12 might play a dual role in both activity and thermostability.
Integration of positive mutations
To further improve the thermostability of HheC, the above eight mutations were combined through several rounds of ISM process. The mutant Ep-6 (F12Y/D182E), which exhibits both the highest enzyme activity and thermostability among six Ep mutants, was chosen as a starting template for ISM randomization and termed as ISM-0. All of the other target sites were iteratively superposed by NNS randomization. The path of the ISM evolution and the mutations are depicted in Fig. 1. To keep the evolutionary advantage of the mutations obtained from the Ep library, all mutations existing in the same mutant were superposed simultaneously. After four rounds of ISM evolution, in total, 2200 clones were screened, and four outstanding colonies were selected, termed ISM-1, ISM-2, ISM-3, and ISM-4. The sequencing results demonstrated that the mutations on four positions (K52E, F136I, N157V, N195R) in the ISM mutants were different compared to the mutants obtained from the above Ep library, which might be due to the compensating effects with the other mutations.

Flowchart describing the mutagenesis pathway of HheC. Thermostability was expressed as residual activity after incubation at 55 °C for 100 min for mutants obtained from error-prone PCR-based random mutagenesis (Ep library) and half-life value t 1/2 at 65 °C for ISM mutants obtained from ISM
The obtained ISM mutants were purified and subsequently subjected to both enzyme activity and thermostability determinations (Table 1). The results demonstrated that the wild-type HheC was completely inactive after incubation at 65 °C for 15 min, while ISM-0 and ISM-1 mutants retained 20 and 60% residual activity, respectively, and the other three mutants did not show significant decreases in activity under the same conditions. In particular, those four ISM mutants are as active as wild-type enzyme at 30 °C.
Thermostability of HheC mutants
To gain more information on the improved thermostability of the HheC mutants, all obtained thermostable mutants were purified and further analyzed. The optimal temperature results demonstrated that all six Ep mutants gained 10 °C higher optimum temperature compared to that of wild-type enzyme (50 °C) (Fig. 2a), and the four ISM mutants displayed an even higher optimum temperature, 65–70 °C (Fig. 2b), indicating that the active site of the stable mutants is more likely to be more stable compared to that of wild-type HheC. Therefore, the residual activity of the ISM mutants after treatment at 65 °C was further tested (Fig. 3). The results showed that wild-type HheC had a half-life (t 1/2) of only 0.9 min, while the t 1/2 values for the mutants ISM-0, ISM-1, ISM-2, ISM-3, and ISM-4 were 3.6, 18.0, 261.2, 1440.1, and 3060.5 min, respectively, resulting in 20-fold to 3400-fold higher half-lives than wild-type HheC.

Temperature optimums of wild-type HheC, mutants from Ep library (a) and ISM randomization (b). The activities were assayed in 50 mM Tris-SO4 buffer (pH 8.0) containing 5 mM 1,3-DCP at various temperatures (30–90 °C). The activity of each HheC mutant at the optimum temperature was defined as 100%. All data presented are the mean values derived from triplicate measurements, and error bars are the standard deviation

Thermal deactivation of wild-type HheC and ISM mutants at 65 °C. Activities were normalized as percentages of the activities of the three enzymes at 30 °C, without being heated. All data presented are the mean values derived from triplicate measurements, and error bars are the standard deviation
To further investigate the thermodynamic stability of these enzymes, the thermal denaturation experiments of the two most stable mutants, ISM-3 and ISM-4 along with wild-type HheC, were performed using a thermofluor-based assay (Fig. 4a). The apparent melting temperature (T m) of ISM-3 and ISM-4 was 68 and 73 °C, respectively, while wild-type enzyme was 55 °C. The thermodynamic stability of the two mutants was also assessed by activity measurements. For this, the \( {T}_{50}^{30} \) values \( \Big({T}_{50}^{30} \) is the temperature at which half of the activity is lost after a 30-min incubation) of the two ISM mutants and wild-type enzyme with 1,3-DCP were determined (Fig. 4b). The \( {T}_{50}^{30} \) values of wild-type HheC, ISM-3, and ISM-4 were 53, 71, and 75 °C, respectively, indicating that the two mutants had 18–22 °C increase in values compared to wild-type HheC.

The thermostability of both wild-type HheC and the two most thermostable mutants ISM-3 and ISM-4. a Melting temperatures of the three enzymes determined by the thermofluor assay and b residual activities of the three enzymes were determined after incubating over the temperature range 40–80 °C for 30 min. Activities were normalized as percentages of the activities of the three enzymes at 37 °C without heating. All data presented are the mean values derived from triplicate measurements, and error bars are the standard deviation
The chemical stability of the tertiary structure of wild-type HheC and the most stable mutant, ISM-4, upon addition of GdnHCl, was also assessed by a fluorescence spectrometry method (Fig. 5). The results showed that the unfolding process of the enzyme could be accurately described as a two-state process with native and unfolded states being the only states to accumulate significantly. Wild-type HheC displayed a GdnHCl concentration of 1.0 M at the midpoint (where 50% of the molecules were unfolded), and the native state was more stable than the unfolded state by 24.2 kcal/mol at 25 °C (Table 2). Although the mutant ISM-4 follows approximately the same trend as wild-type HheC following increased GdnHCl concentrations, the mutant is clearly more stable against chemically induced unfolding with higher ∆G water (34.7 kcal/mol) and midpoint (3.4 M). The m value reflects the degree of protein against denaturants. The lower the m value, the more protected the protein is from the denaturant. The ISM-4 mutant demonstrated a lower m value compared to wild-type enzyme, indicating that ISM-4 was more stable than wild-type enzyme. Taken together, the ISM-4 mutant gained both thermal and chemical stability without comprising the enzyme activity by introducing eight mutations into wild-type HheC.

Equilibrium unfolding of wild-type HheC (WT) and ISM-4 mutant in the presence of guanidinium hydrochloride (GdnHCl). Unfolded fraction (f U) was calculated based on the fluorescence emission. The solid line is the fit according to the Eq. (1) shown in “Materials and methods” section
Enzymatic characterizations
To see whether these mutations beneficial to stability demonstrated a negative effect on the enzyme catalytic properties, the steady-state kinetic parameters of the two most stable mutants, ISM-3 and ISM-4, with 1,3-DCP were determined (Table S2). The results demonstrated that the parameters of the two mutants were comparable to that of wild-type HheC, indicating that the mutations did not cause any negative effect on the catalytic efficiency of HheC. Moreover, the mutants ISM-3 and ISM-4 remained 80% enantioselectivity (E R = 65 (WT), E R = 52 (ISM-3), E R = 50 (ISM-4)) in the kinetic resolution of rac-2-CPE relative to that of wild-type HheC, yielding e.e. values of 91–93% for the product of (R)-styrene oxide (Table S3).
Structural analysis of thermostable mutants
To provide structural information related to the enhanced thermostability in HheC mutants, a homology-based model of the ISM-4 mutants was constructed using an online server, Swiss model. Inspection of the structure model of the ISM-4 mutant revealed that six out of eight mutation sites (except for 136 and 157) were located on the surface of HheC, while only site of 157 was located in the middle of α-helix αF and the others were located in or near the loop region (Fig. 6). Most importantly, the majority of those mutation sites were located in a loop region with a relatively high B-factor value (Fig. S1).

Secondary structure topology diagram of the chain A in the X-ray structure of HheC (PDB code 1ZMT). The crucial residues for thermostability in the selected thermostable Ep mutants are represented as black circles and are numbered
In order to investigate the effect of those mutations on the loop rigidity, the intramolecular interactions of wild-type HheC (PDB code 1ZMT) and the mutant ISM-4, related to those amino acid substitutions, were subsequently analyzed using the PIC server (http://pic.mbu.iisc.ernet.in/index.html) (Tina et al. 2007). The program was designed to reveal interactions such as hydrogen bonds, hydrophobic interactions, and electrostatic interactions, which are well known to be the dominant structural factors responsible for protein thermostability. The results demonstrated that the substitutions of F12Y, K52E, Q87R, and N195R each gained one more putative hydrogen bond relative to wild-type HheC (Fig. 7a–d). In addition, the Q87R mutation generated two new salt bridges with E85 and D87, respectively; one such interaction with E197 was also created by the N195R mutation. Moreover, N195R was located in a loop that bridges the connection of the two helix fragments (W192-T194 and P196-V205), which might lead to the stability of these two helices.

Structure analysis of hydrogen bonding networks of wild-type HheC and ISM-4 mutant. The hydrogen bonding networks formed between the mutation sites at positions 12, 52, 87, and 195 and the surrounding residues are shown in a, b, c, and d, respectively. The hydrogen bonding networks that relate to wild-type HheC are shown in the right side of each panel. Residues involved in the formation of hydrogen bonds are indicated. The mutated sites are shown in red. Hydrogen bonds are depicted as dashed lines
Unlike the above mutations, the N157V/I mutation created a hydrophobic cluster L155-A156-V/I157-A158-L159 (Fig. S2), which was located at the tetrameric interface in HheC mutants that might contribute to the tetramerization of the enzyme and, in turn, enhanced the thermostability of the HheC mutants. To further investigate whether this was the case for the HheC mutants, the oligomeric states of both wild-type HheC and the N157I mutant were analyzed using gel filtration (Fig. S3). The results demonstrated that the fraction of the tetrameric state of wild-type HheC decreased rapidly with prolonged incubation time at 65 °C, while the tetrameric N157I mutant displayed a strong tolerance to temperature, indicating that the tetrameric state of the N157I mutant was more resistant to heat than that of wild-type HheC.
In addition, the effect of each single mutation in the ISM-4 mutant on the folding free energy change (ΔΔG values) was analyzed using the ENCoM server (Frappier et al. 2015) (Table S4), which is available at http://bcb.med.usherbrooke.ca/encom. Surprisingly, the calculated results demonstrated that the two mutants with either N157I or N157V mutation had the two lowest ΔΔG values among all other single-site mutants, indicating that the internal subunit hydrophobic connections might play an important role on the thermostability of HheC, perhaps increasing the resistance of the enzyme to oligomeric dissociation through lowering the protein folding energy. Although the ΔΔG values for the individual mutations K52E, F136I, and N195R in wild-type HheC were unfavorable for protein folding, they were all identified in the mutants that always contained the other mutations, which might compensate for these unfavorable effects.
Discussion
The recently identified 37 HHDHs through database mining using a motif-based enzyme identification strategy greatly increased the numbers of HHDHs (Schallmey et al. 2014). Most importantly, the majority of these HHDHs exhibit novel biochemical and biocatalytic properties compared to the previously studied three types of HHDH (types A, B, and C), representing a group of diverse biocatalysts (Koopmeiners et al. 2016). However, Koopmeiners et al. demonstrated that only 6 of 18 known HHDHs had \( {T}_{50}^{10} \) values above 50 °C, while most had \( {T}_{50}^{10} \) values below 40 °C, including wild-type HheC enzyme, representing a drawback for industrial applications. In this paper, an efficient combinatorial directed evolution strategy was adopted to systematically explore thermostable HheC mutants, and the corresponding structural basis was subsequently analyzed.
After screening 2000 colonies from the constructed Ep library, eight mutation sites that were crucial for the thermostability of HheC were identified, and none of the mutations showed a significant negative effect on the enzyme activity. Among those eight mutations, two tyrosine and two arginine substitutions, proposed to be the preferred amino acids in thermophilic proteins, were obtained, while four of those eight mutation sites bear the proposed unfavorable residues asparagine, glutamine, and methionion in the wild-type HheC. Thus, it is more likely that the improved thermostability of those HheC variants is mainly achieved by deleting those destabilizing factors and favoring the stabilizing ones, and such a strategy is quite commonly adopted by most thermophilic proteins.
Those mutations were subsequently combined through several rounds of ISM approach, resulting in four ISM mutants, which all displayed a higher t 1/2 and apparent T m values than the parental enzyme, confirming a cooperative effect of those mutation sites on the stability of HheC. The more the mutation sites were superposed, the higher the thermostability. To the best of our knowledge, the outstanding thermostable ISM mutant ISM-4 represents the most thermostable HheC variants obtained up to now (Tang et al. 2002; Schallmey et al. 2013; Wang et al. 2015a, b). Moreover, the \( {T}_{50}^{30} \) values of the two most stable ISM mutants were comparable to their corresponding apparent T m values, suggesting that the fraction of activity retained after thermal inactivation might be directly correlated with the fraction of folded tetrameric HheC. Previous studies showed that HheC is only active as a homotetramer and its active site is mainly surrounded by loop regions, one of which is donated from the C-terminal loop of the opposite monomer with W249 protruding into the active site of HheC (de Jong et al. 2003). Taken together, these results implied that the thermal inactivation of HheC might be due to the tetrameric dissociation of HheC.
Structural analysis provides further insight into the structural basis of both the thermostability and activity for these thermostabilizing mutations. All of those identified mutations were located in loop regions, except for the residue at position 157, which lies in the middle of helix αF. Moreover, the majority of these mutation sites (positions 12, 52, 87, 182, 195, and 252) are distributed over the surface of HheC. This correlates well with the fact that both loop and surface regions are often targeted sites for the improvement of protein thermostability (Zhao and Arnold 1999). Such a strategy has been successfully applied in the thermostability enhancement in two Bacillus subtilis enzymes (subtilisin E and LipA) (Zhao and Arnold 1999; Reetz et al. 2006). In the case of LipA, the enzyme stability was proposed to be mainly enhanced by the formation of side chain hydrogen bonds and/or salt bridges that were generated by the side chains of the newly evolved residues, which, in turn, might contribute to the stabilization of loop regions (Reetz et al. 2006).
Analysis of the intramolecular interactions in the mutant ISM-4 related to those newly evolved substitutions revealed extra connections, mainly hydrogen bonds and electrostatic interactions, which might contribute to the stabilization of these local loop regions and, in turn, enhance the thermostability of ISM-4 mutant. Among those new evolved connections, the hydrogen bond formed between Q87R and the backbone carbonyl oxygen atom of K91 was also observed in the X-ray structure of highly engineering HheC-2360 mutant, which was proposed to contribute to the termostability of the mutant enzyme (Schallmey et al. 2013). Moreover, the relocation of the side chain of F136 residue caused by the P135S mutation created additional intramolecular van der Waals interaction between the two F136 residues in the two neighboring monomers of the HheC-2360 mutant. Although no such connection was observed for the F136I mutation in the ISM-4 mutant, the former result implied the importance of the F136 residue on modulating the thermostability of HheC.
Interestingly, the mutation N157V/I, which is not located in a loop or on the surface, actually lies in a hydrophobic cluster (L155-A156-V/I157-A158-L159) near the oligomeric interface of the enzyme. Our results indicated that such a hydrophobic cluster created by the N157V mutation could have an essential effect on both stabilization of the tetrameric state and lowering the folding energy of HheC, further contributing to the thermostability of the enzyme. Moreover, the thermal deactivation kinetics demonstrated that wild-type HheC, ISM-1, and ISM-1 had exponential kinetics, while the deactivation kinetics of the other three ISM mutants, which all carried the mutation at position 157, changed from one-phase to two-phase kinetics. The results emphasize the importance of the N157V mutation for the stabilization of the ISM mutants. Engineered thermostable mutants that are accompanied with alerts regarding thermal deactivation kinetics have been demonstrated in the thermostability evolution of other enzymes (Xiao et al. 2008; Feng et al. 2016). Furthermore, in all four ISM mutants, the N157V mutation contributed (14.5-fold) to the thermostability more than the other mutations (Table 2). Thus, the intersubunit hydrophobic connections might be another crucial factor for the thermostability of HheC by decreasing the probability of dissociation, which further confirmed our hypothesis. Such intersubunit contacts were also proposed to be a prominent force for enzyme stabilization in the highly engineering HheC-2360 mutant. The enzyme was the most stable HheC mutant obtained from previous studies (Schallmey et al. 2013), which carried 37 mutations and demonstrated an increase in the T m value of 8 °C relative to wild-type enzyme. For the ISM-4 mutant, a 22 °C increase in \( {T}_{50}^{30} \) value was obtained by introducing eight mutations, representing a large achievement for the thermostability of HheC. By modulating the interface connections of Thermotoga maritime phosphoribosylanthranilate isomerase using a site-directed mutagenesis approach, Thoma et al. demonstrated that the dimerization of the enzyme is a major stabilization factor (2000). Moreover, Kirino et al. demonstrated that the reconstruction of intersubunit hydrophobic interactions in 3-isopropylmalate dehydrogenase from E. coli could not only stabilize the enzyme but also made the enzyme more resistant to dissociation than wild-type enzyme in the presence of urea (1994).
In addition to the thermostability, enzymatic activity is another important area of interest because an increase in thermostability is often accompanied by a concomitant decrease in enzymatic activity. For this reason, those active site residues are often not targeted. However, in the case of our evolved ISM-4 mutant, although most of the mutations occur far from the active site, we observed that the residue at position 12, which forms part of the halide-binding site in HheC, was also mutated. Interestingly, the F12Y mutation not only enhanced the thermostability of the F12Y mutant but also increased the enzymatic activity with 1,3-DCP by 1.5-fold. A previous study demonstrated that the enzymatic activity of HheC could be improved by decreasing the overall conformational stability of the halide-binding loop because a conformational change induced by the release of halide was identified as rate-limiting (Tang et al. 2003a, 2005), which indicates that the flexibility of the halide-binding loop could facilitate the release of halide and thus improve the enzymatic activity. Alternatively, the loop rigidity is needed for the improved thermostability, in most cases. Inspection of the model of the ISM-4 mutant indeed revealed that the F12Y mutation created one more hydrogen bond with the neighboring residue T131 compared with wild-type HheC, which might stabilize the loop in which F12 is located. Therefore, refinement of the halide release tunnel by the F12Y mutation might be an alternative explanation for the improved activity of F12Y rather than the modification of the halide-binding site. It has been proved that the enzyme activity in haloalkane dehalogenase LinB from Sphingobium japonicum UT26 could be easily modulated by modifying the halide release tunnel (Biedermannová et al. 2012).
In conclusion, our results demonstrated that the combinatorial directed evolution approach in this study represents an efficient way to evolve HheC mutant enzymes with largely improved thermostability at no expense to enzymatic activity. The yielding active thermostable HheC mutants will not only greatly facilitate the applications of the enzyme in the synthesis of chiral drug building blocks but also the biodegradation of emerging halogenated contaminants (Dvorak et al. 2014). Further structural investigation of the mutant revealed that the enhanced thermostability in the ISM-4 mutant could be gained by the improved loop rigidity and intersubunit interactions introduced by the mutations. Thus, both surface loop and interface regions represent hot targets for further evolution of thermostable HheC.
References
Bednar D, Beerens K, Sebestova E, Bendl J, Khare S, Chaloupkova R, Prokop Z, Brezovsky J, Baker D, Damborsky J (2015) FireProt: energy- and evolution-based computational design of thermostable multiple-point mutants. PLoS Comput Biol 11:e1004556. doi:10.1371/journal.pcbi.1004556
Biedermannová L, Prokop Z, Gora A, Chovancová E, Kovács M, Damborsky J, Wade RC (2012) A single mutation in a tunnel to the active site changes the mechanism and kinetics of product release in haloalkane dehalogenase LinB. J Biol Chem 287:29062–29074
Bloom JD, Labthavikul ST, Otey CR, Arnold FH (2006) Protein stability promotes evolvability. Proc Natl Acad Sci U S A 103:5869–5874
Bommarius AS, Paye MF (2013) Stabilizing biocatalysts. Chem Soc Rev 42:6534–6565
Damborsky J, Brezovsky J (2014) Computational methods for designing and engineering biocatalysts. Curr Opin Chem Biol 19:8–16
de Jong RM, Tiesinga JJ, Rozeboom HJ, Kalk KH, Tang L, Janssen DB, Dijkstra BW (2003) Structure and mechanism of a bacterial haloalcohol dehalogenase: a new variation of the short-chain dehydrogenase/reductase fold without an NAD(P)H binding site. EMBO J 22:4933–4944
de Jong RM, Tiesinga JJ, Villa A, Tang L, Janssen DB, Dijkstra BW (2005) Structural basis for the enantioselectivity of an epoxide ring opening reaction catalyzed by haloalcohol dehalogenase HheC. J Am Chem Soc 127:13338–13343
Durão P, Bento I, Fernandes AT, Melo EP, Lindley PF, Martins LO (2006) Perturbations of the T1 copper site in the CotA laccase from Bacillus subtilis: structural, biochemical, enzymatic and stability studies. J Biol Inorg Chem 11:514–526
Dvorak P, Bidmanova S, Damborsky J, Prokop Z (2014) Immobilized synthetic pathway for biodegradation of toxic recalcitrant pollutant 1,2,3-trichloropropane. Environ Sci Technol 48:6859–6866
Ericsson UB, Hallberg BM, DeTitta GT, Dekker N, Nordlund P (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 357:289–298
Feng X, Tang H, Han B, Zhang L, Lv B, Li C (2016) Engineering the thermostability of β-glucuronidase from Penicillium purpurogenum Li-3 by loop transplant. Appl Microbiol Biotechnol. doi:10.1007/s00253-016-7630-5
Fox RJ, Davis SC, Mundorff EC, Newman LM, Gavrilovic V, Ma SK, Chung LM, Ching C, Tam S, Muley S, Grate J, Gruber J, Whitman JC, Sheldon RA, Huisman GW (2007) Improving catalytic function by ProSAR-driven enzyme evolution. Nat Biotechnol 25:338–344
Frappier V, Chartier M, Najmanovich RJ (2015) ENCoM server: exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res 43:W395–W400
Guo C, Chen Y, Zheng Y, Zhang W, Tao Y, Feng J, Tang L (2015) Exploring the enantioselective mechanism of halohydrin dehalogenase from Agrobacterium radiobacter AD1 by iterative saturation mutagenesis. Appl Environ Microbiol 81:2919–2926
Haak RM, Berthiol F, Jerphagnon T, Gayet AJA, Tarabiono C, Postema CP, Ritleng V, Pfeffer M, Janssen DB, Minnaard AJ, Feringa BL, de Vries JG (2008) Dynamic kinetic resolution of racemic β-haloalcohols: direct access to enantioenriched epoxides. J Am Chem Soc 130:13508–13509
Jemli S, Ayadi-Zouari D, Hlima HB, Bejar S (2016) Biocatalysts: application and engineering for industrial purposes. Crit Rev Biotechnol 36:246–258
Karshikoff A, Nilsson L, Ladenstein R (2015) Rigidity versus flexibility: the dilemma of understanding protein thermal stability. FEBS J 282:3899–39917
Kirino H, Aoki M, Aoshima M, Hayashi Y, Ohba M, Yamagishi A, Wakagi T, Oshima T (1994) Hydrophobic interaction at the subunit interface contributes to the thermostability of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur J Biochem 220:275–281
Koopmeiners J, Halmschlag B, Schallmey M, Schallmey A (2016) Biochemical and biocatalytic characterization of 17 novel halohydrin dehalogenases. Appl Microbiol Biotechnol 100:7517–7527
Lutje Spelberg JH, Tang L, van Gelder M, Kellogg RM, Janssen DB (2002) Exploration of the biocatalytic potential of a halohydrin dehalogenase using chromogenic substrates. Tetrahedron Asymmetry 13:1083–1089
Majeric Elenkov M, Hauer B, Janssen DB (2006) Enantioselective ring opening of epoxides with cyanide catalysed by halohydrindehalogenases: a new approach to non-racemic β-hydroxy nitriles. Adv Synth Catal 348:579–585
Majeric Elenkov M, Hoeffken HW, Tang L, Hauer B, Janssen DB (2007) Enzyme-catalyzed nucleophilic ring opening of epoxides for the preparation of enantiopure tertiary alcohols. Adv Synth Catal 349:2279–2285
Nakamura T, Nagasawa T, Yu F, Watanabe I, Yamada H (1994) Characterization of a novel enantioselective halohydrin hydrogen-halide-lyase. Appl Environ Microbiol 60:1297–1301
Reetz MT, Carballeira JD, Vogel A (2006) Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed 45:7745–7751
Schallmey M, Floor RJ, Hauer B, Breuer M, Jekel PA, Wijma HJ, Dijkstra BW, Janssen DB (2013) Biocatalytic and structural properties of a highly engineered halohydrin dehalogenase. Chembiochem 14:870–881
Schallmey M, Koopmeiners J, Wells E, Wardenga R, Schallmey A (2014) Expanding the halohydrin dehalogenase enzyme family: identification of novel enzymes by database mining. Appl Environ Microbiol 80:7303–7315
Schallmey M, Jekel P, Tang L, Majerić-Elenkov M, Höffken HW, Hauer B, Janssen DB (2015) A single point mutation enhances hydroxynitrile synthesis by halohydrin dehalogenase. Enzym Microb Technol 70:50–57
Schmidt-Dannert C, Lopez-Gallego F (2016) A roadmap for biocatalysis-functional and spatial orchestration of enzyme cascades. Microb Biotechnol. doi:10.1111/1751-7915.12386
Siddiqui KS (2016) Defying the activity-stability trade-off in enzymes: taking advantage of entropy to enhance activity and thermostability. Crit Rev Biotechnol. doi:10.3109/07388551.2016.1144045
Tang L, van Hylckama Vlieg JET, Lutje Spelberg JH, Fraaije MW, Janssen DB (2002) Improved stability of halohydrin dehalogenase from Agrobacterium radiobacter AD1 by replacement of cysteine residues. Enzym Microb Technol 30:251–258
Tang L, Lutje Spelberg JH, Fraaije MW, Janssen DB (2003a) Kinetic mechanism and enantioselectivity of halohydrin dehalogenase from Agrobacterium radiobacter. Biochemistry 42:5378–5386
Tang L, van Merode AE, Lutje Spelberg JH, Fraaije MW, Janssen DB (2003b) Steady-state kinetics and tryptophan fluorescence properties of halohydrin dehalogenase from Agrobacterium radiobacter: roles of W139 and W249 in the active site and halide-induced conformational change. Biochemistry 42:14057–14065
Tang L, Torres Pazmino DE, Fraaije MW, de Jong RM, Dijkstra BW, Janssen DB (2005) Improved catalytic properties of halohydrin dehalogenase by modification of the halide-binding site. Biochemistry 44:6609–6618
Tang L, Li L, Wang X (2010) A high-throughput colorimetric assay for screening halohydrin dehalogenase saturation mutagenesis libraries. J Biotechnol 147:164–168
Tang L, Zhu X, Zheng H, Jiang R, Majerić-Elenkov M (2012) Key residues for controlling enantioselectivity of halohydrin dehalogenase from Arthrobacter sp. AD2 revealed by structure-guided directed evolution. Appl Environ Microb 78:2631–2637
Tang L, Zheng K, Liu Y, Zheng Z, Wang H, Song C, Zhou H (2013) Exploring the potential of megaprimer PCR in conjunction with orthogonal array design for mutagenesis library. Biotechnol Appl Biochem 60:190–195
Thoma R, Hennig M, Sterner R, Kirschner K (2000) Structure and function of mutationally generated monomers of dimeric phosphoribosylanthranilate isomerase from Thermotoga maritima. Structure 8:265–276
Tina KG, Bhadra R, Srinivasan N (2007) PIC: protein interactions calculator. Nucleic Acids Res 35:W473–W476
van Hylckama Vlieg JE, Tang L, Lutje Spelberg JH, Smilda T, Poelarends GJ, Bosma T, van Merode AE, Fraaije MW, Janssen DB (2001) Halohydrin dehalogenases are structurally and mechanistically related to short-chain dehydrogenases/reductases. J Bacteriol 183:5058–5066
Wang X, Han S, Yang Z, Tang L (2015a) Improvement of the thermostability and activity of halohydrin dehalogenase from Agrobacterium radiobacter AD1 by engineering C-terminal amino acids. J Biotechnol 212:92–98
Wang X, Lin H, Zheng Y, Feng J, Yang Z, Tang L (2015b) MDC-analyzer-facilitated combinatorial strategy for improving the activity and stability of halohydrin dehalogenase from Agrobacterium radiobacter AD1. J Biotechnol 206:1–7
Wijma HJ, Floor RJ, Jekel PA, Baker D, Siewert MJ, Janssen DB (2014) Computationally designed libraries for rapid by structure-guided directed evolution. Appl Environ Microbiol 78:2631–2637
Xiao Z, Bergeron H, Grosse S, Beauchemin M, Garron M-L, Shaya D, Sulea T, Cygler M, Lau PCK (2008) Improvement of the thermostability and activity of a pectate lyase by single amino acid substitutions, using a strategy based on melting-temperature-guided sequence alignment. Appl Environ Microbiol 74(4):1183–1189
Xue F, Liu Z, Wan NW, Zheng Y (2014) Purification, gene cloning, and characterization of a novel halohydrin dehalogenase from Agromyces mediolanus ZJB120203. Appl Biochem Biotechnol 174:352–364
You Z, Liu Z, Zheng Y (2012) Properties and biotechnological applications of halohydrin dehalogenases: current state and future perspectives. Appl Microbiol Biotechnol 97:9–21
You Z, Liu Z, Zheng Y (2014) Chemical and enzymatic approaches to the synthesis of optically pure ethyl (R)-4-cyano-3-hydroxybutanoate. Appl Microbiol Biotechnol 98:11–21
Zhao H, Arnold FH (1999) Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng 12:47–53
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 21342005, No. 21673034) and sub-project under the National Science and Technology Major Project on Water Pollution Prevention and Control (No. 2012ZX07203-003).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 264 kb)
Rights and permissions
About this article

Cite this article
Wu, Z., Deng, W., Tong, Y. et al. Exploring the thermostable properties of halohydrin dehalogenase from Agrobacterium radiobacter AD1 by a combinatorial directed evolution strategy. Appl Microbiol Biotechnol 101, 3201–3211 (2017). https://doi.org/10.1007/s00253-017-8090-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8090-2