
Abstract
Streptococcus equi ssp. zooepidemicus (SEZ) causes meningitis in both humans and animals. Some dissociative proteins of SEZ are cytotoxic to mouse brain microvascular endothelial cells (mBMECs) and may contribute to the penetration of SEZ across the blood-brain barrier (BBB). In this study, the ability of SEZ to penetrate across an in vitro BBB model was confirmed. We used stable isotope labeling with amino acids in cell culture (SILAC) to label SEZ proteins with heavy or light isotope-tagged amino acids, along with LC-MS/MS to determine which SEZ proteins were involved in interactions with mBMECs. The efficiency of SEZ protein isotope labeling was 94.7 %, which was sufficient for further analysis. Forty-nine labeled peptides were identified as binding to mBMECs, which matched to 25 SEZ proteins. Bioinformatic analysis indicated that most of these proteins were cytoplasmic. These proteins may have functions in breaching the host BBB, and some of them are known virulence factors in other bacteria. Indirect immunofluorescence results indicated that SEZ enolase had binding activity toward mBMECs. Protective test results showed that enolase was a protective antigen against SEZ infection. This research is the first application of SILAC combined with LC-MS/MS to identify SEZ proteins that may contribute to the infection of mBMECs and potentially show functions related to breaching the BBB. The outcomes provide many future avenues for research into the mechanism of SEZ-induced meningitis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Streptococcus equi ssp. zooepidemicus (SEZ) is a Lancefield Group C Streptococcus. This organism can cause septicemia, arthritis, endocarditis, and meningitis in many domesticated animals (Acke et al. 2015; Gruszynski et al. 2015). In China, SEZ is an important swine pathogen that is responsible for serious streptococcal disease in pigs. Since 1975, this bacterium has caused significant economic losses in the Chinese pig farming industry (Feng and Hu 1977). Globally, frequent outbreaks of SEZ have been reported in recent years (Abbott et al. 2010; Byun et al. 2009; Gruszynski et al. 2015; Madzar et al. 2015). This organism can also invade humans who have close contact with an infected animal via wounds or the digestive tract. SEZ-induced meningitis and glomerulonephritis are a great threat to human health and may be fatal (Beres et al. 2008; Jovanovic et al. 2008).
SEZ may induce meningitis in humans and animals by breaking through the blood-brain barrier (BBB) of the host. Many studies have shown that most extracellular pathogens cross the BBB through either the trans- or paracellular pathway (Mook-Kanamori et al. 2011). The penetration of Streptococcus pneumoniae from the blood to the brain through the BBB is the initial step in bacterial meningitis. Pneumolysin is probably involved in this process, and the underlying mechanism may be based on affecting cell viability and cellular structure (Popoff and Poulain 2010). The epsilon toxin of Clostridium perfringens is a potent virulence factor that can damage the endothelial cells of the BBB (Zhu et al. 2001). In previous research conducted by our group using mice as an infection model, SEZ was isolated from both the bloodstream and the brain of a dead mouse. Some SEZ virulence factors may interact with brain microvascular endothelial cells and contribute to SEZ binding to the endothelium of the BBB and crossing the barrier to invade the cerebrospinal fluid and brain.
Stable isotope labeling with amino acids in cell culture (SILAC) is a widely used isobaric protein tagging technique that greatly increases labeling efficiency and reduces sample complexity prior to sample preparation. The relative quantification of proteins can be carried out by mass spectrometry (Mann 2006). In most studies, SILAC is used to study mammalian cell proteomics, but there have also been some successful applications of SILAC in prokaryotic organisms (Soufi et al. 2010; Soufi and Macek 2014). In this study, we employed SILAC to label SEZ with an isobaric tag and combined this technique with LC-MS/MS to detect SEZ proteins that interact with mouse brain microvascular endothelial cells (mBMECs). SEZ possesses many experimentally confirmed virulence factors, but few of them have been reported to be involved in penetration across the BBB. The proteins we identified in this study provide valuable information for further research addressing the mechanism used by SEZ to cross the BBB.
Methods and materials
Cells and bacteria
S. equi ssp. zooepidemicus ATCC35246 was isolated from a dead pig in Sichuan Province, China. The bacterium was cultured in Todd Hewitt Broth (THB) (Bacto, USA) medium at 37 °C. bEnd.3 cells (mouse brain microvascular endothelial cells) were purchased from the ATCC (ATCC number CRL-2299), and human brain microvascular endothelial cells (hBMEC) were purchased from ScienCell Research Laboratories. Mammalian cells were cultured in DMEM (Gibco, USA) with 10 % fetal bovine serum (FBS) (Gibco, USA) at 37 °C under 5 % CO2.
Labeling SEZ with the SILAC reagent
SEZ ATCC35246 was cultured overnight on THB solid medium at 37 °C. A fresh, single colony was inoculated into 5 ml of THB medium and grown at 37 °C to an OD of 0.6 with vigorous shaking (180 rpm). Then, the SEZ culture was inoculated into heavy isotope (Arg13C6, Lys13C6) and light isotope (Arg12C6, Lys12C6) SILAC media (Pierce, USA) at 1:50 (v/v) and incubated at 37 °C with shaking at 180 rpm for 16 h.
Assay of SEZ penetration across the BBB
Human brain microvascular endothelial cells (hBMEC) were used to construct an in vitro BBB model. Cells (1 × 105) were plated on the upside of collagen-coated polytetrafluoroethylene 0.3-μM pore-size membrane transwells (Corning, USA). An Endothelial Cell Medium Kit (ScienCell Research Laboratories, USA) was used to culture hBMEC. After growing for 7–8 days, the resistance of this in vitro BBB model reached 200 Ω cm2. SEZ (5 × 106) were added to each transwell containing hBMECs and cultured at 37 °C under 5 % CO2. The medium downside of the transwells was collected at different time points and spread onto THB agar plates to count bacterial colonies (Ring et al. 1998).
Preparation of the SEZ supernatant and mBMEC treatment
Heavy- and light-SILAC-labeled SEZ cultures were centrifuged at 13,000×g for 2 min at 4 °C, and the supernatants were filtered through 0.22-μm filters to remove all bacterial cells. The filtered supernatants were dialyzed three times in PBS at 4 °C for 4 h. Ultrafiltration tube Amicon Ultra-4 Centrifugal Filter Units with Ultracel-3 membranes (Merck Millipore, USA) were used to concentrate the proteins in the supernatants at 4000×g, 4 °C for 10 min. The resulting protein concentrations were measured using a BCA Protein Assay Kit (Pierce, USA). Plastic cell culture plates were blocked with PBS containing 10 % FBS to prevent nonspecific protein binding. A total of 2 mg of heavy isotope-labeled total protein was added to mBMEC cells cultured in a 10-cm plate. An equal quantity of the light isotope-labeled protein was added to an empty plate. After 2 h incubation at 37 °C under 5 % CO2, the supernatants were collected and mixed together.
Protein digestion
Freeze-dried mixed protein powder was dissolved in 6 M guanidine-HCl. Dithiothreitol was added to a final concentration of 5 mM, and the mixture was incubated at 95 °C for 15–20 min and then cooled to room temperature. IAM was subsequently added to a final concentration of 25 mM, and the mixture was incubated at room temperature for 45 min in the dark. Next, 50 mM NH4HCO3 (pH 7.8) was used to dilute the guanidine-HCl to a final concentration <1 M. In the tryptic digestion step, trypsin was added to the substrate at a ratio of 1:20 to 1:100. Finally, the digest was incubated at 37 °C for 12 h before an equal dose of trypsin solution was added, and the digest was again incubated at room temperature for 24 h.
Mass spectrometry
The resulting peptide products (1.5 μg) were separated by reverse-phase liquid chromatography using a nano-LC system (DIONEX Thermo Scientific, USA) and analyzed by tandem mass spectrometry using an LTQ-Orbitrap mass spectrometer (Thermo Scientific) with a nanoelectrospray ion source. Peptide samples were acidified with 0.1 % formic acid (FA) and automatically loaded into a nano-LC system equipped with a nano-trap column (Acclaim PepMap100 C18, 75 μm × 2 cm, 3 μm, 100 Å, Thermo Scientific) at a flow rate of 4 μl/min in loading buffer (2 % acetonitrile and 0.1 % FA in HPLC-grade water). After 8 min, the peptides were eluted and separated in the analytical column (Acclaim PepMap® RSLC, C18, 75 μm × 15 cm, 3 μm, 100 Å, Thermo Scientific) using a gradient of buffer B (80 % acetonitrile and 0.1 % FA in HPLC-grade water) from 3 to 55 % at 300 nl/min over 112 min. The remaining peptides were eluted with a short gradient of 55 to 98 % buffer B in 5 min. The eluted peptides were analyzed using the LTQ Orbitrap XL. Based on a high-resolution MS prescan, the ten most intense peptide ions were selected for fragment analysis in the linear ion trap if they exceeded an intensity of 5000 counts and were at least doubly charged. The normalized collision energy for the CID was set to 35, and the resulting fragments were detected at normal resolution in the linear ion trap. The lock mass option was activated, and the background signal with a mass of 445.120020 was used as the lock mass. Every ion selected for fragmentation was excluded for 60 s by dynamic exclusion.
MS data analysis
MS data were analyzed using MaxQuant version 1.3.0.5 (Cox and Mann 2008). The MS data were searched against the UniProt S. equi ssp. zooepidemicus group. The settings used for the MaxQuant analysis were as follows: two miscleavages were allowed; carbamidomethylation on cysteine was set as a fixed modification; the enzyme was trypsin; and the variable modifications included in the analysis were methionine oxidation and protein N-terminal acetylation. A mass tolerance of 6 ppm was used for precursor ions, and a tolerance of 20 ppm was used for fragment ions. The search multiplicity was set to two, with each multiplicity denoting one of the SILAC amino acid combinations (light: Lys 0 Arg 0; heavy: Lys 4 Arg 6). Both the match between runs and requantify features of MaxQuant were enabled, and the false discovery rate was 1 % at both the peptide and protein levels. All H/L values of peptides were calculated. Their median was 17.886. The average labeling efficiency was represented by the median and calculated with the formula (median H/L ratio)/(1+ median H/L ratio) × 100 % (Graumann et al. 2008; Soufi and Macek 2014).
Cloning, expression, and purification of recombinant enolase
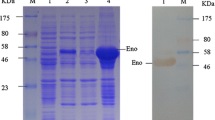
The eno gene was amplified from SEZ ATCC35246 chromosomal DNA through PCR with primers F: GGATCCATGTCAATTATTACTGATGTTTA and R: GTCGACTTTTAAGGTTGTAGAATGATTTG. The amplicon was sequenced to verify the absence of mutations according to the SEZ eno gene (Genbank accession: AEJ25069). The eno gene fragment was cloned into plasmid pET-28a(+) with a His-tag-encoding sequence. Plasmid pET-28a(+)-eno was transformed into Escherichia coli BL21 (DE3) for IPTG-inducible expression of recombinant enolase (rEno). His-tagged rEno was purified by His-Trap chromatography according to the manufacturer’s protocols (GE Healthcare, USA). The purified rEno protein was injected subcutaneously into rabbits three times at 2-week intervals to produce polyclonal antibodies (Zhang et al. 2013).
Bacterial protein extraction and Western blotting
SEZ was inoculated into 500 ml of THB at 1:50 (v/v) and incubated at 37 °C with vigorous shaking at 180 rpm for 12 or 24 h. A 100-μl sample of cell suspension was reserved for total protein extraction, and the bacterial cells were then centrifuged at 13,000×g for 10 min at 4 °C. The proteins in 100-μl sample and the cell pellet were extracted by direct lysis in boiling water, respectively. The supernatant was filtered through a 0.22-μm filter; trichloroacetic acid was added to a final concentration of 10 %, and the mixture was precipitated on ice for 30 min. After centrifugation at 10,000×g for 15 min at 4 °C, the pellet was collected and washed twice with cold acetone. After air-drying, the pellet was dissolved in ddH2O. The proteins were separated using SDS-PAGE and transferred to a PVDF membrane. In Western blot experiments, anti-rEno serum was used as the primary antibody (1:500), and horseradish peroxidase-conjugated goat anti-rabbit IgG (1:10,000) (ABGENT, USA) was used as the secondary antibody. ECL Pico-Detect Western Blotting Substrate (CMCTAG, USA) was used for chemiluminescence detection.
Indirect immunofluorescence
rEno was diluted in DMEM to 100 μg/ml and added to mBMEC grown in a 12-well plate. For the negative control, 0.5 % BSA was used instead of rEno. Following incubation at 37 °C in 5 % CO2 for 2 h, the cells were washed five times with PBS, then fixed with cold 4 % paraformaldehyde at 4 °C for 30 min and washed with PBS three times. Next, the cells were incubated with rabbit anti-rEno polyclonal antibody at 37 °C for 40 min and subsequently washed three times with PBS. The cells were then incubated with Alexa Fluor® 488-conjugated AffiniPure Goat Anti-Rabbit IgG (Jackson ImmunoResearch, USA) at 37 °C for 30 min in the dark and washed three times with PBS. Cell nuclei were stained with DAPI for 10 min at room temperature, and the cells were thoroughly washed with PBS. Finally, the cells were examined using a fluorescence microscope at ×400 magnification (Zeiss, Germany).
Immune protection assays
Fifteen 8-week-old female BALB/c mice were purchased from the Comparative Medicine Center of Yangzhou University and randomly divided into three groups. All experimental protocols involving mice were approved by the Laboratory Animal Monitoring Committee of Jiangsu Province and performed accordingly. Mice in group 1 were inoculated intraperitoneally (i.p.) with 100 μL of purified recombinant enolase (500 μg/mL); mice in group 2 were injected with PBS, while mice in group 3 were used as negative controls (no immunization). The immunization was given three times at 10-day intervals. All mice were challenged i.p. with 0.2 ml of SEZ ATCC35246 (5 × 104 CFU, 5 × LD50) (Hong-Jie et al. 2009). Survival of the mice was recorded daily for 7 days; animals showing signs of irreversible illnesses were humanely euthanized with 100 % CO2.
Passive antibody cell protection test
The passive antibody protection test was conducted with rabbit anti-rEno antibody and mBMECs. SEZ supernatant was prepared as described above. Rabbit serum containing rEno antibody was added to SEZ supernatant at the ratio 1:10 and incubated at 37 °C for 30 min. mBMECs were treated with this mixture, SEZ supernatant and THB medium containing 10 % rabbit anti-rEno serum, respectively. The MTT assay was employed to measure bEnd.3 cell viability at different time points after treatment with the SEZ supernatant, or with THB as a negative control. bEnd.3 cells were cultured in 96-well plates and treated with the SEZ supernatant for 2, 4, or 12 h. Then, a total of 100 μl of fresh medium was added to each cell culture well, followed by 20 μl of a 5-mg/ml stock of MTT. After 4 h incubation at 37 °C, the medium was gently aspirated. Deposited formazan crystals were dissolved in 100 μl DMSO by gently shaking the plate. The absorbance was measured at 570 nm using a microplate reader (Bio-Rad, USA). Cell viability (%) is expressed as a percentage relative to untreated control cells (Wan et al. 2014).
Results
SEZ has BBB penetration ability, and its culture supernatant is cytotoxic to mBMECs
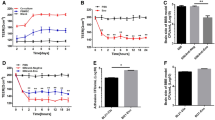
We used hBMECs to construct an in vitro BBB model. After growing for 7–8 days, the resistance of the in vitro BBB model reached 200 Ω cm2 (Fig. 1a) and tight cellular connections formed, so it was suitable for bacterial penetration tests. The number of bacteria that passed across this BBB model to the downside medium of a transwell was counted at different times. Figure 1b shows that 2 h after infection, SEZ started to translocate across the in vitro BBB model and the number of translocated SEZ increased over time. Thus, SEZ was able to translocate across the BBB model by some unknown mechanism.

SEZ has BBB penetration ability, and its supernatant is cytotoxic to mBMECs. a hBMECs were used to construct an in vitro BBB model. The resistance of the in vitro BBB model increased over time, up to 200 Ω cm2 within 8 days. b SEZ started to translocate across the in vitro BBB model from 2 h post-infection, and the number of SEZ CFU in the medium on the transwell downside increased over time. c Cell viability was analyzed using the MTT assay by detecting the absorbance at 570 nm. BMEC cell viability was calculated by using A570 of the treatment groups divided by A570 of the DMEM-treated negative control group. The cell viability of SEZ ATCC35246 supernatant-treated cells (35246) was significantly different compared to that of SEZ supernatant + SEZ enolase (Eno) polyclonal antibody treated cells (35246 + Eno (PA)). Results are shown as means ± SEM (*p < 0.05)
The supernatant of SEZ may contain cytotoxic factors that contribute to the translocation of SEZ across the BBB. To test this speculation, we employed the MTT method to detect the cytotoxicity of the SEZ supernatant towards mBMECs. mBMEC cell viability was calculated by dividing Abs570 nm of the SEZ supernatant-treated group by Abs570 nm of the DMEM-treated negative control group following MTT assay. The cell viability of SEZ ATCC35246 supernatant-treated cells was less than 20 % that of the DMEM negative control group and decreased with time (Fig. 1c, solid bars). This result indicates that the SEZ supernatant was cytotoxic to mBMECs.
Determination of SEZ protein isotope-labeling efficiency by SILAC
In mammalian cells, most cells should be labeled with a given isotope-tagged amino acid after five culture generations in labeling media. However, in bacteria, the isotope-labeling efficiency may be different because of amino acid interconversion (Ping et al. 2013). Theoretically, SEZ grown in SILAC medium should use the isotope-tagged amino acids in its metabolism and biosynthetic processes. Although SEZ doubled more than five times when cultured in either the heavy SILAC medium containing Arg13C6 and Lys13C6 or the light-SILAC medium containing Arg12C6 and Lys12C6, we still measured the isotope-labeling efficiency in SEZ before using its supernatant in further experiments. Equal amounts of the heavy- and light-labeled SEZ total proteins were mixed together. As previously described, LC-MS/MS was employed to characterize this mixture. A total of 193 proteins were identified. The median H/L ratio was calculated as 17.886 (Table S1). Using the formula labeling efficiency = (median H/L ratio)/(1+ median H/L ratio) × 100 %, the labeling efficiency was determined to be 94.7 %. This labeling efficiency was sufficient for further analysis.
Identification of SEZ cytoplasmic proteins that may interact with mBMECs
As Fig. 2 shows, as mBMECs absorb the supernatant of the heavy isotope-tagged SEZ culture, proteins that interact with the mBMECs will be captured by these cells, whereas the supernatant of the light isotope-tagged SEZ culture will not decrease in the empty culture plate. Comparison of the quantity of proteins with a heavy label to those with a light label determined by LC-MS/MS generates the H/L ratio. An H/L ratio ≤0.8 is obtained when heavy-tagged proteins interact with the mBMECs. After searching the SEZ ATCC35246 protein database with the LC-MS/MS raw data using MaxQuant software, 49 peptides with an H/L ratio ≤0.8 were identified as interacting with mBMECs (Table S2) and were matched to 25 different proteins (Table 1), some of which are known virulence factors in other bacteria. Softberry ProtComp Neural Nets identification showed that 22 of these 25 proteins were cytoplasmic and the other three were membrane proteins (Table 1). Structural analysis of these three membrane proteins is shown in Fig. 3. These 25 proteins might be secreted proteins or just come from dead cells lysed in culture.

Diagrammatic representation of generic methodologies used to analyze SEZ proteins that interact with mBMECs. Stable isotope (light and heavy)-labeled amino acids were incorporated into SILAC medium-cultured SEZ bacterial cells and their products. The supernatants of these two labeled bacterial suspensions were collected. Following dialysis with PBS, these two samples were used to treat mBMECs, and the SEZ proteins interacted with the mBMECs. At a suitable time point post-incubation, the SEZ supernatants were harvested and combined for further LC-MS/MS analysis. A decrease in the quantity of any protein indicates that it may potentially interact with mBMECs

Prediction of transmembrane helices in three membrane proteins identified in SILAC assays, analyzed with the TMHMM server, v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). Red peaks represent transmembrane domains of protein; blue peaks represent intercellular domains of protein, and pink peaks represent extracellular domains of protein. These domains are also marked using thick lines in the matching color at the top of the images
Enolase is a SEZ secretory protein and may bind to mBMECs
Enolase existed in many pathogens; it might interact with BMECs through cellular plasminogen (Bergmann et al. 2013b; Sun et al. 2016). After analyzing all 25 proteins identified as interacting with mBMECs from SILAC experiments using the STRING 10 database, enolase was predicted to be located in the center of the 25-protein interaction net (Fig. 4). Moreover, enolase is a known virulence factor in many bacteria. We chose enolase to verify that proteins identified in this study do in fact interact with mBMECs. Recombinant SEZ enolase (rEno) was expressed and purified (Fig. S1). Preparation of anti-rEno serum and cytotoxicity detection were performed using this recombinant protein. As Figure S2 shows, enolase is a SEZ secretory protein and was detected in the SEZ supernatant. Immunofluorescence assay was performed with anti-rEno serum and a FITC-labeled antibody used for immunofluorescence detection (Fig. 5). The upper panel shows that after treating mBMECs with 100 μg/ml rEno, rEno was distributed on the surfaces of most of the mBMECs. No fluorescence was detected when the mBMECs were treated with BSA. Taken together, these results suggest that enolase was able to interact with the mBMECs, and that the proteins detected in this research are valuable targets for further investigation.

Protein-protein interaction network constructed using the STRING database from SEZ proteins that interact with mBMECs (http://string90.embl.de). Seven different colored lines link a number of nodes and represent the seven types of evidence used for predicting associations. A red line indicates the presence of fusion evidence; a green line represents neighborhood evidence; a blue line represents co-occurrence evidence; a purple line represents experimental evidence; a yellow line represents text mining evidence; a light blue line represents database evidence; and a black line represents coexpression evidence

Immunofluorescence detection of the interaction between SEZ enolase and mBMECs. mBMECs were treated with rEno or BSA for 2 h. The treated cells were immunostained with rabbit anti-rEno serum and a FITC-labeled secondary antibody. Cell nuclei were stained with DAPI. The magnification of the fluorescence microscope was ×400
SEZ enolase is a protective antigen, and its antibody decreases SEZ supernatant cytotoxicity
Three groups of five mice were inoculated with purified recombinant enolase, PBS, or were used as a negative control (no immunization). SEZ at 5 × LD50 was used to challenge these groups of mice. At 2 days after inoculation, all mice in the PBS-treated group and negative control group died; before death, most exhibited marked clinical signs such as ruffled hair, difficulties in breathing, and poor appetite. In the rEno-treated group, only one mouse died by day 2; the other four mice all survived for at least 7 days post-inoculation (Fig. 6); this experiment was repeated once; thus, the protection rate reached 80 % (Table 2). These results indicated high protection of mice from SEZ challenge by rEno. When the Eno antibody was added to SEZ supernatant in assays of BMEC cell viability, the supernatant cytotoxicity was significantly decreased at each time point (Fig. 1c), which indicates that the Eno antibody might neutralize Eno in SEZ supernatant and that Eno played a part in the bacterial cytotoxicity.

Survival of immunized mice following challenge with SEZ. Mice in the negative control group and PBS-treated group all died within 2 days post-infection. Only one mouse in the rEno immunized group died (within 2 days post-infection); the four remaining mice survived more than 7 days
Discussion
Meningitis is a common sequela in both humans and animals infected with SEZ and occurs as a consequence of SEZ invasion of the bloodstream. To invade the meninges, SEZ must breach the protective physiological barrier known as the BBB, which is mainly formed by a monolayer of tight-junction-forming brain microvascular endothelial cells and astrocytes (Kniesel and Wolburg 2000). While intracellular pathogens penetrate the BBB using infected leukocytes as “Trojan horses,” extracellular pathogens use different strategies (Drevets and Leenen 2000). Extracellular pathogens that invade meninges via the transcellular pathway require transcytosis, whereas the paracellular pathway requires opening of the tight junctions (Nassif et al. 2002). Moreover, a high degree of bacteremia is necessary, and many virulence factors contribute to the development of meningitis. Recent studies have revealed that the penetration of extracellular pathogens across the BBB involves host cell actin cytoskeletal rearrangements caused by certain virulence factors (Kim 2002; Kim 2006; Shin and Kim 2006). To rearrange the BMEC actin cytoskeleton, the SEZ virulence factors must first interact with these cells, by either binding or invasion. In this study, using an in vitro hBMEC BBB model, SEZ penetration ability of the BBB was confirmed and cytotoxicity of SEZ culture supernatant was also detected. Using SILAC and LC-MS/MS, we readily determined which proteins in the SEZ supernatant could interact with mBMECs; these proteins are considered potential BBB-breaching virulence factors.
SILAC is mainly used to label eukaryotic cells. However, in the present study, we used this technology to label SEZ, a prokaryote. This method provided a convenient way to identify host cell binding proteins of SEZ. Because of the isotope labeling, bacterial proteins and host proteins could be easily distinguished, decreasing background measurements to a very low level, resulting in an accurate list of target proteins. When cells were infected with SEZ, we found that SEZ could grow rapidly in the cellular medium, thus inspiring our use of SILAC. We then attempted to culture SEZ in SILAC medium. Although only the first generation was able to grow successfully in the SILAC medium, the labeling efficiency was >90 %, which was sufficient for further analysis. A total of 25 proteins were identified as binding to or invading mBMECs in this study. Although 22 of these proteins were cytoplasmic, these proteins might be SEZ secretory proteins three were located in the cellular membrane: a sensor kinase, a peptide-binding protein, and VanY, these proteins might come from dead cells lysed. The sensor kinase peptide identified by LC-MS/MS was “LENMDAVSK” (from amino acid position 429 to 437). This region is outside of the transmembrane region (present in the cytoplasm). A very short transmembrane segment is located at the N-terminus of both the peptide-binding protein and VanY to anchor them in the membrane (Fig. 3). The results here indicating an unstable structure suggest that these proteins may exhibit both dissociative and bound states. The SILAC method is able to detect dissociated proteins accurately.
The proteins we identified included some known virulence factors from other streptococci, whose functions and ligands in host cells have been characterized. For example, a group A Streptococcus sensor kinase extracellular domain may interact with the human antimicrobial peptide LL-37 (Velarde et al. 2014), and S. pneumoniae Pgk and PepO may disturb complement function by interacting with complement molecules (Agarwal et al. 2014; Blom et al. 2014). Additionally, GrpE and Eno were observed to participate in bacterial adherence to epithelial cells in previous studies (Bergmann et al. 2013a; Murakami et al. 2012). Further experiments are needed to determine whether these proteins exhibit similar functions in SEZ and how other identified proteins function in SEZ infection.
In this work, we chose enolase for further study to verify our results because this protein is located in the center of the protein interaction net generated from our data by STRING and because of its multiple functions in many pathogenic streptococci, including immunosuppression, neutrophil extracellular trap formation, and the promotion of bacterial adherence (Bergmann et al. 2013a; Mori et al. 2012; Veiga-Malta et al. 2004). SEZ enolase can bind to mBMECs, much like the enolases of Vibrio parahaemolyticus and Streptococcus suis serotype 2. In these two pathogens, enolase has been reported to be an adhesion-related factor that shows plasminogen-binding activity and plays important roles in bacterial pathogenicity (Jiang et al. 2014; Zhang et al. 2009). Further experiments must be performed to determine whether the enolase of SEZ exhibits the same function. As a protective antigen, enolase protected mice from SEZ infection with high efficiency, and its antibody decreased the cytotoxicity of SEZ supernatant towards mBMECs. Detection of the adherence of SEZ enolase to mBMECs verified the SILAC results in this research.
The molecular basis of meningitis pathogenesis is still poorly understood. In this study, we used SILAC and LC-MS/MS to detect proteins from SEZ that can interact with mBMECs and potentially exhibit functions related to breaching the BBB. Enolase was chosen to verify the results. This investigation should help illuminate the mechanism of SEZ-induced meningitis and identify novel therapeutic targets.
References
Abbott Y, Acke E, Khan S, Muldoon EG, Markey BK, Pinilla M, Leonard FC, Steward K, Waller A (2010) Zoonotic transmission of Streptococcus equi subsp. zooepidemicus from a dog to a handler. J Med Microbiol 59(Pt 1):120–123
Acke E, Midwinter AC, Lawrence K, Gordon SJ, Moore S, Rasiah I, Steward K, French N, Waller A (2015) Prevalence of Streptococcus dysgalactiae subsp. equisimilis and S. equi subsp. zooepidemicus in a sample of healthy dogs, cats and horses. N Z Vet J 63(5):265–271
Agarwal V, Sroka M, Fulde M, Bergmann S, Riesbeck K, Blom AM (2014) Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J Biol Chem 289(22):15833–15844
Beres SB, Sesso R, Pinto SW, Hoe NP, Porcella SF, Deleo FR, Musser JM (2008) Genome sequence of a Lancefield group C Streptococcus zooepidemicus strain causing epidemic nephritis: new information about an old disease. PLoS One 3(8):e3026
Bergmann S, Schoenen H, Hammerschmidt S (2013a) The interaction between bacterial enolase and plasminogen promotes adherence of Streptococcus pneumoniae to epithelial and endothelial cells. International Journal of Medical Microbiology 303(8):452–462
Bergmann S, Schoenen H, Hammerschmidt S (2013b) The interaction between bacterial enolase and plasminogen promotes adherence of Streptococcus pneumoniae to epithelial and endothelial cells. Int J Med Microbiol 303(8):452–462. doi:10.1016/j.ijmm.2013.06.002
Blom AM, Bergmann S, Fulde M, Riesbeck K, Agarwal V (2014) Streptococcus pneumoniae phosphoglycerate kinase is a novel complement inhibitor affecting the membrane attack complex formation. J Biol Chem 289(47)
Byun JW, Yoon SS, Woo GH, Jung BY, Joo YS (2009) An outbreak of fatal hemorrhagic pneumonia caused by Streptococcus equi subsp. zooepidemicus in shelter dogs. J Vet Sci 10(3):269–271
Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26(12):1367–1372
Drevets DA, Leenen PJ (2000) Leukocyte-facilitated entry of intracellular pathogens into the central nervous system. Microbes Infect 2(13):1609–1618
Feng ZG, Hu JS (1977) Outbreak of swine streptococcosis in Sichan province and identification of pathogen. Anim Husbandry Vet Med Lett 2:7–12
Graumann J, Hubner NC, Kim JB, Ko K, Moser M, Kumar C, Cox J, Scholer H, Mann M (2008) Stable isotope labeling by amino acids in cell culture (SILAC) and proteome quantitation of mouse embryonic stem cells to a depth of 5,111 proteins. Mol Cell Proteomics 7(4):672–683
Gruszynski K, Young A, Levine SJ, Garvin JP, Brown S, Turner L, Fritzinger A, Gertz RE Jr, Murphy JM, Vogt M, Beall B (2015) Streptococcus equi subsp. zooepidemicus infections associated with guinea pigs. Emerg Infect Dis 21(1):156–158. doi:10.3201/eid2101.140640
Hong-Jie F, Fu-yu T, Ying M, Cheng-ping L (2009) Virulence and antigenicity of the szp-gene deleted Streptococcus equi ssp. zooepidemicus mutant in mice. Vaccine 27(1):56–61
Jiang W, Han X, Wang Q, Li X, Yi L, Liu Y, Ding C (2014) Vibrio parahaemolyticus enolase is an adhesion-related factor that binds plasminogen and functions as a protective antigen. Appl Microbiol Biotechnol 98(11):4937–4948
Jovanovic M, Stevanovic G, Tosic T, Stosovic B, Zervos MJ (2008) Streptococcus equi subsp. zooepidemicus meningitis. J Med Microbiol 57(Pt 3):373–375
Kim KS (2002) Strategy of Escherichia coli for crossing the blood-brain barrier. J Infect Dis 186(Suppl 2):S220–S224
Kim KS (2006) Microbial translocation of the blood-brain barrier. Int J Parasitol 36(5):607–614
Kniesel U, Wolburg H (2000) Tight junctions of the blood-brain barrier. Cell Mol Neurobiol 20(1):57–76
Madzar D, Hagge M, Moller S, Regensburger M, Lee DH, Schwab S, Jantsch J (2015) Endogenous endophthalmitis complicating Streptococcus equi subspecies zooepidemicus meningitis: a case report. BMC Res Notes 8:184
Mann M (2006) Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol 7(12):952–958
Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D (2011) Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev 24(3):557–591
Mori Y, Yamaguchi M, Terao Y, Hamada S, Ooshima T, Kawabata S (2012) alpha-enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem 287(13):10472–10481
Murakami J, Terao Y, Morisaki I, Hamada S, Kawabata S (2012) Group A Streptococcus adheres to pharyngeal epithelial cells with salivary proline-rich proteins via GrpE chaperone protein. J Biol Chem 287(26):22266–22275
Nassif X, Bourdoulous S, Eugene E, Couraud PO (2002) How do extracellular pathogens cross the blood-brain barrier? Trends Microbiol 10(5):227–232
Ping LY, Zhang H, Zhai LH, Dammer EB, Duong DM, Li N, Yan ZL, Wu JZ, Xu P (2013) Quantitative proteomics reveals significant changes in cell shape and an energy shift after IPTG induction via an optimized SILAC approach for Escherichia coli. J Proteome Res 12(12):5978–5988
Popoff MR, Poulain B (2010) Bacterial toxins and the nervous system: neurotoxins and multipotential toxins interacting with neuronal cells. Toxins 2(4):683–737
Ring A, Weiser JN, Tuomanen EI (1998) Pneumococcal trafficking across the blood-brain barrier. molecular analysis of a novel bidirectional pathway. J Clin Invest 102(2):347–360
Shin S, Kim KS (2006) RhoA and Rac1 contribute to type III group B streptococcal invasion of human brain microvascular endothelial cells. Biochem Biophys Res Commun 345(1):538–542
Soufi B, Kumar C, Gnad F, Mann M, Mijakovic I, Macek B (2010) Stable isotope labeling by amino acids in cell culture (SILAC) applied to quantitative proteomics of Bacillus subtilis. J Proteome Res 9(7):3638–3646
Soufi B, Macek B (2014) Stable isotope labeling by amino acids applied to bacterial cell culture. Methods Mol Biol 1188:9–22
Sun Y, Li N, Zhang J, Liu H, Liu J, Xia X, Sun C, Feng X, Gu J, Du C, Han W, Lei L (2016) Enolase of Streptococcus Suis serotype 2 enhances blood-brain barrier permeability by inducing IL-8 release. Inflammation 39(2):718–726
Veiga-Malta I, Duarte M, Dinis M, Tavares D, Videira A, Ferreira P (2004) Enolase from Streptococcus sobrinus is an immunosuppressive protein. Cell Microbiol 6(1):79–88
Velarde JJ, Ashbaugh M, Wessels MR (2014) The human antimicrobial peptide LL-37 binds directly to CsrS, a sensor histidine kinase of group A Streptococcus, to activate expression of virulence factors. J Biol Chem 289(52):36315–36324
Wan WB, Cao L, Liu LM, Kalionis B, Chen C, Tai XT, Li YM, Xia SJ (2014) EGb761 provides a protective Effect against A beta(1–42) oligomer-induced cell damage and blood-brain barrier disruption in an in vitro bEnd.3 endothelial model. Plos One 9(11):8
Zhang A, Chen B, Mu X, Li R, Zheng P, Zhao Y, Chen H, Jin M (2009) Identification and characterization of a novel protective antigen, enolase of Streptococcus suis serotype 2. Vaccine 27(9):1348–1353
Zhang H, Ma Z, Li Y, Zheng J, Yi L, Fan H, Lu C (2013) Identification of a novel collagen type capital I-binding protein from Streptococcus suis serotype 2. Vet J 197(2):406–414
Zhu C, Ghabriel MN, Blumbergs PC, Reilly PL, Manavis J, Youssef J, Hatami S, Finnie JW (2001) Clostridium perfringens prototoxin-induced alteration of endothelial barrier antigen (EBA) immunoreactivity at the blood-brain barrier (BBB). Exp Neurol 169(1):72–82
Acknowledgments
This study was supported by the National Natural Science Foundation of China (31302093, 31272581), the Fundamental Research Funds for the Central Universities of Nanjing Agricultural University, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the National Basic Research Program (973) of China (2012CB518804), the PhD programs of the Foundation of the Ministry of Education of China (20130097120024), the Natural Science Foundation of Jiangsu Province (BK20130676), and the Jiangsu Province Science and Technology Support Program (BE2013433).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
We confirmed that all experiments were performed in accordance with the relevant guidelines and regulations of the Science and Technology Agency of Jiangsu Province.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Ma Zhe and Peng Jie contributed equally to this work.
Electronic supplementary material
ESM 1
(PDF 497 kb)
Rights and permissions
About this article

Cite this article
Zhe, M., Jie, P., Hui, Z. et al. SILAC and LC-MS/MS identification of Streptococcus equi ssp. zooepidemicus proteins that contribute to mouse brain microvascular endothelial cell infection. Appl Microbiol Biotechnol 100, 7125–7136 (2016). https://doi.org/10.1007/s00253-016-7579-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7579-4